
Abstract
Background
Post-ischemic inflammation is a crucial component in stroke pathology in the early phase of cerebral ischemia–reperfusion (I/R) injury. Inflammation caused by microglia, astrocytes, and necrotic cells, produces pro-inflammatory mediators and exacerbates cerebral I/R injury. This study evaluated the effects of the Alpinia oxyphylla Miq [Yi Zhi Ren (YZR)] extract on cerebral infarction at 1 day after 90 min of transient middle cerebral artery occlusion (MCAo) and investigated the molecular mechanisms underlying the regulation of c-Jun N-terminal kinase (JNK)-mediated inflammatory cascades in the penumbral cortex. Rats were intraperitoneally injected with the YZR extract at the doses of 0.2 g/kg (YZR-0.2 g), 0.4 g/kg (YZR-0.4 g), or 0.8 g/kg (YZR-0.8 g) at MCAo onset.
Results
YZR-0.4 g and YZR-0.8 g treatments markedly reduced cerebral infarction, attenuated neurological deficits, and significantly downregulated the expression of phospho-apoptosis signal-regulating kinase 1 (p-ASK1)/ASK1, tumor necrosis factor receptor-associated factor 3 (TRAF3), TRAF3-interacting JNK-activating modulator (T3JAM), ionized calcium-binding adapter molecule 1 (Iba1), p-JNK/JNK, inducible nitric oxide synthase, cyclooxygenase-2, tumor necrosis factor-α, toll-like receptor 4 (TLR4), glial fibrillary acidic protein (GFAP), nuclear factor-kappa B (NF-κB), and interleukin-6 in the penumbral cortex at 1 day after reperfusion. SP600125 (SP), a selective JNK inhibitor, had the same effects. Furthermore, Iba1- and GFAP-positive cells were colocalized with TLR4, and colocalization of GFAP-positive cells was found with NF-κB in the nuclei.
Conclusion
YZR-0.4 g and YZR-0.8 g treatments exerted beneficial effects on cerebral ischemic injury by downregulating JNK-mediated signaling in the peri-infarct cortex. Moreover, the anti-infarction effects of YZR extract treatments were partially attributed to the downregulation of JNK-mediated TLR4/T3JAM- and ASK1-related inflammatory signaling pathways in the penumbral cortex at 1 day after reperfusion.
Similar content being viewed by others

Background
Post-ischemic inflammation is a crucial component in stroke pathology in the early phase of cerebral ischemia–reperfusion (I/R) injury [1]. The increased production of free radicals and the consequent induction of oxidative stress in the ischemic area initiate inflammatory responses [2]. Generally, inflammation is caused by microglia, astrocytes, and necrotic cells, producing pro-inflammatory mediators and aggravating cerebral I/R injury [3]. In the pathological process of cerebral ischemia, toll-like receptor (TLR)-mediated signaling initiates inflammatory cascades and is closely related to the development of cerebral infarction [4].
TLRs play an important role in the innate immune response. TLR4, a member of the TLR family, is expressed on microglia and astrocytes in the ischemic area in cerebral I/R injury [5]. During brain ischemic insult, activated microglia and reactive astrocytes predominantly express TLR4, which recognizes damage-associated molecular patterns (DAMPs), subsequently triggering downstream cascades through myeloid differentiation primary response gene 88 (MyD88)-dependent and toll/interleukin (IL)-1 receptor homology domain-containing adaptor-inducing interferon-β (TRIF)-dependent signaling pathways [6, 7]. These two pathways result in the activation of transcription factor nuclear factor-kappa B (NF-κB), which stimulates the production of pro-inflammatory mediators [6, 8]. The expression of TLR4, ionized calcium-binding adapter molecule 1 (Iba1; a marker of microglia), and glial fibrillary acidic protein (GFAP; a marker of astrocytes) is markedly increased in the ischemic area at 24 h after transient focal cerebral ischemia [9]. In MyD88-dependent signaling, MyD88 interacts with tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), subsequently activating two pathways involving NF-κB- and mitogen-activated protein kinase (MAPK), which includes extracellular signal-regulated kinase 1/2, c-Jun N-terminal kinase (JNK), and p38 MAPK [10, 11]. In TRIF-dependent signaling, TRIF binds to TRIF-related adaptor molecule (TRAM), subsequently activating TRAF3 and resulting in the activation of interferon-β expression [12, 13]. Studies have reported that TRAF3-interacting JNK-activating modulator (T3JAM) (also named TRAF3IP3), a coiled-coil membrane protein, interacts with TRAF3 in the cytosol, subsequently triggering the activation of JNK-mediated signaling and further amplifying TLR4-mediated signaling [14, 15]. In the early phase of cerebral ischemia, oxidative stress triggers the activation of apoptosis signal-regulating kinase 1 (ASK1), which activates downstream MAPK kinase (MKK) 4/JNK signaling cascades, leading to the initiation of apoptosis and inflammation in the ischemic area [16, 17]. JNK signaling plays a crucial role in modulation of multiple cellular activities, including proliferation, differentiation, inflammation, and apoptosis [18]. Furthermore, JNK is considered a major stress-activated protein kinase and a promising candidate for activating microglia, and it induces neuroinflammation in response to I/R injury in in vitro and in vivo models [19].
Generally, JNK pathways are activated by TLR4/MyD88/TRAF6-, TLR4/T3JAM-, and ASK1-mediated signaling stimuli during cerebral ischemia. Phosphorylated JNK positively regulates activated NF-κB, which translocates into the nucleus and induces the expression of genes encoding pro-inflammatory cytokines such as inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), TNF-α, and IL-6 [18, 20]. In post-ischemic inflammatory cascades, NF-κB-mediated iNOS production induces oxidative stress and then disrupts blood–brain barrier (BBB) integrity, aggravating cerebral infarction [21]. COX exists in two isoforms: COX-1 and COX-2. COX-1 is a constitutive enzyme and is expressed in most tissues, where COX-1-derived prostanoids provide stability to the internal environment. By contrast, COX-2 is the inducible isoform; it is highly expressed in the cerebral ischemic area and consequently promotes microglia activation, thereby enlarging the cerebral infarct area [22, 23]. Elevated TNF-α expression causes increased disruption of the BBB integrity and stimulates cytotoxic iNOS production by microglia and astrocytes. In addition, TNF-α, in turn, could induce the production of NF-κB, leading to the augmentation of the inflammatory response and exacerbation of brain injury [12, 21, 24]. IL-6 that is mainly produced by activated microglia contributes to BBB disruption and is closely associated with neuronal damage in the ischemia penumbra during transient middle cerebral artery occlusion (MCAo). Furthermore, IL-6 shows peak expression in the ischemic penumbra at 24 h after MCAo [21, 25].
Alpinia oxyphylla Miq, commonly known as Yi Zhi Ren (YZR), is a traditional Chinese herb that has been widely used to treat intestinal disorders, urosis, diuresis, ulceration, hypertension, dementia, and cerebrovascular disorders [26, 27]. Studies have reported that YZR attenuates memory impairment through the inhibition of neuroinflammation, amyloid-β deposition, and p-tau expression in the cortex and hippocampus in mice with lipopolysaccharide-induced Alzheimer’s disease [28]. In addition, YZR protects against ischemia-induced memory deficits by promoting hippocampal cornu ammonis neuronal survival after transient global cerebral ischemia [29]. Protocatechuic acid, chrysin and nootkatone are the main active components of YZR [30, 31]. Intraperitoneal (IP) administration of protocatechuic acid (5 mg/kg) contributes to the suppression of oxidative stress by increasing superoxide dismutase and glutathione peroxidase activities and decreasing malondialdehyde expression in the brain in aged rats [32]. Chrysin attenuates cerebral I/R injury by downregulating IL-6, TNF-α, NK-κB, COX-2, and iNOS expression in the ischemic area after transient focal [33] and global [34] cerebral ischemia. Intracerebroventricular (ICV) injections of nootkatone (0.02 mg/kg and 0.2 mg/kg) provide beneficial effects against β-amyloid-induced cognitive impairment by upregulating anti-oxidative and anti-acetylcholinesterase activities in the hippocampus [35]. On the basis of the aforementioned findings, we speculate that the YZR extract protects against cerebral I/R injury by modulating JNK-mediated signaling in the acute phase of transient focal cerebral ischemia. Therefore, in this study the effects of the YZR extract on cerebral infarction were assessed, and the potential mechanism through which the extract modulates JNK-mediated inflammatory signaling in the penumbral cortex at 1 day after transient MCAo was verified.
