
Abstract
Deleterious mutations in the BRCA1 and BRCA2 genes have significant therapeutic relevance in clinical settings regarding personalized therapy approaches. BRCA1 and BRCA2 play a pivotal role in homologous recombination (HR) and thus are sensitive for PARP inhibitors (PARPi). Beyond the narrow scope of evaluating only the BRCA mutation status, PARPi can be beneficial for HR deficient (HRD) patients, who harbor mutations in other HR-associated genes. In the present retrospective study, a novel targeted HRD gene panel was validated and implemented for use with FFPE tissue. Samples of patients with ovarian, breast, pancreatic and prostate cancer were included. Variants were robustly detected with various DNA input amounts and the use of test samples showed complete concordance between previously known mutations and HRD panel results. From all the 90 samples included in this cohort, TP53 was the most frequently altered gene (73%). Deleterious BRCA1/2 mutations were found in 20 (22%) of all samples. New pathogenic or likely pathogenic mutations in additional HR-associated genes were identified in 22 (24%) patients. Taken together, the present study proves the feasibility of a new HRD gene panel with reliable panel performance and offers the possibility to easily screen for resistance mutations acquired over treatment time.
Mutations in HR-associated genes, besides BRCA1/2, might represent promising potential targets for synthetic lethality approaches. Thus, a substantial number of patients may benefit from expanding the scope of therapeutic agents like PARPi.
Similar content being viewed by others

Background
Over the past years, the clinical and therapeutic significance of evaluating the mutational status of genes accounting for homologous recombination deficiency (HRD) has increased. Various studies demonstrated the prevalence of HRD in multiple tumor types like ovarian, breast, pancreatic and prostate cancer [1,2,3]. Those findings suggest an increasing number of patients who could benefit from specific therapies of HR deficient solid tumors.
Homologous recombination (HR) is a cellular mechanism used to restore DNA following double strand breaks (DSB), which considers the original sequence through using a homologous sister chromatid as template. Several genes like ATM, NBN, RAD50, PALB2 but especially BRCA1 and BRCA2 are involved in HR and represent targets for therapy. Defects and disrupted function in recombination genes result in the accumulation of mutations and in the necessity to rely on non-homologous end joining (NHEJ) for DSB repair. Repairing damaged DNA by NHEJ leads to fatal errors and subsequently to cell death [4,5,6].
Tumors deficient in HR are sensitive to platinum-based chemotherapy and inhibitors of Poly (ADP-ribose) polymerase (PARP). PARP1 regulates the restoration of DNA by Base Excision Repair (BER). Normal cells are able to use both reparation mechanisms. Thus, due to dysfunctional proteins in those pathways, HR deficient tumor cells treated with PARP inhibitors (PARPi) subsequently accumulate lethal DNA defects referred to as synthetic lethality [7]. The cell is eventually no longer able to tolerate the damage, which results in cell death [8, 9].
In 2014, the U.S. Food and Drug Administration (FDA) approved Lynparza (olaparib) as the first PARP inhibitor in advanced ovarian cancer for maintenance therapy with germline mutations in BRCA1 and BRCA2 genes, which function as predictive biomarkers. In the following years, the FDA and the European Medicines Agency (EMA) approved additional PARPi naming rucaparib, niraparib, and talazoparib for treatment in several cancer types [8].
In following studies demonstrating the proficient effect of PARP inhibition in breast, prostate and pancreatic cancers, PARP1 emerged as a new target for platinum-sensitive tumors, thereby helping personalized therapy and HRD testing in routine diagnostics to receive increased relevance and importance. In addition to defects in BRCA1/2, also loss-of-function mutations in ATM, RAD50, PALB2 or other crucial HR genes equally may contribute to HR deficiency [4, 10, 11].
Mutations in BRCA1/2 genes (BRCAm) were most frequently identified in ovarian (OvCa, 15.2%) followed by prostate (PC, 10.7%), breast (BC, 8.8%) and pancreatic cancer (PaC, 5.2%) [12]. But the HRD phenotype can also manifest independent of BRCAm in those cancer types with mutations in other HR associated genes that occur with a prevalence of 9.2% for OvCa [13], 11% for BC [14], 6% for PaC [15] and 16% for PC [16]. The consequences of HRD can be assessed at the DNA level by the identification of genomic scars resulting in a genomic instability score or HRD score.
