
Abstract
Mammalian sterile 20-like (Ste20-like) protein kinase 3 (MST3) or serine/threonine-protein kinase 24 (STK24) is a serine/threonine protein kinase that belongs to the mammalian STE20-like protein kinase family. MST3 is a pleiotropic protein that plays a critical role in regulating a variety of events, including apoptosis, immune response, metabolism, hypertension, tumor progression, and development of the central nervous system. The MST3-mediated regulation is intricately related to protein activity, post-translational modification, and subcellular location. Here, we review the recent progress on the regulatory mechanisms against MST3 and its-mediated control of disease progression.
Similar content being viewed by others

Introduction
Sterile 20 (STE20) is a serine/threonine protein kinase family that was originally discovered in the budding yeast. Presently, 28 mammalian STE20-like (MST) kinases, homologs to yeast STE20, have been identified [1]. According to the relative location of the kinase domains, these are divided into two families, namely, the p21-activated kinase (PAK) family (COOH terminal kinase domain) and germinal center kinase (GCK) family (NH2 terminal kinase domain). In mammals, the five characterized MST family kinases can be divided into two subgroups, namely, GCK-II (MST1 and MST2) and GCK-III (MST3, MST4, and YSK1) [2]. It is reported that the MST family numbers are closely related to the regulation of a variety of biological activities, such as cytoskeletal organization, cell motility, apoptosis, and central nervous system (CNS) development (Table 1). At present, increasing attention has been paid to MST3 regarding its roles in modulating apoptosis, immune response, metabolism, hypertension, tumor progression, and CNS development [2,3,4,5]. The MST3-mediated regulation of disease progression is closely associated with protein activity, which is affected by protein cleavage, subcellular distribution, and post-transcriptional modification [6,7,8]. Therefore, in this review, we summarize the recent progress on the regulatory mode against MST3 and the mechanisms underlying the MST3-mediated control of disease development.
Homology of MST kinases
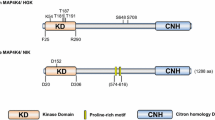
The MST kinases contain an N-terminal kinase domain and a C-terminal regulatory domain. In human MST3, the N-terminal kinase domain is located at 36–286 amino acids, whereas the C-terminal regulatory domain is located at 287–443 amino acids [22] (Fig. 1). Sequence alignment revealed that human MST3 shares a nearly 70% sequence identity with MST4 and YSK1 while sharing a nearly 40% identity with MST1 and MST2 (Fig. 2). The MST proteins contain high sequence identity at the N-terminal kinase domain but not at the C-terminal domain. Human MST3 has five variants, whereas mouse MST3 has four isoforms. The high rate of the identity of canonical sequences of MST3 among humans, mice and rats is up to 93% (Fig. 3).

Protein domain and kinase sites of MSTs

Sequence alignment of human MST family protein. Multiple alignments were carried out using UniProt-Align (https://www.uniprot.org/align). The alignment was drawn using ESPript 3.0 (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi). MST1 (accession no. UniProtKB-Q13043); MST2 (accession no. Q13188); MST3 (accession no. Q9Y6E0); MST4 (accession no. Q9P289); YSK1 (accession no. O00506)

Sequence alignment of MST3 isoforms from human and mouse species. Multiple alignments were carried out using UniProt-Align (https://www.uniprot.org/align). The alignment was drawn using ESPript 3.0 (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi). Human MST3 variant 1(a) (canonical sequence; accession no. NP_003567.2); MST3 variant 2(b) (accession no. NP_001027467.2); MST3 variant 3 (accession no. NP_001273578.1); MST3 variant X1 (accession no. XP_016876283.1); MST3 variant X2 (accession no. XP_024305194.1); mouse MST3 (canonical sequence; accession no. NM_145465.2); mouse MST3 variant X1 (accession no. XM_017315988.2); mouse MST3 variant X2 (accession no. XM_011245043.4); mouse MST3 variant X3 (accession no. XM_006518918.3); rat MST3 (accession no.NP_001120966.1); rat MST3 variant X1 (accession no. XP_038949467.1)
Subcellular distribution
Under normal conditions, MST3 is localized predominantly in the cytoplasm. During apoptosis, the activated caspase-3 cleaves MST3 at the junction of the N- and C-terminal domain (AETD313G), following which the truncated MST3 (MST3/N) is translocated into the nucleus [22]. The nuclear localization sequence (NLS) at the C-terminus of its kinase domain (residues 278–292) is required for the intranuclear translocation of MST3 [23], whereas a nuclear export signal (NES) is postulated to be in the C-terminal regulatory domain (amino acids 335–386) (Fig. 1) [23]. It is reported that the myristoylation of MST3 induces the diffusion in the cytosol or translocation into the nucleus via its nuclear localization sequence [24]. These results suggest that the subcellular location of MST3 can be modulated by the diverse cleavage or post-translational modification.