Methods
Experimental animals
A total of 151 adult male Sprague–Dawley rats, 8–9 weeks old and weighting 290–330 g (purchased from BioLASCO Co., Ltd., Yilan, Taiwan), were used in the present study. They were housed under the conditions of controlled temperature (22–24 °C), humidity (50–55%), and lighting cycle (12/12-h light/dark). All experimental procedures were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of China Medical University (Permit Number: CMUIACUC-2019-312). Twenty three rats died during the experiments and 7 rats subjected to incomplete MCAo were excluded from this study.
YZR extract preparation
YZR extract powder was obtained from Chuang Song Zong Pharmaceutical Co., Ltd. (Kaohsiung, Taiwan). Two grams of YZR extract powder was dissolved with 8 mL of double-distilled water. The concentration measurements were performed as described previously [36]. The final concentration of the YZR aqueous extract was maintained at 0.1 g/mL.
High-performance liquid chromatography assessment of the indicators of the YZR extract
The standards comprising protocatechuic acid [purity: 99.9%, National Institutes for Food and Drug Control (NIFDC), China], chrysin (purity: 100%, NIFDC, China) and nootkatone (purity: 99.5%, NATURE STANDARD, Shanghai, China) were precisely weighed and dissolved in absolute methanol to prepare standard solutions. Two grams of the YZR extract powder was dissolved in 100 mL of absolute methanol and the solution was then shaken using an ultrasonic cleaner at room temperature (RT) for 30 min. After filtration of the solution, the filtrate was collected as the sample solution. Subsequently, high-performance liquid chromatography (HPLC) measurements were conducted as described previously [37]. In brief, 20 μL of the standard or sample solution was injected into the Waters HPLC system (Waters Corp., Miford, MA, USA), which consists of the Waters 2690 Separations Module and Waters 2996 Photodiode Array Detector. The HPLC profile of the YZR extract was determined using a C18 column (Cosmosil 5C18-AR-II, 4.6 mm I.D. × 250 mm, 5 μm). The mobile phase consisted of water with 0.1% phosphoric acid (A) and acetonitrile with 0.1% phosphoric acid (B). In gradient elution processes, the proportion of mobile phase A was decreased from 93 to 30%, whereas the proportion of mobile phase B was increased from 7 to 70%. The flow-rate of the mobile phase was 1.0 mL/min, and the total run time was 85 min. The effluent was monitored by a photodiode array detector at 254 nm.
Transient middle cerebral artery occlusion
Transient MCAo was performed in the rats by using the intraluminal suture occlusion technique described previously [37]. In Brief, all rats were anesthetized with isoflurane (5% and 2% isoflurane for induction and maintenance, respectively). The rat’s head was fixed in the stereotaxic frame and a burr hold was drilled into the skull (2.0 mm posterior and 2.5 mm lateral to the right from the bregma) to expose the distal territory of the middle cerebral artery (MCA). A 3-cm midline neck incision was made to expose the right external carotid artery (ECA) and internal carotid artery (ICA). A 3–0 nylon suture with a heat-blunted tip was carefully inserted into the lumen of the right ICA through the stump of the ECA and was advanced up to the origin of the MCA. After 90 min of MCAo, the suture was gently withdrawn to permit reperfusion. Blood flow in the MCA was monitored using a Laser-Doppler flowmetry (DRT4, Moor Instruments Inc., Wilmington, USA) in the MCAo procedure. Successful establishment of MCAo was defined as a reduction in the MCA blood flow to 20–30% of baseline in the ischemic period and an increase in MCA blood flow to 60% of baseline in the reperfusion period. The rats subjected to incomplete MCAo were excluded for further study.
Assessment of neurological function
Modified neurological severity score (mNSS) tests were performed to determine neurological function at 1 day after reperfusion. The mNSS tests listed in Table 1 are divided into four components: motor, sensory, beam balance, and reflex tests, as described previously [38]. The neurological deficit scores (NDSs) for each rat were obtained using the mNSS, which ranges from 0 to 18. A normal score is 0, and the maximal deficit score is 18.
Experiment A
Grouping
The rats were randomly divided into five groups (n = 5–6): Sham, Control, YZR-0.2 g, YZR-0.4 g, and YZR-0.8 g groups. The rats in the YZR-0.2 g, YZR-0.4 g, and YZR-0.8 g groups were IP injected with the YZR extract at the doses of 0.2, 0.4, and 0.8 g/kg, respectively, after the initiation of MCAo. After 90 min of ischemia followed by 1 day of reperfusion, the rats were euthanized by CO2 inhalation, and their brains were immediately removed. The rats in the Control group were subjected to the identical protocols of the YZR-0.8 g group, except that the rats were injected with normal saline instead of the YZR extract. The rats in the Sham group were subjected to the identical protocols of the Control group, except that the MCA was not occluded.
Measurement of cerebral infarction
After 1 day of reperfusion, the rats were euthanized, and their brains were immediately removed. The fresh brains were placed at − 20 °C for 5 min and were then cut into six coronal sections of 2-mm thickness. The brain sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Merck, Germany) at 37 °C for 5 min and consequently fixed in 4% paraformaldehyde (PFA) solution at RT overnight. In each TTC-stained brain section, the white portion in the ipsilateral hemisphere indicates the infarcted area, whereas the deep red portion indicates the healthy region. The percentage of cerebral infarct areas was determined by dividing the infarct area by the total coronal sectional area; the calculation was conducted using ImageJ software (NIH, MD, USA).
Experiment B
Grouping
The rats were randomly divided into five groups (n = 5): Sham, Control, YZR-0.2 g, YZR-0.4 g, and YZR-0.8 g groups. The rats in these groups were subjected to protocols identical to those for Experiment A.
Western blot analysis
After 1 day of reperfusion, the rats were euthanized, and their brains were immediately removed. The right penumbral cortices of the brain samples were divided into cytosolic and mitochondrial fractions, as described previously [37]. Equal amounts of protein samples (15 μg/lane) were loaded and then separated through 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred onto nitrocellulose (NC) membranes, and incubated with primary antibodies listed in Table 2 at 4 °C overnight. Subsequently, appropriate secondary antibodies (1:5000 dilution) (Table 2) were used to detect the primary antibodies presented in the NC membranes, with incubation at RT for 1 h. The images were scanned using a luminescence image analyzer (LAS-3000, FujiFilm), and the data were analyzed using ImageJ software.
Experiment C
Grouping
The rats were randomly divided into four groups (n = 5): D+Sham, D+Control, D+YZR-0.8 g, and SP groups. The rats in the SP group were subjected to the Control group protocols identical to those for Experiment A, but they were given an ICV injection of SP600125, a selective JNK inhibitor, 20 min before MCAo. The rats in the D+Sham, D+Control, and D+YZR-0.8 g groups were subjected to protocols identical to those for the Sham, Control, and YZR-0.8 g groups, as described in Experiment A, respectively; however, all of them were given ICV injections of 1% dimethyl sulfoxide (DMSO) 20 min before MCAo.
Intracerebroventricular injection of SP600125 or 1% DMSO
The rats were maintained under anesthesia with 2% isoflurane, and a burr hole located 0.8 mm posterior to the bregma and 1.5 mm lateral to the midline was drilled into the right side of the skull. The rats were given an ICV injection of 10 μL of SP600125 solution (2 mM in DMSO, ab120065 abcam) or 1% DMSO solution. The solution was injected at a depth of 3.5 mm from the skull using a 10-µL Hamilton syringe (Hamilton Company, Reno, NV, USA).
Measurement of neurological function and cerebral infarction
One day after reperfusion, the rats were subjected to mNSS tests and were subsequently euthanized for measurement of cerebral infarction. The cerebral infarction measurement procedures are the same as those in Experiment A.
Experiment D
Grouping
The rats were randomly divided into four groups (n = 5): D+Sham, D+Control, D+YZR-0.8 g, and SP groups. The rats in these groups were subjected to protocols identical to those for Experiment C.