Due to the FDAs original approval of olaparib based on the BRCA1/2 status in ovarian cancer, current standard testing does not always include other HR genes, despite the fact that the FDAs approval for olaparib as treatment for castration-resistant PC was based on the findings of the PROfound trail which also included ATM as a predictive biomarker [17]. This demonstrates that further HR associated genes might be able to act as predictive biomarkers and therefore, more patients could benefit from therapy with PARPi or platinum-based chemotherapy.
The purpose of this retrospective study was to identify ovarian, breast, prostate and pancreatic cancer patients who may have additional mutations and were previously only tested for BRCA1 and BRCA2 but may benefit from PARPi. In total, 42 h-associated genes were included in the evaluation by means of a novel targeted next-generation sequencing (NGS) panel. Formalin-fixed, paraffin-embedded (FFPE) samples of 90 patients, which were previously screened for BRCA1/2 mutations, were analyzed. Furthermore, including the analysis of additional genes in routine diagnostic testing for HRD may generate new insights into PAPRi resistance mechanisms and represents the basis of personalized oncology approaches.
Materials and methods
Patient selection
For validation, 24 formalin-fixed, paraffin-embedded (FFPE) samples with known mutation status of BRCA1/2 or ATM, consisting of 11 ovarian, three breast cancer samples and ten colorectal liver metastases (CRLM) previously evaluated by whole exome sequencing [18] were used as a training cohort. In addition to those samples, a commercially available BRCA Somatic Multiplex FFPE standard (HD810, Horizon Discovery LTD., Group, Cambridge, UK) with 16 mutations in BRCA1, BRCA2, BRIP1, BARD1 and NBN was included.
For the subsequent retrospective analysis of HRD prevalence, samples, which were tested for BRCA1/2 mutations earlier were examined. Of those, 90 fit the inclusion criteria defined as follows: tumor cell count of at least 30%, sufficient DNA concentration and volume for an input of 100 ng that would allow for library preparation. The amount of tumor cells was quantitatively assessed by means of hematoxylin and eosin staining. Imitating the circumstances of a routine diagnostic setting, primary tumors and metastases of ovarian (n = 63), breast (n = 19), pancreatic (n = 9) and prostate (n = 5) cancer were included regardless of the histological subtype of the tumor. The local ethics committee (Ethics Committee of the Rhineland-Palatinate State Medical Association, Mainz, Germany) approved the study design and all samples were handled in compliance with the standards proposed by the Declaration of Helsinki.
DNA extraction and quantification
Tumor-specific genomic DNA extraction from unstained tissue slides was performed using Maxwell RSC FFPE Plus DNA Kit (Promega, Madison, WI, USA) according to the manufacturer’s technical manual, with proteinase K digestion overnight. Quantification of DNA samples was conducted with Qubit DNA HS assay Kit (Thermo Fisher Scienfific, Waltham, MA, USA).
Targeted next-generation sequencing
The QIAseq targeted DNA workflow (Qiagen, Hilden, Germany) was used to generate NGS libraries. The novel QIAseq panel includes 42 genes that are involved in DNA damage repair (ARID1A, ATM, ATR, ATRX, BAP1, BARD1, BLM, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, ERCC3, FAM175A, FANCA, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, HDAC2, MLH3, MRE11A, NBN, PALB2, PPP2R2A, RAD50, RAD51, RAD51B, RAD51C, RAD51D, RAD52, RAD54L, RNASEH2A, RNASEH2B, RNASEH2C, TP53, WRN, XRCC2). The panel contains 799 amplicons with 1893 primer pairs and covers the complete coding sequence of all 42 genes and flanking intronic sequences. Typically, 100 ng genomic DNA was used as input and library preparation was performed according to the QIAseq Targeted DNA Panel Handbook. Briefly, in a multi-enzyme reaction, genomic DNA samples were fragmented, end repaired and A-tailed. Following sample indexing with unique molecular index (UMI), the targets were enriched by single primer extension. The final library was amplified by a universal primer.
For sequencing, the libraries were pooled equivalently, denatured and 8 million reads were assessed to one sample. The final loading concentration varied from 1.1 pM to 1.3 pM. Paired-end libraries were sequenced on an Illumiina NextSeq 500 with v2.5 reagent kit chemistry (Illumina, San Diego, Ca, USA) using 2 × 150 bp output with usage of a custom sequencing primer for read 1 (QIAseq A Read1 Primer I).
