
Abstract
Axenfeld–Rieger Syndrome (ARS) is a rare autosomal dominant genetic disease with considerable expressive variability, characterized by ocular and non-ocular manifestations, cardiovascular, mild craniofacial abnormalities and dental malformations. Current data report an incidence of Xenfeld-Rieger syndrome in the population of 1: 200,000.
The case described is that of a 14-year-old female patient whose ARS is suspected and investigated following a dental specialist visit for orthodontic reasons, acquired the patient’s family and clinical data following a medical approach multidisciplinary, we proceed to the orthodontic involved the use of the Rapid Palatal Expander (RPE) and a fixed orthodontic treatment.
The aim of this study is to report the case of the orthopaedic and orthodontic treatment in a patient affected by ARS and with facial dysmorphism and teeth anomalies associated to ocular anomalies.
Similar content being viewed by others


Introduction
Axenfeld–Rieger Syndrome (ARS) is a rare autosomal dominant genetic disease with considerable expressive variability [1], characterized by ocular and non-ocular manifestations (cardiovascular, mild craniofacial abnormalities and dental malformations). In some cases, patients may have short stature, mental retardation and finger malformations [2,3,4,5,6]. An abnormal migration and differentiation of neural crest cells are considered responsible for anomalies in ocular, craniofacial and dental development. In this syndrome, some authors trace various clinical conditions such as Axenfeld’s anomaly, Rieger’s anomaly and Rieger’s syndrome, even if analysis of the genetic profiles suggests different pathological conditions [7].
It is a heterogeneous disorder inherited in an autosomal dominant manner and shows significant expressive variability both between families and within the same family [8] .
Current data report an incidence of Xenfeld-Rieger syndrome in the population of 1: 200,000 while there is no definitive data on a gender and racial prevalence [9, 10]. The mutation in genes of two transcription factors, FOXC1, FOXC2, FKHL7, PANCR and PITX2 [11,12,13,14], represent almost half of the known cases while the genetic defect is not known in 60%.
Defects of the ectodermal tissue with involvement of the cells of the neuronal crest, which arise at the 3rd trimest of pregnancy, can be the cause of the facial morphological abnormalities of the maxilla, jaw, hypertelorism, teeth, saddle and non-involution of the skin periumbilical [15].
Clinical aspects may concern ocular alterations (malformations of the ocular anterior segment) and pathologies such as: heterochromia, aniridia, coloboma of the iris [16], an a prominent anteriorly displaced Schwalbe’s line. Pupil abnormalities in form, position and number occur. Additional ocular problems of ARS cause increased intraocular pressure resulting in glaucoma in approximately 50% of children with ARS. Other ocular anomalies include corneal opacities, lens defects, conjunctival xerosis, blue sclera, chroidal hypoplasia and retinal detachment [17]. Craniofacial malformations include maxillary hypoplasia hyperplastic maxillary, cleft palate, mandibular protrusion with anterior open bite, hypertelorism a broad nasal bridge and an enlarged sella turcica. The face appears to be flattened, with a prominent forehead and a flat, broad nasal root. Widely spaced eyes, a broad flat bridge of the nose and / or a protruding lower lip. Dental alterations include hypodontia or partial anodontia, microdontia, taurodontia, abnormally shaped teeth, peg-shaped teeth, enamel hypoplasia, hyperplastic maxillary labial frenum [18].
In addition, the presence of periumbilical skin, ear malformations with conductive deafness, anomalies of the pituitary gland, congenital heart defects renal malformation, kyphosis, scoliosis, maldevelopment of the sternum, alterations of the extremities, myotonic dystrophy with muscular atrophy, growth retardation, lypodystrophy, lack of vision, drooping eyelids may be present.
Some patients present mental retardation with problems with learning, language and behavior.