Western blot analysis
One day after transient MCAo, the rats were euthanized, and their brains were immediately removed for Western blot analysis of p-ASK1, ASK1, Iba1, T3JAM, TRAF3, p-JNK, JNK, iNOS, COX-2, and TNF-α (listed in Table 2) expression. The procedures of Western blot assay were the same as those for Experiment B.
Experiment E
Grouping
The rats were randomly divided into six groups (n = 5): Sham, Control, YZR-0.2 g, YZR-0.4 g, YZR-0.8 g, and SP groups. The rats in these groups were subjected to protocols identical to those for Experiment A and C.
Immunohistochemical analysis
After completing neurological examinations at 1 day of reperfusion, the rats were euthanized. They were transcardially perfused with cold 0.9% saline, and their brains were removed quickly. Subsequently, the brains were embedded in optimal cutting temperature compound in small aluminum foil paper boxes, frozen at − 35 ± 5 °C using dry ice, and cut into 15 µm thick coronal brain sections using a cryostat (Leica CM3050 S, Wetzlar, Germany), as previously described [36]. The brain sections were rinsed in phosphate buffered saline/Tween 20 (0.01%; PBST) and post-fixed with 4% PFA at RT for 15 min. After washing with PBST, the brain sections were immersed with 3% hydrogen peroxidase/methanol for 20 min to inhibit endogenous peroxidase activity and then incubated with Iba1, GFAP, TLR4, NF-κB, iNOS, and IL-6 (listed in Table 2) at 4 °C overnight. The brain sections were subsequently stained with appropriate secondary antibodies (Table 2) and labeled with avidin–biotin–peroxidase complexes (Leica Biosystems Newcastle Ltd., UK). Immunopositive cells in the selected penumbral cortex were detected using a light microscope (Axioskop 40, Zeiss). The negative control slides from the Control group were stained without the primary antibodies.
Immunofluorescence staining
The brain sections adjacent to those used in immunohistochemical (IHC) analysis were used for immunofluorescence (IF) staining. The brain sections were rinsed in PBST and post-fixed with 4% PFA at RT for 15 min. After washing with PBST, the brain sections were blocked with a blocking buffer containing 1% bovine serum albumin in PBST at RT for 1 h, and the sections were subsequently incubated with mouse and rabbit primary antibodies (listed in Table 2) at 4 °C overnight. After being washed with PBST for 5 min, the brain sections were incubated with anti-mouse and anti-rabbit immunoglobulin G secondary antibodies (Table 2) at 37 °C for 1.5 h and then counterstained with 4',6-diamidino-2-phenylindole (DAPI, ab4139 abcam) at RT for 3 min, as described previously [36]. Immunopositive cells in the selected penumbral cortex were evaluated using a fluorescence microscope (CKX53, Olympus, Tokyo, Japan). In addition, the percentage of activated forms of Iba1 (GFAP) was evaluated by dividing activated forms of Iba1 (GFAP)-positive cells by total Iba1 (GFAP)-positive cells in the selected penumbral cortex.
Statistical analysis
The normality test was performed on all data by Kolmogorov–Smirnov test with a significance level of 0.05. All numeric data, except for neurological function scores, follow the normal distribution (P > 0.05). The data acquired from cerebral infarction, Western blot, IHC, and IF analyses among the experimental groups were evaluated using one-way analysis of variance (ANOVA) followed by Bonferroni post-hoc test, and the data were expressed as mean ± standard deviation. The data acquired from neurological tests among the experimental groups were analyzed using Kruskal–Wallis one-way ANOVA, and the data were expressed as median (min−max). P values less than 0.05 were considered as statistically significant.
Results
HPLC analysis of the YZR extract
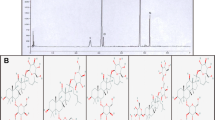
After the detection of the standard and YZR extract solutions using HPLC analysis, the retention times of protocatechuic acid, chrysin, and nootkatone were 10.0, 58.7, and 75.0 min, respectively. The contents of protocatechuic acid, chrysin, and nootkatone in the YZR extract were 0.064, 0.017, and 0.083 mg/g, respectively (Fig. 1A and B).

HPLC chromatograms of protocatechuic acid, chrysin, and nootkatone in the YZR extract. HPLC chromatograms A and B denote the standard and YZR extract solutions, respectively. AU: absorbance unit
Effects of YZR extract treatments on cerebral infarction
After 1 day of reperfusion, TTC staining revealed that the percentage of cerebral infarct areas was markedly higher in the Control group than in the Sham group (P < 0.05) and was markedly lower in the YZR-0.4 g and YZR-0.8 g groups than in the Control group (both P < 0.05; Fig. 2A and B) (F4,21 = 45.700, P = 0.000). The percentage of cerebral infarct areas did not differ significantly between the Control and YZR-0.2 g groups (P > 0.05).

Effects of YZR-0.4 g and YZR-0.8 g treatments on cerebral infarction at 1 day after reperfusion. A Representative TTC staining images (S1–S6) of the Sham, Control, YZR-0.2 g, YZR-0.4 g, and YZR-0.8 g groups (n = 5–6) showed that normal tissues turned deep red, whereas infarcted tissues remained pale white. B The percentage of cerebral infarct areas was measured among the experimental groups at 1 day after reperfusion. Data are expressed as mean ± standard deviation. *P < 0.05 vs. the Sham group; #P < 0.05 vs. the Control group. Scale bar represents 1 cm
Effects of YZR extract treatments on neurological function
The mNSS tests revealed that the NDSs of motor, sensory, and beam balance functions were markedly higher in the Control group than in the Sham group (all P < 0.05) and were markedly lower in the YZR-0.4 g and YZR-0.8 g groups than in the Control groups (all P < 0.05; Table 3). However, the NDSs of motor, sensory, and beam balance functions did not differ significantly between the Control and YZR-0.2 g groups (P > 0.05). In addition, the rats in the experimental groups did not lose the reflex function at 1 day after reperfusion.
Effects of YZR extract treatments on the cytosolic expression of p-ASK1, ASK1, MyD88, TRAF6, T3JAM, TRAF3, Iba1, p-JNK, JNK, p-p38 MAPK, p38 MAPK, iNOS, COX-2, and TNF-α
The cytosolic expression of p-ASK1/ASK1, T3JAM/actin, TRAF3/actin, Iba1/actin, p-JNK/JNK, iNOS/actin, COX-2/actin, and TNF-α/actin in the penumbral cortex was markedly higher in the Control group (2.3-, 2.4-, 1.7-, 2.2-, 1.7-, 2.6-, 2.0-, and 1.8-fold, respectively) than in the Sham group (all P < 0.05) and was markedly lower in the YZR-0.4 g (0.5-, 0.5-, 0.7-, 0.6-, 0.6-, 0.4-, 0.5-, and 0.6-fold, respectively) and YZR-0.8 g (0.5-, 0.5-, 0.6-, 0.6-, 0.7-, 0.4-, 0.5-, and 0.6-fold, respectively) groups than in the Control group at 1 day after reperfusion (all P < 0.05; Figs. 3A, B, E–G, 4A, B, and D–F) [(F4,20 = 12.359, P = 0.000), (F4,20 = 27.241, P = 0.000), (F4,20 = 19.613, P = 0.000), (F4,20 = 21.942, P = 0.000), (F4,20 = 26.072, P = 0.000), (F4,20 = 26.183, P = 0.000), (F4,20 = 48.012, P = 0.000), and (F4,20 = 52.574, P = 0.000), respectively]. However, the cytosolic expression of these proteins did not differ significantly between the Control and YZR-0.2 g groups (P > 0.05). In addition, the cytosolic expression of MyD88/actin, TRAF6/actin, and p-p38 MAPK/p38 MAPK in the penumbral cortex did not differ significantly among the experimental groups (P > 0.05; Figs. 3A, C, D, 4A and C) [(F4,20 = 0.456, P = 0.767), (F4,20 = 0.325, P = 0.858), and (F4,20 = 1.788, P = 0.171), respectively].