Kinase activity sites, modifications, and interactions
The MSTs are serine/threonine protein kinases that promote phosphorylation or activation of substrate proteins by transferring phosphate groups from GTP or ATP to the serine or threonine residue of target proteins. The mutants of MST1 K59R, MST2 K56R, MST3/MST4 K53R and YSK1 K49R and D158A display deficient kinase activity [22, 25, 26]. In contrast, the phosphorylation of MST2 Ther117 or Ther384 by Akt renders the kinase inactive [27] (Fig. 1). The activation of MST3 is associated with post-translational modifications, including autophosphorylation, phosphorylation, and myristoylation. Thr178 is the conserved autophosphorylation site of STE20-like kinases [28]. The mutation of threonine to alanine at codon 178 of MST3 or MST4 leads to the deficiency of kinase activity [12, 28]. Although MST3 also autophosphorylates at codon Thr328, the phosphorylation at this residue does not affect the kinase activity of MST3 [29]. In addition, the phosphorylation of MST3 at Lys53 [19, 30] or Ser79 [31] is essential for the kinase activity of MST3 (Fig. 1).
Recently, several upstream kinases and regulators have been reported to regulate the activation of MST3. The cyclin-dependent kinase 5 (Cdk5) that phosphorylates MST3 at Ser79 is essential for the activity of MST3 [31]. A novel isoform of MST3 (MST3b) with a different 5' coding region from MST3 (strictly expressed in the brain), is effectively phosphorylated by cyclic AMP-dependent protein kinase (PKA) at Thr18 (this residue is absent in MST3) [32]. Although MST3 can also be phosphorylated at tyrosine following treatment with a tyrosine phosphatase inhibitor (PV), the tyrosine modification does not alter the activity of MST3 [33]. In addition, MST3 can be myristoylated, which could avoid the binding of its negative regulatory domain with the catalytic domain, resulting in a constitutively active enzyme [24].
Apart from post-translational modification, the activity of MST3 is also modulated by a series of cellular activities, such as caspase-mediated cleavage and interaction with regulators (Table 2). Caspase 3-mediated cleavage of MST3 at the junction of the N-terminal kinase domain and C-terminal regulatory domain activates its intrinsic kinase activity by removing the negative regulatory domain [22]. The binding of MST3 with its master regulator MO25 scaffolding protein stimulates its kinase activity three- to four-fold [34]. In contrast, striatin-interacting phosphatase and kinase (STRIPAK) complex components, protein phosphatase 2A (PP2A) [35] or FAM40A [36], inactivates MST3 by dephosphorylating its activation loop.
Roles and regulatory mechanisms of MST3 in disease progression
MST3 and apoptosis
Apoptosis is a form of programmed cell death that is critical for maintaining cellular physiologic homeostasis. Apoptosis is triggered by a series of effectors (e.g., caspases 3 and 8) and regulators through two major pathways, namely, extrinsic (death receptor-mediated) and intrinsic (mitochondria-mediated) pathways. Researchers have demonstrated that MST3 is closely related to the regulation of apoptosis (Fig. 4 and Table 3). In response to staurosporine, MST3 triggers apoptosis by activating caspase 3 (cleavage of caspase 3), a key player in activating both extrinsic and intrinsic apoptotic pathways by cleaving several downstream cell survival-associated proteins [3]. Moreover, Wu et al. reported that MST3 is overexpressed in placental trophoblasts during labor-induced oxidative stress. The overexpression of MST3 in the human trophoblast cell line 3A-sub-E promotes caspase 3-mediated apoptosis [30]. In line with this result, researchers reported that MST3 triggers cell death in the hydrogen peroxide (H2O2)-treated human colon carcinoma HCT116 cell line by suppressing the JNK survival pathway and up-regulating cytoprotective HO-1 (heme oxygenase-1) [6]. Further analysis demonstrated that the MST3 kinase activity is essential for H2O2-induced apoptosis of cells because the kinase-dead mutant of MST3 (Lys53 to Arg53) displayed an impaired ability to induce apoptosis than the wild-type MST3 [30]. In addition to the caspase-mediated canonical apoptosis, MST3 activates the caspase-independent apoptotic pathway in human cervix HeLa cells by promoting the nuclear translocation of apoptosis-inducing factor and endonuclease G by disrupting the mitochondrial membrane potential (Δψm) [3].