There are few reports concerning case presentations of patients with ARS and few papers [19,20,21,22] concerning orthopedic-orthodontic treatment relative to the patients with this syndrome. The aim of this study is to report the case of the orthopedic and orthodontic treatment in a patient affected by ARS and with facial dysmorphism and teeth anomalies associated to ocular anomalies.
Case report
An Italian female patient of 14 years came to our observation from the Ophthalmology Department of the University of Bari for dental evaluation and orthodontic therapy to correct problems of bad occlusion with misalignment of the teeth.
The patient was examined clinically and thoroughly and a family and medical history recorded. Clinical and medical findings led to the clinical diagnosis of Axenfeld-Rieger syndrome.
The family pedigree is shown in Fig. 1.

Pedigree of three generations of the family of the patient with Axenfeld-Rieger Syndrome
Family tree show that most of the family does not affected by Reiger syndrome. The cousins are affected by celiac disease, moreover the family history shows how the parents had 3 spontaneous abortions.
Specialist medical examinations report the following clinical data: psycho-intellectual and motor retardation, skeletal age retardation with vertebral lordosis and joint laxity (Figs. 2 and 3), speech delay, severe bilateral sensorineural hearing loss and the presence of bilateral congenital glaucoma.

Extraoral photo (a) frontal view before treatment; b lateral right view before treatment

Profile image of the patient at 8 years of age with evidence of joint laxity and lordosis
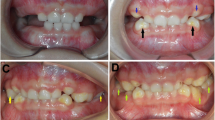
The intra-oral examination shows the presence of open bite with enamel hypoplasia affecting the upper central incisors, lower diastema due to lingual thrust with non-coincident intericisive lines (Fig. 4) the presence of conoid-like teeth and tarurodontism with some elements that radiographically showed signs of ankylosis, transverse contraction of the upper jaw and dental crowding. The laterolateral teleradiography (L-L rx) (Fig. 5a) and the relative cephalometric analysis according to Jarabak revealed the presence of a skeletal Class III (subspinale(A)-nasion(N)-suprmanetlae (B) angle ANB = − 5°), skeletal open-bite, hyperdivergence, mandibular growth with vertical growth pattern (Table 1). The aim of the treatment was to correct or at least to avoid the worsening of the skeletal relationship between the upper and lower jaws and to resolve the crowding in the upper arch. The therapy for malocclusion involved the use of the Rapid Palatal Expander (RPE) and a fixed orthodontic treatment.

Presence of open bite with enamel hypoplasia on elements 1.1 and 2.1

a-b Latero-lateral teleradiography of the head before treatment; Latero-lateral teleradiography of the head with RPE
After a plurispecialized medical examination, an oral hygiene and prophylaxis program with recurrent medical checks, the patient underwent conservative therapies.
Then the patient was subjected an orthopedic-orthodontic treatment divided in two steps:
-
Orthopedic treatment with RPE with a protocol of two activations per day for 15 days in order to correct the transversal contraction of the upper jaw, maintained for 1 year.
Therefore, the fixed orthodontic treatment was started.
-
Orthodontic treatment of the upper arch with Straight Wire technique (orthodontic archwire sequence: NiTi .014; NiTi .016; NiTi .018; NiTi .016x.022; Stainless steel .016x.022) was performed. This treatment allowed the dental alignment, increased the space in the dental arch to solve dental crowding and improved the cross-bite and the open bite through strategic positioning of the brackets (Figs. 5b and 6).

Panoramic X-ray
Discussion
The malformations of the anterior chamber of the eye and the presence of dental anomalies are fundamental elements that characterize ARS. The only involvement of the eyes is called Rieger anomaly or Axenfeld anomaly or hypoplasia of the iris, while the presence of symptoms and of extraocular signs preponderate towards the ARS syndrome. These 2 conditions can be superimposed suggesting a single pathological entity, but genetic analyzes on patients diagnosed with Rieger and ARS anomaly provide information on the presence of 2 slightly different conditions.