Effects of YZR-0.4 g and YZR-0.8 g treatments on cytosolic p-ASK1, ASK, MyD88, TRAF6, T3JAM, TRAF3, and Iba1 expression in the penumbral cortex. A Representative Western blot images showed cytosolic p-ASK1, ASK, MyD88, TRAF6, T3JAM, TRAF3, and Iba1 expression in the penumbral cortex in the Sham, Control, YZR-0.2 g, YZR-0.4 g, and YZR-0.8 g groups (n = 5) at 1 day after reperfusion. Actin was used as an internal control in the Western blot analysis. The ratios of B p-ASK1/ASK1, C MyD88/actin, D TRAF6/actin, E T3JAM/actin, F TRAF3/actin, and G Iba1/actin were measured in the penumbral cortex among the experimental groups. cyto, cytosolic fraction. *P < 0.05 vs. the Sham group; #P < 0.05 vs. the Control group

Effects of YZR-0.4 g and YZR-0.8 g treatments on cytosolic p-JNK, JNK, p-p38 MAPK, p38 MAPK, iNOS, COX-2, and TNF-α expression in the penumbral cortex. A Representative Western blot images showed cytosolic p-JNK, JNK, p-p38 MAPK, p38 MAPK, iNOS, COX-2, and TNF-α expression in the penumbral cortex in the Sham, Control, YZR-0.2 g, YZR-0.4 g, and YZR-0.8 g groups (n = 5) at 1 day after reperfusion. The ratios of B p-JNK/JNK, C p-p38 MAPK/p38 MAPK, D iNOS/actin, E COX-2/actin, and F TNF-α/actin were measured in the penumbral cortex among the experimental groups. *P < 0.05 vs. the Sham group; #P < 0.05 vs. the Control group
Effects of D+YZR-0.8 g and SP treatments on cerebral infarction
The percentage of cerebral infarct areas was markedly higher in the D+Control group than in the D+Sham group (P < 0.05) and was markedly lower in the D+YZR-0.8 g and SP groups than in the D+Control group at 1 day after reperfusion (both P < 0.05; Fig. 5A and B) (F3,16 = 128.346, P = 0.000).

Effects of D+YZR-0.8 g and SP treatments on cerebral infarct areas at 1 day after reperfusion. A Representative TTC staining images (S1–S6) in the D+Sham, D+Control, D+YZR-0.8 g, and SP groups (n = 5) showed cerebral infarction. Scale bar represents 1 cm. B The percentage of cerebral infarct areas was measured among the experimental groups at 1 day after reperfusion. *P < 0.05 vs. the D+Sham group; #P < 0.05 vs. the D+Control group

Effects of D+YZR-0.8 g and SP treatments on cytosolic p-ASK1, ASK1, T3JAM, TRAF3, and Iba1 expression in the penumbral cortex. A Representative Western blot images showed cytosolic p-ASK1, ASK1, T3JAM, TRAF3, and Iba1 expression in the penumbral cortex in the D+Sham, D+Control, D+YZR-0.8 g, and SP groups (n = 5) at 1 day after reperfusion. The ratios of B p-ASK1/ASK1, C T3JAM/actin, D TRAF3/actin, and E Iba1/actin were measured in the penumbral cortex among the experimental groups. *P < 0.05 vs. the D+Sham group; #P < 0.05 vs. the D+Control group

Effects of D+YZR-0.8 g and SP treatments on cytosolic p-JNK, JNK, iNOS, COX-2, and TNF-α expression in the penumbral cortex. A Representative Western blot images showed cytosolic p-JNK, JNK, iNOS, COX-2, and TNF-α expression in the penumbral cortex in the D+Sham, D+Control, D+YZR-0.8 g, and SP groups (n = 5) at 1 day after reperfusion. The ratios of B p-JNK/JNK, C iNOS/actin, D COX-2/actin, and E TNF-α/actin were measured in the penumbral cortex among the experimental groups. *P < 0.05 vs. the D+Sham group; #P < 0.05 vs. the D+Control group
Effects of D+YZR-0.8 g and SP treatments on neurological function
The NDSs of motor, sensory, and beam balance functions were markedly higher in the D+Control group than in the D+Sham group (all P < 0.05) and were markedly lower in the D+YZR-0.8 g and SP groups than in the D+Control groups (all P < 0.05; Table 4).
Effects of D+YZR-0.8 g and SP treatments on the cytosolic expression of p-ASK1, ASK1, T3JAM, TRAF3, Iba1, p-JNK, JNK, iNOS, COX-2, and TNF-α
The cytosolic expression of p-ASK1/ASK1, T3JAM/actin, TRAF3/actin, Iba1/actin, p-JNK/JNK, iNOS/actin, COX-2/actin, and TNF-α/actin in the penumbral cortex was markedly higher in the D+Control group (1.9-, 2.3-, 1.9-, 2.3-, 1.9-, 2.7-, 2.0-, and 1.8-fold, respectively) than in the D+Sham group (all P < 0.05) and was markedly lower in the D+YZR-0.8 g (0.6-, 0.5-, 0.6-, 0.5-, 0.5-, 0.4-, 0.6-, and 0.6-fold, respectively) and SP (0.6-, 0.4-, 0.5-, 0.5-, 0.6-, 0.4-, 0.5-, and 0.6-fold, respectively) groups than in the D+Control group at 1 day after reperfusion (all P < 0.05; Figs. 6A–E and 7A–E) [(F3,16 = 7.963, P = 0.002), (F3,16 = 32.593, P = 0.000), (F3,16 = 19.315, P = 0.000), (F3,16 = 42.457, P = 0.000), (F3,16 = 27.482, P = 0.000), (F3,16 = 37.871, P = 0.000), (F3,16 = 20.725, P = 0.000), and (F3,16 = 10.819, P = 0.000), respectively].
Effects of YZR extract and SP treatments on the expression of Iba1, TLR4, GFAP, NF-κB, iNOS, and IL-6
In the present study, the immunopositive cells were detected in the selected penumbral cortex (Fig. 8B). The numbers of Iba1-, TLR4-, GFAP-, NF-κB-, iNOS-, and IL-6-positive cells in the penumbral cortex were markedly higher in the Control group than in the Sham group (all P < 0.05) and were markedly lower in the YZR-0.4 g, YZR-0.8 g, and SP groups than in the Control group at 1 day after reperfusion (all P < 0.05; Figs. 9A–D, 10A–D, and 11A–D) [(F5,24 = 38.712, P = 0.000), (F5,24 = 34.085, P = 0.000), (F5,24 = 33.126, P = 0.000), (F5,24 = 126.919, P = 0.000), (F5,24 = 46.320, P = 0.000), and (F5,24 = 24.066, P = 0.000), respectively]. However, no significant differences were found in immunopositive cell numbers between the Control and YZR-0.2 g groups (P > 0.05).