Schematic diagram of MST3-mediated regulation
Although MST3 has been proved to trigger apoptosis through the caspase effector, it can also be cleaved by caspase 3 at the domain (AETD313G) during anti-Fas antibody- or staurosporine-induced apoptosis in Jurkat cells [22]. The cleavage of MST3 by caspase 3 causes nuclear accumulation of the active kinase domain of MST3 and increased apoptosis induced by the truncated MST3 [22]. These results suggest that MST3 and caspase 3 form a feedback loop during the initial stages of apoptosis by modulating the protein cleavage-mediated enhanced activity. However, this speculation requires further investigation.
MST3 and immune regulation
Reports have revealed that the MST kinase family members, namely, MST1 and MST2, are the important components of the immune-associated Hippo pathway [14]. MST1 and MST2 play crucial roles in regulating both innate and adaptive immune response-related activities, such as T cell homeostasis, lymphocyte trafficking, antiviral immune signaling [46, 47], and CD8α+ dendritic cell activation [14]. Neutrophils are the first responders to inflammation and infection; they migrate to inflammatory sites and execute the program of degranulation to release antimicrobial molecules or cytotoxic agents [48]. Zhang et al. reported that the deficiency of STK24 enhances the degranulation of neutrophils. The STK24 binds to UNC13D and suppresses UNC13D-lipid binding and granule docking, thus inhibiting the exocytic process of neutrophil degranulation [37], thereby indicating a critical function of MST3 in promoting host immune response. In addition, the number of immune inhibitory myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) were increased, whereas the number of tumoricidal CD4+ T cells was decreased in the STK24 knockout gastric tumor sections, indicating that MST3 promotes antitumoral immune response [38, 39]. Nevertheless, the underlying mechanisms need further investigation.
MST3 and metabolism
Recently, a series of studies have unveiled a previously unknown function of GCKIII kinases in metabolic regulation [2]. As a member of the GCKIII family protein, MST3 has lately been demonstrated to increase insulin resistance and blood glucose levels in mice fed with an obesity-promoting high-fat diet (HFD) [40]. Knockout of MST3 in these mice led to impaired hyperglycemia, hyperinsulinemia, and insulin resistance. Mechanistic analysis revealed that a lack of MST3 in both cultured liver cells and the livers of animals after HFD activates the insulin signaling pathway downstream of IRS1 by inhibiting forkhead box (FOX)O1-mediated downregulation of genes encoding gluconeogenic enzymes [40]. In addition to the regulation of insulin signaling, studies have reported that MST3, MST4, and STK25 are exclusively localized around intracellular lipid droplets and increase fat accumulation in human hepatocytes as well as the initiation and progression of nonalcoholic fatty liver disease (NAFLD) [9, 41]. Mice treated with antisense oligonucleotides (ASOs) targeting MST3 effectively ameliorated HFD-induced nonalcoholic fatty liver disease (NAFLD)-associated liver steatosis, inflammation, fibrosis, and hepatocellular damage [41]. Mechanistically, MST3 ASOs inhibit the expression of lipogenic genes, as well as acetyl-CoA carboxylase (ACC) protein abundance, leading to reduced lipotoxicity-mediated oxidative and endoplasmic reticulum stress in the liver of obese mice [41]. Similarly, researchers unveiled that MST3 modulates the dynamic metabolic balance of liver lipid catabolism versus lipid anabolism. Knockdown of MST3 decreased the accumulation of lipids in human hepatocytes by stimulating β-oxidation and triacylglycerol secretion while suppressing fatty acid influx and lipid synthesis [42]. Moreover, recent study reported that all 28 STE20 kinases including MST3 phosphorylate the energy metabolism-related protein kinases, AMP-activated protein kinase (AMPK) and the salt-inducible kinase 3 (SIK3) [49]. The MST3 or the brain expressed MST3b isoform phosphorylates AMPKα1-T183 and SIK3-T221 [50].