The disease is autosomal dominant with variable penetrance; in our case report the genealogical survey did not show the presence of relatives diagnosed with ARS even if they showed a predisposition to neoformative pathologies [23], therefore a reduced penetrance of the syndrome is assumed. Another feature is the absence of dental agenesis while only dental anomalies (enamel hypoplasia localized on some teeth) were present. The shape of the sella turcica in the present case was apparently normal, an open bite and a skeletal class III was present (Fig. 5). In fact, maxillary hypoplasia was observed in about 90.5% of cases, with a concave facial profile and a flat medial face [24]. This patient did not have the classic dental presentation of ARS, such as the lack of central and lateral incisors and canines described by other authors. In fact, the treatment only involved the use of RPE and fixed orthodontic treatment for alignment of the dental elements. However, the fundamental orthodontic treatment to partially respond to aesthetic needs can be problematic due to the presence of dental anomalies (short and curved roots, ankylosis and poor vertical dimension of the bone alveolar ridge). Some authors recommend a post adolescent orthodontic surgical approach [22]. Patients with rare diseases often presented specific orthodontic risks associated with the systemic disorders. Therefore, orthodontists must understand the orthodontic risks associated with these conditions and must carefully design an orthodontic treatment plan to minimize the risks. In patients with ARS, the risks of orthodontic treatment have not been clear in part because few cases of orthodontic treatment have been reported [21]. A priority aspect for this type of patient is the maintenance of correct oral hygiene as for all patients suffering from rare diseases [25,26,27,28,29,30,31] in order to safeguard the periodontal condition having a reduced size of the mandibular and maxillary bone crest and the presence of short roots with little presence of adherent gingiva [32, 33]. These factors can determine the premature loss of tooth following periodontal disease, furthermore the maintenance of periodontal health is fundamental for implant rehabilitation in sectors with agenesis or where there has been the loss of dental elements, not easy to plan as described by Pirih et al. for the reduction of bone thickness [34].
Waldron et al. report the presence of osteopenia of the bones in association with this syndrome (reduced calcification, decreased density or reduced bone mass) with predisposition to osteoporosis, recommending the intake of vitamin D and physical exercise for the prevention of this complication [35].
Dental and orthodontic treatment plays a fundamental role so that patients suffering from the syndrome improve the functional and phonetic aspects compromised by malocclusion and hypodontics. Furthermore, improving the aesthetics of the smile as well as the support of the upper lip and the general appearance helps patients in their relationship life. In fact, delaying aesthetic dental rehabilitation can lead to discomfort in young patients with impaired social life at school age [19].
Genetic aspects
The genes, mainly involved in the mutation responsible for 50% of ARS cases, are forkhead box C1 (FOXC1) and pituitary homeobox 2 (PITX2).
DNA mutations involving PITX2, located at 4q25 [36], result in ARS in which patients have ocular phenotypes often seen with craniofacial and dental abnormalities, while mutations in FOXC1 located at 6p25 result in ARS defined by ocular phenotypes observed with cardiovascular defects and sensorineural hearing loss.
The presence of the mutation of these two genes was investigated in a multicentric study conducted by Souzeau et al. which included 34 individuals from 18 families: FOXC1 variants were present in 64.7% of individuals and PITX2 variants in 35.3% of individuals [37].
Other genes have however been taken into consideration and investigated in the pathogenesis of ARS; in fact, there seems to be an involvement of a sequence between the PITX2 gene and the noncoding PANCR gene as reported by Qui et al. [13] whose mutation is involved in congenital glaucoma and other forms of glaucoma [38]. heterozygous missense variant in the PRDM5 gene localizes to human chromosome 4q26 is reported by Micheal et al. associated with PITX2 [39].
While more and more frequently mutations of the FKHL7 gene located at 6p25 is reported in the association ARS [40,41,42,43], also some sporadic cases of ARS have been related to deletion of the PAX6 gene (still controversial and to be definitively demonstrated [44]) at 11p13 and deletion of the 16q23-q24 region; a second locus for Rieger syndrome located on chromosome 13q14 has also recently been identified [45].