Effects of YZR-0.4 g and YZR-0.8 g treatments on neuronal protection at 1 day after reperfusion. A Representative images showed NeuN expression in the penumbral cortex in the Sham, Control, YZR-0.2 g, YZR-0.4 g, YZR-0.8 g, and SP groups (n = 5) at 1 day after reperfusion. B A representative TTC staining image showed a coronal brain section. The dashed line square indicates the region where immunopositive cells were counted. PC: penumbral cortex. Dashed line square = 1 mm2. C The bar graphs showed the number of NeuN-positive cells in the penumbral cortex among the experimental groups. *P < 0.05 vs. the Sham group; #P < 0.05 vs. the Control group. Arrows in A indicate NeuN-positive cells. Scale bars represent A 50 μm and B 2 mm

Effects of YZR-0.4 g, YZR-0.8 g, and SP treatments on Iba1 and TLR4 expression in the penumbral cortex. Representative images showed A Iba1 and B TLR4 expression in the penumbral cortex in the Sham, Control, YZR-0.2 g, YZR-0.4 g, YZR-0.8 g, and SP groups (n = 5) at 1 day after reperfusion. The bar graphs showed the numbers of C Iba1- and D TLR4-positive cells in the penumbral cortex among the experimental groups. *P < 0.05 vs. the Sham group; #P < 0.05 vs. the Control group. Arrows in A and B indicate Iba1- and TLR4-positive cells, respectively. Scale bar represents 40 μm

Effects of YZR-0.4 g, YZR-0.8 g, and SP treatments on GFAP and NF-κB expression in the penumbral cortex. Representative images showed A GFAP and B NF-κB expression in the penumbral cortex in the Sham, Control, YZR-0.2 g, YZR-0.4 g, YZR-0.8 g, and SP groups (n = 5) at 1 day after reperfusion. The bar graphs showed the numbers of C GFAP- and D NF-κB-positive cells in the penumbral cortex among the experimental groups. *P < 0.05 vs. the Sham group; #P < 0.05 vs. the Control group. Arrows in A and B indicate GFAP- and NF-κB-positive cells, respectively. Scale bar represents 40 μm

Effects of YZR-0.4 g, YZR-0.8 g, and SP treatments on iNOS and IL-6 expression in the penumbral cortex. Representative images showed A iNOS and B IL-6 expression in the penumbral cortex in the Sham, Control, YZR-0.2 g, YZR-0.4 g, YZR-0.8 g, and SP groups (n = 5) at 1 day after reperfusion. The bar graphs showed the numbers of C iNOS- and D IL-6-positive cells in the penumbral cortex among the experimental groups. *P < 0.05 vs. the Sham group; #P < 0.05 vs. the Control group. Arrows in A and B indicate iNOS- and IL-6-positive cells, respectively. Scale bar represents 40 μm
Expression of neuronal nuclei-, TLR4/Iba1-, TLR4/GFAP-, and NF-κB/GFAP-positive cells, and activated forms of Iba1 and GFAP in the penumbral cortex
TLR4-, Iba1-, GFAP-, and NF-κB-positive cells were predominantly expressed in the selected penumbral cortex (Figs. 12A-1, A-2, B-1, B-2, C-1, and C-2). Iba1- and GFAP-positive cells colocalized with TLR4 (Fig. 12A-3 and B-3). GFAP-positive cells colocalized with NF-κB (Fig. 12C-3). NF-κB double labeling with GFAP was detected in the nucleus (Fig. 12C-4 and C-5). The numbers of TLR4/Iba1-, TLR4/GFAP-, and NF-κB/GFAP-positive cells in the penumbral cortex were markedly higher in the Control group than in the Sham group (all P < 0.05) and were markedly lower in the YZR-0.4 g, YZR-0.8 g, and SP groups than in the Control group at 1 day after reperfusion (all P < 0.05; 12F–H) [(F5,24 = 39.640, P = 0.000), (F5,24 = 34.769, P = 0.000), and (F5,24 = 113.297, P = 0.000), respectively]. By contrast, the number of neuronal nuclei (NeuN)-positive cells in the penumbral cortex was markedly lower in the Control group than in the Sham group (P < 0.05) and was markedly higher in the YZR-0.4 g, YZR-0.8 g, and SP groups than in the Control group at 1 day after reperfusion (P < 0.05; Fig. 8A and C) (F5,24 = 47.439, P = 0.000). However, no significant differences were found in immunopositive cell numbers between the Control and YZR-0.2 g groups (P > 0.05). The activated forms of Iba1 (activated microglia)- and GFAP (reactive astrocytes)-positive cells are morphologically characterized by swollen processes with amoeboid cell bodies and hypertrophy of main cellular processes, respectively (Fig. 12A-2 and C-2). The percentages of activated forms of Iba1 and GFAP in the penumbral cortex were markedly higher in the Control group than in the Sham group (both P < 0.05), and were markedly lower in the YZR-0.4 g, YZR-0.8 g, and SP groups (all P < 0.05; Fig. 12D and E) [(F5,24 = 37.792, P = 0.000) and (F5,24 = 77.384, P = 0.000), respectively]. No significant differences were found in the percentages of activated forms of Iba1 and GFAP between the Control and YZR-0.2 g groups (P > 0.05).

Expression of TLR4/Iba1, TLR4/GFAP, and NF-κB/GFAP, and activated forms of Iba1 and GFAP in the penumbral cortex. Arrows in A-1, A-2, B-1, B-2, C-1, C-2, and C-4 indicate TLR4-, Iba1-, TLR4-, GFAP-, NF-κB-, GFAP-, and DAPI-positive cells, respectively. Arrows in A-3, B-3, and C-3 indicate TLR4/Iba1, TLR4/GFAP, and NF-κB/GFAP double-labeled cells, respectively. Arrows in C-5 indicate NF-κB/GFAP-positive cells labeled with DAPI. Arrowheads in A-2 and C-2 indicate activated forms of Iba1 and GFAP, respectively. The bar graphs showed the percentages of activated forms of D Iba1 and E GFAP in the penumbral cortex among the experimental groups (n = 5) at 1 day after reperfusion. In addition, the bar graphs showed the numbers of F TLR4/Iba1- G TLR4/GFAP-, and H NF-κB/GFAP-positive cells in the penumbral cortex among the experimental groups (n = 5). *P < 0.05 vs. the Sham group; #P < 0.05 vs. the Control group. Scale bars in B-3 and C-5 represent 50 μm
Discussion
Post-ischemic inflammation, one of the main pathological features in the early stage of cerebral I/R injury, contributes to the release of pro-inflammatory mediators, which exacerbate cerebral infarction [39]. Studies have reported that TLR4 expressed on microglia and astrocytes plays a crucial role in the generation of pro-inflammatory cytokines in the initial stage of cerebral I/R injury, whereas pharmacological interventions alleviate cerebral infarction by inhibiting TLR4-mediated microglial and astrocytic activation in the ischemic area in the acute phase of transient MCAo [40]. In the present study, the TTC-stained brain sections revealed that cerebral infarction was predominantly distributed in the right cerebral hemisphere involving the cortex and striatum at 1 day after 90 min of MCAo. However, the YZR extract administered at doses of 0.4 g/kg (YZR-0.4 g) and 0.8 g/kg (YZR-0.8 g), but not 0.2 g/kg (YZR-0.2 g), significantly reduced infarct areas and effectively alleviated behavioral deficits (including motor, sensory, and beam balance functions). TTC staining results also revealed that YZR-0.8 g treatment could fully reverse cortical infarction in cerebral I/R injury. In addition, our Western blot, IHC, and IF results revealed that the expression of Iba1 (a marker of microglia), TLR4, and GFAP (a marker of reactive astrocytes) was markedly increased in the penumbral cortex at 1 day after reperfusion. In cerebral ischemia, the transformation of microglia morphology into an amoeboid cell shape in the ischemic area is widely utilized to determine microglia activation [41]. Moreover, activated microglia and reactive astrocytes release inflammatory mediators in the ischemia area, leading to the exacerbation of cerebral infarction in the acute phase after cerebral ischemia [42]. Our results further revealed that amoeboid microglia and reactive astrocytes were predominantly expressed in the penumbral cortex, and activated microglia and reactive astrocytes were double labeled with TLR4, whereas YZR-0.4 g and YZR-0.8 g treatments effectively downregulated the increased expression of TLR4, TLR4/Iba1, and TLR4/GFAP, and inhibited microglial and astrocytic activation in the peri-infarct region. By contrast, the expression of NeuN (neuronal marker) was downregulated in the cortical penumbra, whereas YZR extract treatments effectively rescued cortical neurons in the peri-infarct zone. On the basis of these findings, we infer that YZR-0.4 g and YZR-0.8 g treatments effectively reduced cerebral infarct areas and alleviated neurological deficits at 1 day after reperfusion. Moreover, YZR extract treatments exert neuroprotective effects against cerebral infarction partially through the downregulation of TLR4-mediated inflammatory signaling in the acute phase of transient MCAo.