MST3 and hypertension
A combination of defective renal salt and water excretion and increased salt intake frequently contributes to hypertension. Lu et al. disclosed that MST3 is a stress-regulated kinase that maintains sodium homeostasis after a high-salt diet and protects the development of hypertension in mice. The MST3 protein expression is markedly reduced in the kidneys of spontaneously hypertensive rat (SHR) kidneys, whereas this level was elevated when normal mice were administered a high-salt diet [19]. In vitro study unveiled that under hypertonic stress (900 mOsm/L hyperosmolar NaCl medium), the wild type-MST3-MDCK (Madin–Darby Canine Kidney) cells survived. In contrast, the KD-MST3-MDCK (K53R kinase-dead MST3) cells could not resist the hypertonic stress [19], suggesting that MST3-mediated maintenance of sodium homeostasis requires its kinase activity. Further analysis using mice with MST3 hypomorphic mutation demonstrated that the MST3−/− mice exhibit hypernatremia, hypokalemia, and hypertension, and MST3 maintained Na+ homeostasis and blood pressure stability by regulating epithelial Na+ channel (ENaC) [20]. Moreover, Chan et al. reported that MST3/STK24 is expressed primarily in the medullary thick ascending tubule (TAL) and at lower levels in the late distal convoluted tubules (DCTs) [43]. The hypertension and lower urinary Na+ excretion found in MST3−/− mice is associated with increased ENaC activity, WNK4 (with-no-lysine 4) expression, and NKCC2 (Na–K-Cl cotransporter) S130 phosphorylation [43], indicating that MST3 participates in maintaining the Na+/K+ homeostasis in response to K+ loading by inhibiting WNK4 expression, NKCC2, and ENaC activity.
MST3 and tumor progression
Recently, studies have shown that MST3 deregulation is associated with cancer cell migration and metastasis. MST3 is overexpressed in the tumor tissues of patients suffering from the human breast [4] and gastric cancer [7]. The overexpression of MST3 predicts poor prognosis in these cancer patients [4, 7], suggesting that MST3 promotes tumor development and progression. Mechanistically, the overexpression of MST3 increases the phosphorylation of VAV2, and subsequently promotes VAV2-mediated activation of the Rac1-cyclin D1 signaling pathway that is required for the growth of breast cancer cells. Further investigation demonstrated that the proline-rich region of MST3 (K353DIPKRP359) interacts with the SH3 domain of VAV2, which is required for MST3-mediated promotion of proliferation of these cancer cells [4]. Lee et al. reported that the inhibition of MST3 expression led to enhanced expression of cyclin-dependent kinase inhibitor p21, resulting in p21-mediated inhibition of cell cycle in human gastric cancer cell line MKN45, but not NCIN87 [7], suggesting that MST3 promotes tumor cell growth in a cell type-dependent manner. In addition, studies have reported an indirect role of MST3 in regulating the development of cancer. Nuclear Dbf2-related (NDR), a serine/threonine protein kinase, which directly phosphorylates p21 at S146, increases the progression of G1 by stabilizing c-Myc and preventing the accumulation of p21. Because NDR is the first identified substrate for MST3 (phosphorylates NDR at Thr444/Thr442) [51], it suggests that MST3 plays the oncogenic role by activating NDR. Furthermore, MST3 interacts with the evolutionarily conserved MO25 scaffolding protein [52], which is the master regulator of the LKB1 (serine-threonine liver kinase B1) tumor suppressor [53], indicating the possibility of MST3 in regulating tumor progression by targeting the MO25-LKB1 pathway. In view of the role of MST3 in promoting cancer development, Olesen et al. have discovered fourteen chemical compounds as MST3 inhibitors by using the kinase domain of MST3 (residues 1–303) to screen against the kinase inhibitor library from Selleck Chemicals (Table 4) [54]. This finding indicates that targeting MST3 with small-molecule inhibitors may be beneficial for controlling disease progression.
Although MST3 has been previously recognized as a pro-tumoral protein, the function of MST3 to inhibit tumor progression has been reported. Luo et al. reported that the down-expression of MST3 by the oncogene MiR-222 that directly binds to the promoter region of MST3 promotes the migration and invasion of colorectal cancer cell line [55]. Mechanistically, the suppression of endogenous MST3 enhances the cellular migration in human adenocarcinoma cells, MCF-7, by increasing the phosphorylation of paxillin by a protein tyrosine phosphatase PTP-PEST [28]. The suppression of STK24 increased cell migration by inhibiting CDH1 (E-Cadherin) and enhancing the levels of CD44 in gastric cancer cells [38]. In addition, an in vivo study reported that the suppression of CD4+ T cells increased tumor metastasis and growth in an STK24-silenced mouse model of gastric cancer while enhancing the expansion of CD11b+Ly6C+ MDSCs and F4/80+ TAMs [38, 39].