Data analysis and variant calling
Generated bcl files were converted to FASTQ format and imported into the CLC Genomics Workbench version 12.0 (Qiagen Bioinformatics). This software was used to analyze and annotate sequenced samples on the basis of the human reference genome GRCh37 (hg19). Variant annotation was based on RefSeq accession numbers. The detection limit of variants was set to 10% allele frequency. Polymorphisms were excluded from further analysis by filtering against dbSNP. Furthermore, intron variants with potential splice effect were only taken into account if they were located one or two base pairs outside the exon. To ensure sufficient quality and sensitivity of the sequenced samples, only targets with a minimum coverage above 100 reads were considered. Targets with a read depth below 100x were excluded from the analysis.
Data interpretation
The interpretation and classification of the detected variants was based on the guidelines of the American College of Medical Genetics and Genomics (ACMG) [19]. Pathogenicity was assessed by the five-tier system of classification: [1] pathogenic, [2] likely pathogenic, [3] uncertain significance, [4] likely benign, or [5] benign. Detected variants, which lead to a premature stop codon, a frameshift in the open reading frame or disruption of the splice site, were classified as likely pathogenic and therefore included in the tier 2 category. Remaining variants of uncertain significance (VUS) with no entry in the common databases were submitted to the prediction tools SIFT [20] and CADD [21] for functional analysis. Based on the predicted result, VUS were processed as likely pathogenic or likely benign. All Variants in tier 4 or 5 were dismissed and excluded from further analysis.
Results
Validation and implementation of a custom next generation sequencing panel for selected HRD associated genes
All 42 genes included in this panel play a pivotal role in HR or stand in close functional relation to the DNA repair pathway [22, 23]. Genes included in this panel were chosen based on publications listed in Supplementary Table S1.
To implement and assess the sequencing output of the new HRD panel, previously analyzed samples and a BRCA Somatic Multiplex FFPE standard were used. Only tumour samples with > 30% tumor cellularity were included and only mutations with > 10% frequency were considered. Large-scale deletions of several hundred or thousand base pairs cannot be identified with the current NGS panel. After initial sequencing, it became apparent that the coverage in specific regions was insufficient throughout all samples. In order to allow for appropriate evaluation of the variance with high predictive confidence, a cut-off coverage of at least 100 reads per target region was applied for the HRD panel. Therefore, a booster panel (containing 34 primers) was designed, which improved the coverage in those specific areas and increased the number of included primers from originally 1859 to 1893 (Supplementary Fig. S1).
The optimal DNA input amount was determined with a BRCA Somatic Multiplex FFPE standard by using decreasing amounts of gDNA from 100 ng to 5 ng as input for library preparation. Here, the detection limit was reached at 15 ng DNA input as one variant was undetectable (Supplementary Fig. S2). Thus, 100 ng of DNA was used as input for HRD library preparation in further analyses.
Variant detection performance was evaluated by 24 samples of the training cohort with known mutations in either BRCA1/2 or other HR-associated genes. Comparative analyses of the previous results and the HRD panel outcome showed complete concordance regarding variant detection throughout all samples (Supplementary Fig. S3). Those corresponding findings concluded the validation and implementation process of the new panel.
Effective mutation analysis with the new HRD panel
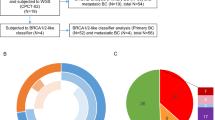
In this retrospective analysis, 98 patients were included who were previously analyzed for BRCA1/2 status (Table 1). In total, eight samples had to be excluded after sequencing (dropout rate 8.2%) due to poor DNA quality and, consequently, insufficient sequencing performance and target sample coverage. Hence, the following molecular pathological analysis with the new HRD panel consisted of 90 samples including 14 from the 24 validation samples. The main and therefore most meaningful sample size was made up by ovarian carcinomas (n = 63).
The heatmap (Fig. 1) only shows likely pathogenic or pathogenic alterations and their biological consequences. A detailed list of all identified mutations is provided in Supplementary Table S2. Mutations in the gene TP53 were most abundant and accounted for 73% (n = 66) of modifications throughout all samples. Besides previously detected BRCA1/2 variants (22%), ATM was the second most altered gene (9%). In BRCA1/2 mutated samples, additional variants, were identified in eight cases (BC_3, BC_4, OvCa_2, OvCa_4, OvCa_6, OvCa_9, OvCa_11 and OvCa_65). In prostate cancer, deleterious variants were found in three out of five samples. Three samples of pancreatic cancer (n = 9) presented variants in genes other than TP53. In ovarian cancer deletions, which lead to a frameshift in the open reading frame, occurred in potentially targetable HR genes RAD51D (OvCa_1), NBN (OvCa_24), BRIP1 (OvCa_32) and FANCL (OvCa_65), besides deletions in BRCA1/2 and TP53.