In the pathology of cerebral I/R injury, TLR4 that is mainly expressed in microglia and astrocytes recognizes DAMPs and subsequently elicits downstream inflammatory signaling cascades through MyD88- and TRIF-dependent pathways [3, 4]. In the MyD88-dependent pathway, TLR4 interacts with MyD88 and subsequently stimulates TRAF6, thereby triggering the activation of the downstream targets JNK, p38 MAPK, and NF-κB [3, 43, 44]. Moreover, JNK and p38 MAPK contribute to post-ischemic inflammation and are considered the upstream kinases of NF-κB [18]. In the TRIF-dependent pathway, TLR4 binds to TRIF by interacting with TRAM, and TLR4/TRIF signaling subsequently activates TRAF3. Furthermore, TRAF3 located on the cell membrane cooperates with T3JAM and then promotes the activation of JNK and TLR4, creating a vicious circle and amplifying TLR4-mediated inflammatory signaling [10, 13,14,15]. Previous studies have reported that TLR4-mediated signaling causes JNK, p38 MAPK, and NF-κB activation, which induces the production of pro-inflammatory factors, including iNOS, COX-2, TNF-α, and IL-6, in the ischemic area, further exaggerating BBB disruption and cerebral infarction. By contrast, pharmacological reduction of the aforementioned inflammatory mediators and cytokines effectively attenuates cerebral infarction at 1 day after MCAo [3, 8, 40, 45]. The current findings indicated that the expression levels of TRAF3, T3JAM, p-JNK, NF-κB, iNOS, COX-2, TNF-α, and IL-6 were markedly higher in the penumbral cortex. However, YZR-0.4 g and YZR-0.8 g treatments effectively reversed the increased expression of the aforementioned proteins but did not affect the expression of MyD88, TRAF6, and p-p38 MAPK in the penumbral cortex at 1 day after reperfusion. In addition, the IF assay revealed that reactive astrocytes colocalized with NF-κB located in the nucleus in the peri-infarct region, indicating the activation and translocation of NF-κB into the nucleus following cerebral I/R injury. In addition, YZR extract treatments effectively downregulated NF-κB activation in the nuclei of reactive astrocytes. The present results indicate that YZR-0.4 g and YZR-0.8 g treatments exert neuroprotective effects against cerebral I/R injury possibly by downregulating the TLR4-mediated TRAF3/T3JAM/JNK, but not MyD88/TRAF6/JNK (p38 MAPK), signaling pathway in the peri-infarct cortex. Furthermore, the effects of YZR extract treatments on cerebral infarction are partially due to the suppression of JNK/NF-κB-mediated iNOS, COX-2, TNF-α, and IL-6 expression in the penumbral cortex at 1 day after reperfusion.
JNK, a member of the MAPK family, is considered a major stress-responsive kinase and is closely involved in the induction of inflammation and apoptosis in the penumbra region after transient focal cerebral ischemia [19]. In the initial stage of cerebral ischemia, phosphorylated JNK induces NF-κB activation, which causes the production of excessive amounts of pro-inflammatory mediators in the ischemic area, subsequently exacerbating cerebral infarction. Thus, JNK plays a critical role in the regulation of post-ischemic inflammation [46]. In addition to JNK activation by TLR4/MyD88- and TLR4/T3JAM-mediated signaling, JNK is activated by ASK1 signaling in ischemic brain injury [16]. In ASK1/JNK signaling, ASK1 activates the downstream MKK4/MKK7-JNK signaling pathway in response to ischemia-induced oxidative stress after transient cerebral ischemia. Moreover, reactive oxygen species and the pro-inflammatory cytokine TNF-α induce ASK1 phosphorylation at Thr-845, which is required for ASK1 kinase activity [16, 47]. Related studies have reported that phosphorylated ASK1 (Thr-845) activates downstream JNK signaling and activated JNK then translocates into the nucleus and modulates stress-responsive transcription factors (such as NF-κB and activator protein 1), which induce gene transcription, resulting in inflammation and apoptosis in the ischemic area following cerebral I/R injury [17, 46]. In a previous study, increased activation of ASK1/JNK signaling in the penumbra worsened neurological deficits and exaggerated infarct size at 1 day after transient MCAo [48]. By contrast, pharmacological inhibition of ASK1/JNK signaling markedly reduced the cerebral infarct volume in acute perinatal hypoxic-ischemic cerebral injury [49]. Our Western blot findings revealed that the expression levels of p-ASK1/ASK1 and p-JNK/JNK ratios were markedly upregulated in the penumbral cortex, whereas YZR-0.4 g and YZR-0.8 g treatments effectively reduced the p-ASK1/ASK1 and p-JNK/JNK ratios at 1 day after reperfusion. On the basis of these results, we suggest that YZR extract treatments attenuate cerebral ischemic injury partially through the suppression of ASK1/JNK signaling activation in the peri-infarct cortex. Furthermore, the anti-infarct effects of YZR extract treatments may be partially attributed to the downregulation of TLR4/T3JAM/JNK- and ASK1/JNK-mediated inflammatory signaling in the penumbral cortex at 1 day after reperfusion.
JNK-mediated inflammatory signaling leads to the production of excessive amounts of the pro-inflammatory mediators, such as TNF-α, which in turn induces ASK1 activation, promoting the activation of ASK1/JNK signaling in cerebral I/R injury [50]. By contrast, pharmacological downregulation of p-JNK attenuated TLR4, Iba1, NF-κB, COX-2, and TNF-α expression in the peri-infarct cortex and striatum; reduced cerebral infarct areas; and ameliorated neurobehavioral deficits in a rat model of permanent MCAo [51]. On the basis of these findings, we propose that activated JNK could in turn regulate its upstream factor expression and play a key role in the regulation of TLR4/JNK- and ASK1/JNK-mediated inflammatory signaling after focal cerebral ischemia. Thus, in the present study, to identify the possible role of JNK in the effects of YZR extract treatments on cerebral infarction after transient MCAo, pretreatment with SP600125, an inhibitor of the JNK pathway, and pretreatment with 1% DMSO were performed in the SP (as the positive control group) and D+YZR-0.8 g groups (as the treatment group), respectively. SP600125, a non-protein synthetic inhibitor of JNK enzymatic activity, inhibits interactions between JNK and its substrates [52]. In the acute phase of cerebral I/R injury, SP600125 treatment reduced cerebral infarction by inhibiting the expression of inflammatory mediators including TNF-α, IL-1β, IL-6, and matrix metalloproteinase-9 and downregulating the mitochondria-mediated apoptotic pathway in the ischemic area [53,54,55]. In the present study, the Western blotting, IHC, and TTC staining results revealed that SP and D+YZR-0.8 g treatments effectively reversed the increased p-ASK1/ASK1 and p-JNK/JNK ratios, and the increased expression of TLR4, Iba1, GFAP, T3JAM, TRAF3, NF-κB, iNOS, COX-2, TNF-α, and IL-6 in the penumbral cortex and subsequently reduced the percentage of cerebral infarct areas and NDSs at 1 day after reperfusion. In addition, the aforementioned effects of the YZR extract treatment (D+YZR-0.8 g group) on cerebral I/R injury were similar to those of the SP600125 treatment (SP group). According to these findings, we reasonably assume that YZR extract treatments protect against cerebral I/R injury by downregulating JNK-mediated signaling in the penumbral cortex. The inhibitory effect of YZR extract treatments on JNK activation contributes to the suppression of TLR4/Iba1 (GFAP)/TRAF3/T3JAM- and NF-κB/ASK1-mediated signaling, and then abrogates the vicious circles of TLR4/JNK and ASK1/JNK signaling in the penumbral cortex. Thus, YZR extract treatments reduce cerebral infarction partially through the downregulation of JNK-mediated TLR4/T3JAM- and ASK1-related inflammatory signaling at 1 day after reperfusion (Fig. 13).

Schematic representation of the possible effects of YZR extract treatments on cerebral infarction induced by the downregulation of the JNK-mediated TLR4/T3JAM- and ASK1-related inflammatory signaling pathways in the penumbral cortex. P: phosphorylated
Conclusions
The findings of this study indicated that the YZR extract administered at doses of 0.4 g/kg and 0.8 g/kg significantly reduced cerebral infarction and alleviated neurological deficits in the early stage of MCAo; that is, after 90 min of MCAo. Further analysis revealed that YZR extract treatments exert neuroprotective effects against cerebral I/R injury by downregulating the JNK-mediated signaling pathway in the peri-infarct cortex. Furthermore, the anti-infarct effects of YZR extract treatments are partially attributed to the downregulation of the JNK-mediated TLR4/T3JAM- and ASK1-related NF-κB signaling pathways in the penumbral cortex at 1 day after reperfusion. Thus, the results of the present study suggest that the A. oxyphylla Miq extract may reduce cerebral infarction in the early phase of cerebral I/R injury. However, to elucidate the precise mechanisms underlying the anti-infarct effects of the YZR extract treatment, further research is needed to clarify the effects of the A. oxyphylla Miq extract on the regulation of JNK-mediated apoptotic signaling in the acute phase of transient focal cerebral ischemia.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author upon request.