MST3 and CNS development
Proper neuronal migration during cortical development is required for normal neuronal function. It is reported that MST4 (STK25), a GCKIII family member, promotes neuronal migration in the neocortex by balancing the Rac1 activity and RhoA levels through the formation of complexes with α-PIX and β-PIX, GTPase regulatory enzymes, and Cullin3-Bacurd1/Kctd13 [8]. Although the conditional knockout of the STK25 gene during mouse embryogenesis causes anomalous neuronal migration in the neocortex, it did not cause a cortical phenotype, indicating the existence of a complementary mechanism [8]. In their subsequent study, they found that MST3 compensates MST4 to regulate neuronal migration and polarization by modulating the activity of Rho GTPases [8]. Another study reported that MST3 is highly expressed in the developing mouse brain. The overexpression of MST3 contributed to radial neuronal migration and final neuronal positioning in the developing mouse neocortex. Mechanistically, MST3 regulates neuronal migration by negatively regulating Rho-GTPase activity of RhoA, because RhoA plays a critical role in actin cytoskeletal reorganization by phosphorylating RhoA at Ser26 [31]. The phosphorylation of MST3 by Cdk5 at Ser79 is essential for its kinase activity and function in neuronal morphogenesis and migration [31]. Moreover, MST3 is necessary for the proper development of filopodia, dendritic spine, and excitatory synapse in the CNS. The MST3-mediated promotion of dendritic filopodia and spine synapse development occurs through the phosphorylation of TAO1/2 kinases [44].
Recently, a neuron-specific homolog of MST3 (isoform a), termed MST3b (isoform b), was reported. Amino acid sequence alignment revealed that the six N-terminal amino acids of MST3b are different from that of the canonical MST3. The further functional investigation demonstrated that the MST3b is essential for the development and repair of brain circuitry by promoting axon outgrowth [56]. Injured spinal cord neurons have increased levels of MST3b which promotes neuronal regeneration through the activation of P42/44MAPK and LIMK/Cofilin signaling pathways [45].
Conclusion
MST3 has emerged as a pleiotropic regulator in modulating a variety of biological functions, such as apoptosis, immune signaling, metabolism, hypertension, tumor progression, and CNS development. The function of MST3-mediated regulation is closely related to protein activity-associated activities, such as protein cleavage, post-transcriptional modification, subcellular distribution, and interactions with its several adaptor proteins. Therefore, targeting MST3 and its activity-associated characteristics can be used as a potential therapeutic strategy for controlling the progression of various disease processes.
Availability of data and materials
The data will be provided on contacting corresponding author.
References
Garland B, Delisle S, Al-Zahrani KN, Pryce BR, Sabourin LA. The Ste20-like kinase—a Jack of all trades? J Cell Sci. 2021. https://doi.org/10.1242/jcs.258269.
Pombo CM, Iglesias C, Sartages M, Zalvide JB. MST kinases and metabolism. Endocrinology. 2019;160(5):1111–8.
Lin CY, Wu HY, Wang PL, Yuan CJ. Mammalian Ste20-like protein kinase 3 induces a caspase-independent apoptotic pathway. Int J Biochem Cell Biol. 2010;42(1):98–105.
Cho CY, Lee KT, Chen WC, Wang CY, Chang YS, Huang HL, Hsu HP, Yen MC, Lai MZ, Lai MD. MST3 promotes proliferation and tumorigenicity through the VAV2/Rac1 signal axis in breast cancer. Oncotarget. 2016;7(12):14586–604.
Chen S, Fang Y, Xu S, Reis C, Zhang J. Mammalian sterile20-like kinases: signalings and roles in central nervous system. Aging Dis. 2018;9(3):537–52.
Chen CB, Ng JK, Choo PH, Wu W, Porter AG. Mammalian sterile 20-like kinase 3 (MST3) mediates oxidative-stress-induced cell death by modulating JNK activation. Biosci Rep. 2009;29(6):405–15.
Lee KT, Chang CL, Li CY, Song H, Shan YS, Lai MD. The oncogenic role of MST3 in human gastric cancer. Am J Cancer Res. 2018;8(10):2130–9.
Matsuki T, Iio A, Ueda M, Tsuneura Y, Howell BW, Nakayama A. STK25 and MST3 have overlapping roles to regulate rho GTpases during cortical development. J Neurosci. 2021;41(43):8887–903.