Conclusions
Early diagnosis of ARS is essential to avoid complications caused by glaucoma, and patients should also undergo repeated eye, pediatric, physiotherapy and speech therapy checks to maintain psychophysical health and for a more favorable insertion into social life. While, the dental management of these patients must be aimed at the maintenance of dental hygiene and must be aimed to the morpho-functional restoration with a multidisciplinary approach that includes orthodontic, oral and maxillofacial surgery, implant surgery and restorative treatments together with speech and language therapy.
Availability of data and materials
Not aplicable.
References
Cunningham ET Jr, Eliott D, Miller NR, Maumenee IH, Green WR. Familial Axenfeld-Rieger anomaly, atrial septal defect, and sensorineural hearing loss: a possible new genetic syndrome. Arch Ophthalmol. 1998;116(1):78–82.
Bausz M, Csidey M, Csakany B, Nemeth O, Nagy ZZ, Maka E. Axenfeld-Rieger syndrome: ophthalmological and dental diagnostic and therapeutic options. Orv Hetil. 2021;162(5):192–9.
Rao A, Padhy D, Sarangi S, Das G. Unclassified Axenfeld-Rieger syndrome: a CASE SERIES and review of literature. Semin Ophthalmol. 2018;33(3):300–7.
Kleinmann RE, Kazarian EL, Raptopoulos V, Braverman LE. Primary empty Sella and Rieger's anomaly of the anterior chamber of the eye: a familial syndrome. N Engl J Med. 1981;304(2):90–3.
Golaszewska K, Dub N, Saeed E, Mariak Z, Konopinska J. Axenfeld-Rieger syndrome combined with a foveal anomaly in a three-generation family: a case report. BMC Ophthalmol. 2021;21(1):154.
Tsai JC, Grajewski AL. Cardiac valvular disease and Axenfeld-Rieger syndrome. Am J Ophthalmol. 1994;118(2):255–6.
Grosso S, Farnetani MA, Berardi R, Vivarelli R, Vanni M, Morgese G, et al. Familial Axenfeld-Rieger anomaly, cardiac malformations, and sensorineural hearing loss: a provisionally unique genetic syndrome? Am J Med Genet. 2002;111(2):182–6.
Perveen R, Lloyd IC, Clayton-Smith J, Churchill A, van Heyningen V, Hanson I, et al. Phenotypic variability and asymmetry of Rieger syndrome associated with PITX2 mutations. Invest Ophthalmol Vis Sci. 2000;41(9):2456–60.
O'Dwyer EM, Jones DC. Dental anomalies in Axenfeld-Rieger syndrome. Int J Paediatr Dent. 2005;15(6):459–63.
Seifi M, Walter MA. Axenfeld-Rieger syndrome. Clin Genet. 2018;93(6):1123–30.
French CR. Mechanistic insights into Axenfeld-Rieger syndrome from Zebrafish foxc1 and pitx2 mutants. Int J Mol Sci. 2021;22(18):10001.
Fan Z, Sun S, Liu H, Yu M, Liu Z, Wong SW, et al. Novel PITX2 mutations identified in Axenfeld-Rieger syndrome and the pattern of PITX2-related tooth agenesis. Oral Dis. 2019;25(8):2010–9.
Qin Y, Gao P, Yu S, Li J, Huang Y, Jia D, et al. A large deletion spanning PITX2 and PANCR in a Chinese family with Axenfeld-Rieger syndrome. Mol Vis. 2020;26:670–8.
Hjalt TA, Semina EV. Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med. 2005;7(25):1–17.
Shields MB. Axenfeld-Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc. 1983;81:736–84.