Heatmap of all variants found by means of the HRD panel. Investigated samples (n = 90) include prostate (n = 5, PC), pancreatic (n = 7, PaC), breast (n = 13, BC) and ovarian cancers (n = 63, OvCa) and are arranged according to their tumor type. All HRD genes with at least one variant are included and listed in the first row. Squares incorporating a white dot symbolize an additional variant in this gene. Below, the mutation rate in all samples per gene is shown in percent. Colored rectangles represent different variant classes: grey = missense variants, deep pink = deletions that lead to a frameshift in the open reading frame, olive = variants effecting the splice site, yellow = variants that lead to a premature stop codon, light blue = insertions that lead to a frameshift and aquamarine = deletions or insertions that do not lead to a frameshift. Only pathogenic, likely pathogenic and variants that were predicted likely pathogenic are presented. Likely benign or benign variants were excluded from further analysis
Missense variants accounted for 54% of all alterations found in this study. Deletions, which lead to a frameshift in the open reading frame represented the second most abundant variants (17%); closely followed by stopgain variants (14%). The variant distribution was further investigated in Fig. 2 for pancreatic (a + b), breast (c + d) and ovarian (e + f) cancer samples. Most variants per tumor entity were detected in TP53. In ovarian cancer samples, 15% of all detected variants were deletions leading to a frameshift. Of all filtered SNVs, transitions were outweighed by transversions resulting in a ratio of transitions to transversions of 2.33 (pancreatic cancer), 2.25 (breast cancer) and 2.36 (ovarian cancer) with G > A being the most frequent base substitution in all entities.

Summary of SNV class and variant distribution per tumor entity. The number of variant classes (a-c), variant distribution in percent (d-f) and the number of variants per gene (g-i) are shown for pancreatic (PaC: a, d, g (n = 7)), breast (BC: b (n = 10), e, h (n = 11)) and ovarian (OvCa: c (n = 53), f, I (n = 58)) cancer samples. Samples of prostate cancer are excluded from variant categorization due to the limited number of alterations found. Only pathogenic, likely pathogenic and variants that were predicted likely pathogenic are incorporated in this chart
All analyzed samples of this study are summarized in Table 2 and assigned to categories based on the detected variants. In addition to 20 (22.2%) patients with mutations in BRCA1/2, 22 (24.4%) of this cohort harbored mutations in additional HRD-associated genes, which were classified as pathogenic (tier 1) or likely pathogenic (tier 2) based on ACMG guidelines. Variants of unknown significance (VUS), which lead to a premature stop codon, a frameshift in the open reading frame, or disruption of the splice site, were classified as likely pathogenic and therefore included in the tier 2 category. The remaining VUS were evaluated by prediction tools and categorized accordingly. This subsequently lead to 5.6% of variants (5 patients), which were predicted as damaging, and 12.2% of variants (11 cases) with no significant effect on the protein function. Furthermore, 32 (35.6%) of patients have no indication of a HR dysfunction and present likely benign/benign variants and wild type sequences in the genes evaluated. Taken together, our data reveals that 22 (24.4%) of all 90 patients, who did not harbor a BRCA1/2 mutation, exhibit deleterious variants in HRD-associated genes. Those findings highlight that a substantial amount of previously BRCA1/2 WT patients were then identified as potential HRD-positive, which might be relevant for PARPi therapy. In total, 42/90 (48%) patients had tumors with mutation in at least one HR gene.
Discussion
Over the last decade, it has become apparent that both, germline and somatic, deleterious mutations in BRCA1/2 (BRCAm) have disruptive effects on DNA damage repair by HR mechanisms and might subsequently be susceptible to PARP inhibition (PARPi) [4, 22, 24]. Especially patients with germline or somatic variants in HR-associated genes with functional BRCA1/2 represent a group of clinical significance, which is disregarded by the narrow scope of solely determining BRCAm status. Different approaches (mutational and methylation analysis, genomic scar identification, functional assays, and combined tests) of assessing the HRD status are currently used in clinical settings to in order broaden the patient population who might benefit from PARPi therapy [25]. ESMO recommendations for HRD testing in ovarian cancer describe good clinical validity for BRCAm analysis and genomic scar assays [25]. The Myriad myChoice test (Myriad Genetics, Salt Lake City, UT) is the only test approved by the FDA for therapy with PARPi (olaparib and niraparib) in ovarian cancer and calculates an HRD score to define genomic scares and genomic instability by loss of heterozygosity (LOH), telomeric allelic imbalance (TAI), and large-scale state transitions (LST) [26]. Another approach by Foundation Medicine is to determine HRD by using genome-wide LOH as a biomarker in association with BRCA1/2 [12]. Based on mutational signatures (somatic substitution, insertion/deletion, and rearrangement patterns) as well as genomic scars, HRDetect is an additional assay to predict BRCA1/2 deficiency and was tested successfully in independent cohorts of patients with BC, OvCa and PaC [27]. Apart from that, the detection of RAD51 nuclear foci correlates with PARPi sensitivity and is also a feasible method to predict the response to PARPi beyond BRCA1/2 pathogenic variants in BC. The lack of RAD51 foci formation is associated with the sensitivity to PARPi and distinguishes between responsive and resistant tumors with an improved predictive power compared to the HRD score [28, 29]. Standardization of HRD evaluation could be useful to ensure comparability as currently no gold standard for HRD assessment has been specified and approaches used in clinical practice are heterogeneous.