Mahlapuu M, Caputo M, Xia Y, Cansby E. GCKIII kinases in lipotoxicity: roles in NAFLD and beyond. Hepatol Commun. 2022;6(10):2613–22.
Turunen SP, von Nandelstadh P, Öhman T, Gucciardo E, Seashore-Ludlow B, Martins B, Rantanen V, Li H, Höpfner K, Östling P, et al. FGFR4 phosphorylates MST1 to confer breast cancer cells resistance to MST1/2-dependent apoptosis. Cell Death Differ. 2019;26(12):2577–93.
Fallahi E, O’Driscoll NA, Matallanas D. The MST/Hippo pathway and cell death: a non-canonical affair. Genes (Basel). 2016;7(6):28.
Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, Song X, Shi T, Yang Y, Sastry N, Horbinski CM, et al. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell. 2017;32(6):840-855.e848.
Bata N, Chaikuad A, Bakas NA, Limpert AS, Lambert LJ, Sheffler DJ, Berger LM, Liu G, Yuan C, Wang L, et al. Inhibitors of the hippo pathway kinases STK3/MST2 and STK4/MST1 have utility for the treatment of acute myeloid leukemia. J Med Chem. 2022;65(2):1352–69.
Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, Li Y, Guy C, Nguyen TM, Dhungana Y, et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α(+) dendritic cells. Nature. 2018;558(7708):141–5.
Zhang M, Tao W, Yuan Z, Liu Y. Mst-1 deficiency promotes post-traumatic spinal motor neuron survival via enhancement of autophagy flux. J Neurochem. 2017;143(2):244–56.
Wu X, Wu J, Hu W, Wang Q, Liu H, Chu Z, Lv K, Xu Y. MST4 kinase inhibitor hesperadin attenuates autophagy and behavioral disorder via the MST4/AKT pathway in intracerebral hemorrhage mice. Behav Neurol. 2020;2020:2476861.
Liu Q, Li J, Zhang W, Xiao C, Zhang S, Nian C, Li J, Su D, Chen L, Zhao Q, et al. Glycogen accumulation and phase separation drives liver tumor initiation. Cell. 2021;184(22):5559-5576.e5519.
Cheung P, Xiol J, Dill MT, Yuan WC, Panero R, Roper J, Osorio FG, Maglic D, Li Q, Gurung B, et al. Regenerative reprogramming of the intestinal stem cell state via hippo signaling suppresses metastatic colorectal cancer. Cell Stem Cell. 2020;27(4):590-604.e599.
Lu TJ, Chan CH, Ling P, Chao YM, Bao BY, Chiang CY, Lee TH, Weng YP, Kan WC, Lu TL. MST3 (mammalian Ste20-like protein kinase 3), a novel gene involved in ion homeostasis and renal regulation of blood pressure in spontaneous hypertensive rats. Int Urol Nephrol. 2018;50(12):2299–307.
Lu TJ, Kan WC, Yang SS, Jiang ST, Wu SN, Ling P, Bao BY, Lin CY, Yang ZY, Weng YP, et al. MST3 is involved in ENaC-mediated hypertension. Am J Physiol Renal Physiol. 2019;317(7):F30-f42.
Matsuki T, Matthews RT, Cooper JA, van der Brug MP, Cookson MR, Hardy JA, Olson EC, Howell BW. Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell. 2010;143(5):826–36.
Huang CY, Wu YM, Hsu CY, Lee WS, Lai MD, Lu TJ, Huang CL, Leu TH, Shih HM, Fang HI, et al. Caspase activation of mammalian sterile 20-like kinase 3 (Mst3). Nuclear translocation and induction of apoptosis. J Biol Chem. 2002;277(37):34367–74.
Lee WS, Hsu CY, Wang PL, Huang CY, Chang CH, Yuan CJ. Identification and characterization of the nuclear import and export signals of the mammalian Ste20-like protein kinase 3. FEBS Lett. 2004;572(1–3):41–5.
Martin DD, Vilas GL, Prescher JA, Rajaiah G, Falck JR, Bertozzi CR, Berthiaume LG. Rapid detection, discovery, and identification of post-translationally myristoylated proteins during apoptosis using a bio-orthogonal azidomyristate analog. Faseb j. 2008;22(3):797–806.
Callus BA, Verhagen AM, Vaux DL. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. Febs j. 2006;273(18):4264–76.