Verloes A, Dodinval P. Rieger anomaly and uveal coloboma with associated anomalies. Third observation of a rare oculo-palato-osseous syndrome--the Abruzzo-Erikson syndrome. Ophthalmic Paediatr Genet. 1990;11(1):41–7.
Cella W, de Vasconcellos JP, de Melo MB, Kneipp B, Costa FF, Longui CA, et al. Structural assessment of PITX2, FOXC1, CYP1B1, and GJA1 genes in patients with Axenfeld-Rieger syndrome with developmental glaucoma. Invest Ophthalmol Vis Sci. 2006;47(5):1803–9.
Berenstein-Aizman G, Hazan-Molina H, Drori D, Aizenbud D. Axenfeld-Rieger syndrome: dentofacial manifestation and oral rehabilitation considerations. Pediatr Dent. 2011;33(5):440–4.
Siddiqui HP, Sennimalai K, Samrit VD, Duggal R, Yadav R. Adjunctive orthodontic therapy for prosthetic rehabilitation in a growing child with Axenfeld-Rieger syndrome: a case report. Spec Care Dentist. 2021;41(3):423–30.
Dunbar AC, McIntyre GT, Laverick S, Stevenson B. Axenfeld–Rieger syndrome: a case report. J Orthod. 2015;42(4):324–30.
Seki D, Takeshita N, Seiryu M, Deguchi T, Takano-Yamamoto T. Improvement of open bite and stomatognathic function in an Axenfeld-Rieger syndrome patient by orthodontic sectional arch mechanics: clinical considerations and the risk of orthodontic tooth movement. Acta Med Okayama. 2019;73(3):255–62.
Bender CA, Koudstaal MJ, Van Elswijk JF, Prahl C, Wolvius EB. Two cases of axenfeld-rieger syndrome, report of the complex pathology and treatment. Cleft Palate Craniofac J. 2014;51(3):354–60.
Gilding LN, Somervaille TCP. The diverse consequences of FOXC1 deregulation in Cancer. Cancers (Basel). 2019;11(2):184.
Dressler S, Meyer-Marcotty P, Weisschuh N, Jablonski-Momeni A, Pieper K, Gramer G, et al. Dental and craniofacial anomalies associated with Axenfeld-Rieger syndrome with PITX2 mutation. Case Rep Med. 2010;2010:621984.
Cazzolla AP, Lo Muzio L, Di Fede O, Lacarbonara V, Colaprico A, Testa NF, et al. Orthopedic-orthodontic treatment of the patient with Turner's syndrome: review of the literature and case report. Spec Care Dentist. 2018;38(4):239–48.
Cazzolla A, Zhurakivska K, Ciavarella D, Lacaita M, Favia G, Testa N, et al. Primary hyperoxaluria: orthodontic management in a pediatric patient: a case report. Spec Care Dentist. 2018;38(4):259–65.
Cazzolla AP, Lacaita MG, Lacarbonara V, Zhurakivska K, De Franco A, Gissi I, et al. Orthopedic and orthodontic management in a patient with DiGeorge syndrome and familial Mediterranean fever: a case report. Spec Care Dentist. 2019;39(3):340–7.
Arriagada-Vargas C, Abeleira-Pazos MT, Outumuro-Rial M, Garcia-Mato E, Varela-Aneiros I, Limeres-Posse J, et al. Rare disorders: diagnosis and therapeutic planning for patients seeking orthodontic treatment. J Clin Med. 2022;11(6):1527.
Hanisch M, Hanisch L, Kleinheinz J, Danesh G, Benz K, Jackowski J. Orthodontically relevant manifestations in people with rare diseases. Med Princ Pract. 2019;28(3):216–21.
Taddei M, D'Alessandro G, Amunni F, Piana G. Orthodontic treatment of a particular subgroup of children with special health care needs, children with craniofacial anomalies: an analysis of treatment length and clinical outcome. Angle Orthod. 2016;86(1):115–20.