Abbreviations
- I/R:
-
Ischemia–reperfusion
- TLR:
-
Toll-like receptor
- DAMP:
-
Damage-associated molecular pattern
- MyD88:
-
Myeloid differentiation primary response gene 88
- IL:
-
Interleukin
- TRIF:
-
Toll/interleukin 1 receptor homology domain-containing adaptor-inducing interferon-β
- NF-κB:
-
Nuclear factor-kappa B
- SP:
-
SP600125
- Iba1:
-
Ionized calcium-binding adapter molecule 1
- GFAP:
-
Glial fibrillary acidic protein
- TNF:
-
Tumor necrosis factor
- TRAF6:
-
TNF receptor-associated factor 6
- MAPK:
-
Mitogen-activated protein kinase
- JNK:
-
C-Jun N-terminal kinase
- TRAM:
-
TRIF-related adaptor molecule
- T3JAM:
-
TRAF3-interacting JNK-activating modulator
- ASK1:
-
Apoptosis signal-regulating kinase 1
- MKK:
-
MAPK kinase
- iNOS:
-
Inducible nitric oxide synthase
- COX-2:
-
Cyclooxygenase-2
- BBB:
-
Blood–brain barrier
- MCAo:
-
Middle cerebral artery occlusion
- YZR:
-
Yi Zhi Ren
- IP:
-
Intraperitoneal
- ICV:
-
Intracerebroventricular
- NIFDC:
-
National Institutes for Food and Drug Control
- RT:
-
Room temperature
- HPLC:
-
High-performance liquid chromatography
- MCA:
-
Middle cerebral artery
- ECA:
-
External carotid artery
- ICA:
-
Internal carotid artery
- mNSS:
-
Modified neurological severity score
- NDS:
-
Neurological deficit score
- TTC:
-
2,3,5-Triphenyltetrazolium chloride
- PFA:
-
Paraformaldehyde
- NC:
-
Nitrocellulose
- DMSO:
-
Dimethyl sulfoxide
- PBST:
-
Phosphate buffered saline/Tween 20
- IHC:
-
Immunohistochemical
- IF:
-
Immunofluorescence
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- ANOVA:
-
One-way analysis of variance
- NeuN:
-
Neuronal nuclei
- AU:
-
Absorbance unit
- PC:
-
Penumbral cortex
References
Hwang JW, Jeon YT, Lim YJ, Park HP. Sevoflurane postconditioning-induced anti-inflammation via inhibition of the toll-like receptor-4/nuclear factor kappa B pathway contributes to neuroprotection against transient global cerebral ischemia in rats. Int J Mol Sci. 2017;18:2347.
Xu L, Li Y, Fu Q, Ma S. Perillaldehyde attenuates cerebral ischemia-reperfusion injury-triggered overexpression of inflammatory cytokines via modulating Akt/JNK pathway in the rat brain cortex. Biochem Biophys Res Commun. 2014;454:65–70.
Huang L, Chen C, Zhang X, Li X, Chen Z, Yang C, et al. Neuroprotective effect of curcumin against cerebral ischemia-reperfusion via mediating autophagy and inflammation. J Mol Neurosci. 2018;64:129–39.
Wang YC, Lin S, Yang QW. Toll-like receptors in cerebral ischemic inflammatory injury. J Neuroinflammation. 2011;8:134.
Lee KM, Bang J, Kim BY, Lee IS, Han JS, Hwang BY, et al. Fructus mume alleviates chronic cerebral hypoperfusion-induced white matter and hippocampal damage via inhibition of inflammation and downregulation of TLR4 and p38 MAPK signaling. BMC Complement Altern Med. 2015;15:125.
Zhang P, Guo ZF, Xu YM, Li YS, Song JG. N-Butylphthalide (NBP) ameliorated cerebral ischemia reperfusion-induced brain injury via HGF-regulated TLR4/NF-kappaB signaling pathway. Biomed Pharmacother. 2016;83:658–66.
Wang Y, Li L, Deng S, Liu F, He Z. Ursolic acid ameliorates inflammation in cerebral ischemia and reperfusion injury possibly via high mobility group box 1/toll-like receptor 4/NFkappaB pathway. Front Neurol. 2018;9:253.
Lei JR, Tu XK, Wang Y, Tu DW, Shi SS. Resveratrol downregulates the TLR4 signaling pathway to reduce brain damage in a rat model of focal cerebral ischemia. Exp Ther Med. 2019;17:3215–21.
Khamchai S, Chumboatong W, Hata J, Tocharus C, Suksamrarn A, Tocharus J. Morin protects the blood-brain barrier integrity against cerebral ischemia reperfusion through anti-inflammatory actions in rats. Sci Rep. 2020;10:13379.
Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med. 2007;13:460–9.
Wang PF, Xiong XY, Chen J, Wang YC, Duan W, Yang QW. Function and mechanism of toll-like receptors in cerebral ischemic tolerance: from preconditioning to treatment. J Neuroinflamm. 2015;12:80.
Wang Y, Ge P, Zhu Y. TLR2 and TLR4 in the brain injury caused by cerebral ischemia and reperfusion. Mediat Inflamm. 2013;2013:124614.
Li M, Liu J, Bi Y, Chen J, Zhao L. Potential medications or compounds acting on toll-like receptors in cerebral ischemia. Curr Neuropharmacol. 2018;16:160–75.
Dadgostar H, Doyle SE, Shahangian A, Garcia DE, Cheng G. T3JAM, a novel protein that specifically interacts with TRAF3 and promotes the activation of JNK(1). FEBS Lett. 2003;553:403–7.
Li Y, Guan J, Wang W, Hou C, Zhou L, Ma J, et al. TRAF3-interacting JNK-activating modulator promotes inflammation by stimulating translocation of Toll-like receptor 4 to lipid rafts. J Biol Chem. 2019;294:2744–56.
Wang Q, Zhang QG, Wu DN, Yin XH, Zhang GY. Neuroprotection of selenite against ischemic brain injury through negatively regulating early activation of ASK1/JNK cascade via activation of PI3K/AKT pathway. Acta Pharmacol Sin. 2007;28:19–27.
Plotnikov MB, Chernysheva GA, Aliev OI, Smol’iakova VI, Fomina TI, Osipenko AN, et al. Protective effects of a new C-Jun N-terminal kinase inhibitor in the model of global cerebral ischemia in rats. Molecules. 2019;24:1722.
Chen X, Li X, Zhang W, He J, Xu B, Lei B, et al. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-kappaB pathway. Metabolism. 2018;83:256–70.
Zheng J, Dai Q, Han K, Hong W, Jia D, Mo Y, et al. JNK-IN-8, a c-Jun N-terminal kinase inhibitor, improves functional recovery through suppressing neuroinflammation in ischemic stroke. J Cell Physiol. 2020;235:2792–9.
Guo RB, Wang GF, Zhao AP, Gu J, Sun XL, Hu G. Paeoniflorin protects against ischemia-induced brain damages in rats via inhibiting MAPKs/NF-kappaB-mediated inflammatory responses. PLoS ONE. 2012;7:e49701.
Yang C, Hawkins KE, Dore S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. 2019;316:C135–53.
Lu H, Wang B. SIRT1 exerts neuroprotective effects by attenuating cerebral ischemia/reperfusion-induced injury via targeting p53/microRNA-22. Int J Mol Med. 2017;39:208–16.
Vaibhav K, Shrivastava P, Javed H, Khan A, Ahmed ME, Tabassum R, et al. Piperine suppresses cerebral ischemia-reperfusion-induced inflammation through the repression of COX-2, NOS-2, and NF-kappaB in middle cerebral artery occlusion rat model. Mol Cell Biochem. 2012;367:73–84.
Wang X, An F, Wang S, An Z. Orientin attenuates cerebral ischemia/reperfusion injury in rat model through the AQP-4 and TLR4/NF-kappaB/TNF-alpha signaling pathway. J Stroke Cerebrovasc Dis. 2017;26:2199–214.
Block F, Peters M, Nolden-Koch M. Expression of IL-6 in the ischemic penumbra. NeuroReport. 2000;11:963–7.
Shi SH, Zhao X, Liu AJ, Liu B, Li H, Wu B, Bi KS, et al. Protective effect of n-butanol extract from Alpinia oxyphylla on learning and memory impairments. Physiol Behav. 2015;139:13–20.
Kim EJ, Lee YJ, Ahn YM, Lee H, Kang DG, Lee HS. Renoprotective effect of Alpiniae oxyphyllae Fructus on ischemia/reperfusion-induced acute renal failure. Arch Pharm Res. 2013;36:1004–12.