Preisinger C, Short B, De Corte V, Bruyneel E, Haas A, Kopajtich R, Gettemans J, Barr FA. YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3zeta. J Cell Biol. 2004;164(7):1009–20.
Romano D, Matallanas D, Weitsman G, Preisinger C, Ng T, Kolch W. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res. 2010;70(3):1195–203.
Lu TJ, Lai WY, Huang CY, Hsieh WJ, Yu JS, Hsieh YJ, Chang WT, Leu TH, Chang WC, Chuang WJ, et al. Inhibition of cell migration by autophosphorylated mammalian sterile 20-like kinase 3 (MST3) involves paxillin and protein-tyrosine phosphatase-PEST. J Biol Chem. 2006;281(50):38405–17.
Fuller SJ, McGuffin LJ, Marshall AK, Giraldo A, Pikkarainen S, Clerk A, Sugden PH. A novel non-canonical mechanism of regulation of MST3 (mammalian Sterile20-related kinase 3). Biochem J. 2012;442(3):595–610.
Wu HY, Lin CY, Lin TY, Chen TC, Yuan CJ. Mammalian Ste20-like protein kinase 3 mediates trophoblast apoptosis in spontaneous delivery. Apoptosis. 2008;13(2):283–94.
Tang J, Ip JP, Ye T, Ng YP, Yung WH, Wu Z, Fang W, Fu AK, Ip NY. Cdk5-dependent Mst3 phosphorylation and activity regulate neuronal migration through RhoA inhibition. J Neurosci. 2014;34(22):7425–36.
Zhou TH, Ling K, Guo J, Zhou H, Wu YL, Jing Q, Ma L, Pei G. Identification of a human brain-specific isoform of mammalian STE20-like kinase 3 that is regulated by cAMP-dependent protein kinase. J Biol Chem. 2000;275(4):2513–9.
Kan WC, Lu TL, Ling P, Lee TH, Cho CY, Huang CY, Jeng WY, Weng YP, Chiang CY, Wu JB, et al. Pervanadate induces Mammalian Ste20 Kinase 3 (MST3) tyrosine phosphorylation but not activation. J Inorg Biochem. 2016;160:33–9.
Filippi BM, de los Heros P, Mehellou Y, Navratilova I, Gourlay R, Deak M, Plater L, Toth R, Zeqiraj E, Alessi DR: MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases. Embo j 2011, 30(9):1730-1741
Gordon J, Hwang J, Carrier KJ, Jones CA, Kern QL, Moreno CS, Karas RH, Pallas DC. Protein phosphatase 2a (PP2A) binds within the oligomerization domain of striatin and regulates the phosphorylation and activation of the mammalian Ste20-Like kinase Mst3. BMC Biochem. 2011;12:54.
Madsen CD, Hooper S, Tozluoglu M, Bruckbauer A, Fletcher G, Erler JT, Bates PA, Thompson B, Sahai E. STRIPAK components determine mode of cancer cell migration and metastasis. Nat Cell Biol. 2015;17(1):68–80.
Zhang Y, Tang W, Zhang H, Niu X, Xu Y, Zhang J, Gao K, Pan W, Boggon TJ, Toomre D, et al. A network of interactions enables CCM3 and STK24 to coordinate UNC13D-driven vesicle exocytosis in neutrophils. Dev Cell. 2013;27(2):215–26.
Chen YL, Wang CY, Fang JH, Hsu HP. Serine/threonine-protein kinase 24 is an inhibitor of gastric cancer metastasis through suppressing CDH1 gene and enhancing stemness. Am J Cancer Res. 2021;11(9):4277–93.
Hsu HP, Wang CY, Hsieh PY, Fang JH, Chen YL. Knockdown of serine/threonine-protein kinase 24 promotes tumorigenesis and myeloid-derived suppressor cell expansion in an orthotopic immunocompetent gastric cancer animal model. J Cancer. 2020;11(1):213–28.
Iglesias C, Floridia E, Sartages M, Porteiro B, Fraile M, Guerrero A, Santos D, Cuñarro J, Tovar S, Nogueiras R, et al. The MST3/STK24 kinase mediates impaired fasting blood glucose after a high-fat diet. Diabetologia. 2017;60(12):2453–62.
Caputo M, Kurhe Y, Kumari S, Cansby E, Amrutkar M, Scandalis E, Booten SL, Ståhlman M, Borén J, Marschall HU, et al. Silencing of STE20-type kinase MST3 in mice with antisense oligonucleotide treatment ameliorates diet-induced nonalcoholic fatty liver disease. Faseb j. 2021;35(5): e21567.