Blanck-Lubarsch M, Hohoff A, Wiechmann D, Stamm T. Orthodontic treatment of children/adolescents with special health care needs: an analysis of treatment length and clinical outcome. BMC Oral Health. 2014;14:67.
Cervino G, Cicciù M, De Stefano R, Falcomatà D, Bianchi A, Crimi S, et al. Oral health in patients with Marfan syndrome. Arch Oral Biol. 2020;116:104745.
Marra PM, Parascandolo S, Fiorillo L, Cicciù M, Cervino G, D'Amico C, et al. Dental trauma in children with autistic disorder: a retrospective study. Biomed Res Int. 2021;2021:3125251.
Pirih FQ, Casarin M, Perussolo J, Ozaki M, Carnio JG, Camargo PM, et al. Rieger syndrome: rehabilitation with dental implants. Clin Adv Periodontics. 2019;9(4):172–6.
Waldron JM, McNamara C, Hewson AR, McNamara CM. Axenfeld-Rieger syndrome (ARS): a review and case report. Spec Care Dentist. 2010;30(5):218–22.
Zhang F, Zhang L, He L, Cao M, Yang Y, Duan X, et al. A PITX2 splice-site mutation in a family with Axenfeld-Rieger syndrome leads to decreased expression of nuclear PITX2 protein. Int Ophthalmol. 2021;41(4):1503–11.
Souzeau E, Siggs OM, Pasutto F, Knight LSW, Perez-Jurado LA, McGregor L, et al. Gene-specific facial dysmorphism in Axenfeld-Rieger syndrome caused by FOXC1 and PITX2 variants. Am J Med Genet A. 2021;185(2):434–9.
Tanwar M, Dada T, Dada R. Axenfeld-Rieger syndrome associated with congenital Glaucoma and cytochrome P4501B1 gene mutations. Case Rep Med. 2010;2010:212656.
Micheal S, Siddiqui SN, Zafar SN, Venselaar H, Qamar R, Khan MI, et al. Whole exome sequencing identifies a heterozygous missense variant in the PRDM5 gene in a family with Axenfeld-Rieger syndrome. Neurogenetics. 2016;17(1):17–23.
Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, et al. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet. 1998;63(5):1316–28.
Mirzayans F, Gould DB, Héon E, Billingsley GD, Cheung JC, Mears AJ, et al. Axenfeld-Rieger syndrome resulting from mutation of the FKHL7 gene on chromosome 6p25. Eur J Hum Genet. 2000;8(1):71–4.
Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, et al. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998;19(2):140–7.
Suzuki T, Takahashi K, Kuwahara S, Wada Y, Abe T, Tamai M. A novel (Pro79Thr) mutation in the FKHL7 gene in a Japanese family with Axenfeld-Rieger syndrome. Am J Ophthalmol. 2001;132(4):572–5.
Riise R, D'Haene B, De Baere E, Grønskov K, Brøndum-Nielsen K. Rieger syndrome is not associated with PAX6 deletion: a correction to Acta Ophthalmol Scand 2001: 79: 201-203. Acta Ophthalmol. 2009;87(8):923.
Phillips JC, del Bono EA, Haines JL, Pralea AM, Cohen JS, Greff LJ, et al. A second locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet. 1996;59(3):613–9.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
M.D. and A.P.C. writing; bibliographic research, M.D.;review and editing: A.C. and F.S.; data analysis: A.P.C, N.F.T. and M.D.C.; revising the work critically for important intellectual content: A.B., S.C., supervision and project administration: L.L.M., D.C.and V.C.; The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained from all subjects involved in the study.
Consent for publication
All authors have read and agreed to the published version of the manuscript.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cazzolla, A.P., Testa, N.F., Spirito, F. et al. Axenfeld–Rieger syndrome: orthopedic and orthodontic management in a pediatric patient: a case report. Head Face Med 18, 25 (2022). https://doi.org/10.1186/s13005-022-00329-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13005-022-00329-y