Wang Y, Wang M, Fan K, Li T, Yan T, Wu B, et al. Protective effects of Alpinae oxyphyllae Fructus extracts on lipopolysaccharide-induced animal model of Alzheimer’s disease. J Ethnopharmacol. 2018;217:98–106.
Koo BS, Lee WC, Chang YC, Kim CH. Protective effects of alpinae oxyphyllae fructus (Alpinia oxyphylla MIQ) water-extracts on neurons from ischemic damage and neuronal cell toxicity. Phytother Res. 2004;18:142–8.
Qi Y, Cheng X, Jing H, Yan T, Xiao F, Wu B, et al. Comparative pharmacokinetic study of the components in Alpinia oxyphylla Miq.-Schisandra chinensis (Turcz) Baill. herb pair and its single herb between normal and Alzheimer’s disease rats by UPLC-MS/MS. J Pharm Biomed Anal. 2020;177:112874.
An LJ, Guan S, Shi GF, Bao YM, Duan YL, Jiang B. Protocatechuic acid from Alpinia oxyphylla against MPP+-induced neurotoxicity in PC12 cells. Food Chem Toxicol. 2006;44:436–43.
Shi GF, An LJ, Jiang B, Guan S, Bao YM. Alpinia protocatechuic acid protects against oxidative damage in vitro and reduces oxidative stress in vivo. Neurosci Lett. 2006;403:206–10.
Yao Y, Chen L, Xiao J, Wang C, Jiang W, Zhang R, et al. Chrysin protects against focal cerebral ischemia/reperfusion injury in mice through attenuation of oxidative stress and inflammation. Int J Mol Sci. 2014;15:20913–26.
El Khashab IH, Abdelsalam RM, Elbrairy AI, Attia AS. Chrysin attenuates global cerebral ischemic reperfusion injury via suppression of oxidative stress, inflammation and apoptosis. Biomed Pharmacother. 2019;112:108619.
He B, Xu F, Xiao F, Yan T, Wu B, Bi K, et al. Neuroprotective effects of nootkatone from Alpiniae oxyphyllae Fructus against amyloid-beta-induced cognitive impairment. Metab Brain Dis. 2018;33:251–9.
Cheng CY, Kao ST, Lee YC. Angelica sinensis extract protects against ischemia-reperfusion injury in the hippocampus by activating p38 MAPK-mediated p90RSK/p-Bad and p90RSK/CREB/BDNF signaling after transient global cerebral ischemia in rats. J Ethnopharmacol. 2020;252:112612.
Cheng CY, Ho TY, Hsiang CY, Tang NY, Hsieh CL, Kao ST, et al. Angelica sinensis exerts angiogenic and anti-apoptotic effects against cerebral ischemia-reperfusion injury by activating p38MAPK/HIF-1[Formula: see text]/VEGF-A signaling in rats. Am J Chin Med. 2017;45:1683–708.
Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–8.
Fang T, Zhou D, Lu L, Tong X, Wu J, Yi L. LXW7 ameliorates focal cerebral ischemia injury and attenuates inflammatory responses in activated microglia in rats. Braz J Med Biol Res. 2016;49:e5287.
Zhang W, Song J, Li W, Kong D, Liang Y, Zhao X, et al. Salvianolic acid D alleviates cerebral ischemia-reperfusion injury by suppressing the cytoplasmic translocation and release of HMGB1-triggered NF-kappaB activation to inhibit inflammatory response. Mediators Inflamm. 2020;2020:9049614.
Heindl S, Gesierich B, Benakis C, Llovera G, Duering M, Liesz A. Automated morphological analysis of microglia after stroke. Front Cell Neurosci. 2018;12:106.
Liu M, Xu Z, Wang L, Zhang L, Liu Y, Cao J, et al. Cottonseed oil alleviates ischemic stroke injury by inhibiting the inflammatory activation of microglia and astrocyte. J Neuroinflamm. 2020;17:270.
Luo SY, Li R, Le ZY, Li QL, Chen ZW. Anfibatide protects against rat cerebral ischemia/reperfusion injury via TLR4/JNK/caspase-3 pathway. Eur J Pharmacol. 2017;807:127–37.
Wang Y, Chen G, Yu X, Li Y, Zhang L, He Z, et al. Salvianolic acid B ameliorates cerebral ischemia/reperfusion injury through inhibiting TLR4/MyD88 signaling pathway. Inflammation. 2016;39:1503–13.
Shah FA, Kury LA, Li T, Zeb A, Koh PO, Liu F, et al. Polydatin attenuates neuronal loss via reducing neuroinflammation and oxidative stress in Rat MCAO models. Front Pharmacol. 2019;10:663.
Shvedova M, Anfinogenova Y, Atochina-Vasserman EN, Schepetkin IA, Atochin DN. c-Jun N-terminal kinases (JNKs) in myocardial and cerebral ischemia/reperfusion injury. Front Pharmacol. 2018;9:715.
Zhang Q, Zhang G. Activation and autophosphorylation of apoptosis signal-regulating kinase 1 (ASK1) following cerebral ischemia in rat hippocampus. Neurosci Lett. 2002;329:232–6.
Wu X, Li L, Zhang L, Wu J, Zhou Y, Zhao Y, et al. Inhibition of thioredoxin-1 with siRNA exacerbates apoptosis by activating the ASK1-JNK/p38 pathway in brain of a stroke model rats. Brain Res. 2015;1599:20–31.
Hao H, Li S, Tang H, Liu B, Cai Y, Shi C, et al. NQDI-1, an inhibitor of ASK1 attenuates acute perinatal hypoxic-ischemic cerebral injury by modulating cell death. Mol Med Rep. 2016;13:4585–92.
Liu DH, Yuan FG, Hu SQ, Diao F, Wu YP, Zong YY, et al. Endogenous nitric oxide induces activation of apoptosis signal-regulating kinase 1 via S-nitrosylation in rat hippocampus during cerebral ischemia-reperfusion. Neuroscience. 2013;229:36–48.
Mohsin Alvi A, Tariq Al Kury L, Umar Ijaz M, Ali Shah F, Tariq Khan M, Sadiq Sheikh A, et al. Post-treatment of synthetic polyphenolic 1,3,4 oxadiazole compound A3, attenuated ischemic stroke-induced neuroinflammation and neurodegeneration. Biomolecules. 2020;10:816.
Plotnikov MB, Chernysheva GA, Smolyakova VI, Aliev OI, Trofimova ES, Sherstoboev EY, et al. Neuroprotective effects of a novel inhibitor of c-Jun N-terminal kinase in the rat model of transient focal cerebral ischemia. Cells. 2020;9:1860.
Murata Y, Fujiwara N, Seo JH, Yan F, Liu X, Terasaki Y, et al. Delayed inhibition of c-Jun N-terminal kinase worsens outcomes after focal cerebral ischemia. J Neurosci. 2012;32:8112–5.
Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712.
Zhu Y, Li S, Liu J, Wen Q, Yu J, Yu L, et al. Role of JNK signaling pathway in dexmedetomidine post-conditioning-induced reduction of the inflammatory response and autophagy effect of focal cerebral ischemia reperfusion injury in rats. Inflammation. 2019;42:2181–91.
Acknowledgements
We thank Professor Yu-Chuen Huang and Professor Wen-Ling Liao at China Medical University, Taichung, Taiwan, for kindly helping with statistical analysis.
Funding
This study was supported by grants from the Ministry of Science and Technology of Taiwan (MOST 109-2320-B-039-047-) and China Medical University Hospital (DMR-109-010), Taichung, Taiwan. These funding sources had no role in the design of the study and collection, analysis, and interpretation of the data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
CYC and SCH designed experiments. CYC performed experiments, analyzed data and wrote the manuscript. SYC and STK helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental procedures were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of China Medical University (Permit Number: CMUIACUC-2019-312).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cheng, CY., Chiang, Sy., Kao, ST. et al. Alpinia oxyphylla Miq extract reduces cerebral infarction by downregulating JNK-mediated TLR4/T3JAM- and ASK1-related inflammatory signaling in the acute phase of transient focal cerebral ischemia in rats. Chin Med 16, 82 (2021). https://doi.org/10.1186/s13020-021-00495-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13020-021-00495-2