Cansby E, Kulkarni NM, Magnusson E, Kurhe Y, Amrutkar M, Nerstedt A, Ståhlman M, Sihlbom C, Marschall HU, Borén J, et al. Protein kinase MST3 modulates lipid homeostasis in hepatocytes and correlates with nonalcoholic steatohepatitis in humans. Faseb j. 2019;33(9):9974–89.
Chan CH, Wu SN, Bao BY, Li HW, Lu TL. MST3 involvement in Na(+) and K(+) homeostasis with increasing dietary potassium intake. Int J Mol Sci. 2021;22(3):999.
Ultanir SK, Yadav S, Hertz NT, Oses-Prieto JA, Claxton S, Burlingame AL, Shokat KM, Jan LY, Jan YN. MST3 kinase phosphorylates TAO1/2 to enable Myosin Va function in promoting spine synapse development. Neuron. 2014;84(5):968–82.
Zhang Y, Hu H, Tian T, Zhang L, Zhao D, Wu Q, Chang Y, Wang Q, Zhou S, Feng G, et al. Mst3b promotes spinal cord neuronal regeneration by promoting growth cone branching out in spinal cord injury rats. Mol Neurobiol. 2015;51(3):1144–57.
Shi Z, Zhou Z. MST kinases in innate immune signaling. Cell Stress. 2017;2(1):4–13.
Wang Y, Jia A, Cao Y, Hu X, Wang Y, Yang Q, Bi Y, Liu G. Hippo kinases MST1/2 regulate immune cell functions in cancer, infection, and autoimmune diseases. Crit Rev Eukaryot Gene Expr. 2020;30(5):427–42.
Mollinedo F. Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol. 2019;40(3):228–42.
Liu Y, Wang TV, Cui Y, Li C, Jiang L, Rao Y. STE20 phosphorylation of AMPK-related kinases revealed by biochemical purifications combined with genetics. J Biol Chem. 2022;298(5): 101928.
Liu Y, Wang TV, Cui Y, Gao S, Rao Y. Biochemical purification uncovers mammalian sterile 3 (MST3) as a new protein kinase for multifunctional protein kinases AMPK and SIK3. J Biol Chem. 2022;298(5): 101929.
Stegert MR, Hergovich A, Tamaskovic R, Bichsel SJ, Hemmings BA. Regulation of NDR protein kinase by hydrophobic motif phosphorylation mediated by the mammalian Ste20-like kinase MST3. Mol Cell Biol. 2005;25(24):11019–29.
Mehellou Y, Alessi DR, Macartney TJ, Szklarz M, Knapp S, Elkins JM. Structural insights into the activation of MST3 by MO25. Biochem Biophys Res Commun. 2013;431(3):604–9.
Li N, Wang Y, Neri S, Zhen Y, Fong LWR, Qiao Y, Li X, Chen Z, Stephan C, Deng W, et al. Tankyrase disrupts metabolic homeostasis and promotes tumorigenesis by inhibiting LKB1-AMPK signalling. Nat Commun. 2019;10(1):4363.
Olesen SH, Zhu JY, Martin MP, Schönbrunn E. Discovery of diverse small-molecule inhibitors of mammalian sterile20-like kinase 3 (MST3). ChemMedChem. 2016;11(11):1137–44.
Luo F, Zhou J, Wang S, Sun Z, Han Q, Bai C. microRNA-222 promotes colorectal cancer cell migration and invasion by targeting MST3. FEBS Open Bio. 2019;9(5):901–13.
Irwin N, Li YM, O’Toole JE, Benowitz LI. Mst3b, a purine-sensitive Ste20-like protein kinase, regulates axon outgrowth. Proc Natl Acad Sci U S A. 2006;103(48):18320–5.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 31872634).
Author information
Authors and Affiliations
Contributions
QJ, XJ, contributed to writing the manuscript. XJ, JL and WX prepared Figs. 1, 2, 3, 4 and Tables 1, 2, 3, 4. ZK revised the language of the manuscript. YH designed and modified the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors contributed to the article and approved the submitted version.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qiu, J., Xiong, J., Jiang, L. et al. Molecular mechanisms involved in regulating protein activity and biological function of MST3. Cell Div 18, 8 (2023). https://doi.org/10.1186/s13008-023-00090-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13008-023-00090-x