This study evaluated HRD status by targeted NGS for 42 h-associated genes. However, this technique does not allow for assessment of LOH, TAI and LST or the distinguishing between somatic and germline variants. There is an ongoing discussion about the contribution of mutations in non-BRCA HR-associated genes to the HRD status determined by genomic scars. Recent studies found no, little or good correlation between HRD status determined by mutations in non-BRCA HR-associated genes and determined by genomic scars [30,31,32]. In future, it will be important to define those genes and specific mutations that positively contribute to the HRD status. Here, the new HRD gene panel was validated and implemented under routine conditions and a low dropout rate of 8.2% was observed. All previously reported variants were detected in the validation process by two independent methods in FFPE samples and reference material.This proves the usability and feasibility of the new HRD panel in practice.
Due to the fact that ovarian cancer was the first cancer to be approved for PARPi therapy by the FDA in 2014 [33], ovarian cancer (OvCa) contributed the largest sample size. In 2018, 2019, and 2020 olaparib was approved for breast cancer (BC) [34], pancreatic (PaC) [35] and prostate cancer (PC) [22], respectively. Now, a variety of PARPi are approved in OvCa (olaparib, rucaparib and niraparib), BC (olaparib and ralazoparib), PaC (olaparib) and PC (olaparib and rucaparib) [36]. Thus, the cohort in this study included breast, ovarian, pancreatic and prostate cancer. The majority of samples were ovarian cancers, because one inclusion criteriion was the known BRCAm status. Unexpectedly, this cohort exhibited a high prevalence of mutations in HR-associated genes (46.6%). Among these samples, the BRCAm rate was 22.2% (20 patients) and an additional 24.4% (22 patients) harbored mutations in other HR-associated genes. In OvCa, BRCA1/2 mutation rate was reported to be around 13–20% [1, 13] but those numbers vary widely (8.1 − 47.7%) depending on the histological subtype and the method used for HR evaluation [37]. For BC, BRCAm rate was reported to be 7% and the accumulated HRD prevalence was 11% [14]. The proportion of BRCAm to HR mutated genes, besides BRCA1/2, for pancreatic and prostate cancer was 5–6% and 4–16%, respectively [15, 16]. According to a different study, mutations in BRCA1/2 were most commonly detected in ovarian (15.2%), prostate, (10.7%) breast (8.8%) and pancreatic cancer (5.2%) [12]. Another Pan-cancer study described a combined HRD frequency of 85% for OvCa, BC, PC, PaC. The landscape of HRD was also affected by tumor type, status of metastasis and cancer stage [38]. In in the present study, the samples resemble a heterogeneous cohort (including primary tumors, metastases and pre-treated patients) which is common under routine circumstances. Taken this into consideration, the data collected in this study are consistent with the current literature. Whether all 22 (24.4%) patients, who harbor at least one mutation in HRD-associated genes besides BRCA1/2, are susceptible for PARP inhibition needs to be determined. Furthermore, several HR associated genes (ATM, ATR, NBN, RAD51, RAD53, CHEK1, CHEK2, FANC genes, CDK12, FAM175A and BARD1) have been identified to contribute to synthetic lethality in combination with PARG or PARP inhibitors in vitro [39,40,41,42]. Thus, genes that were frequently mutated in this cohort were evaluated in the following regarding their significance in HR and therapeutic relevance.
Throughout all 90 patients included in the present study, TP53 was the most frequently altered gene and was mutated in 73% of tumors. The TP53 gene is the most commonly mutated gene in several types of human cancer [43, 44] and is therefore of constant interest in research projects. Although, TP53 might be a relevant predictor in treatment response to PARPi because mutations are able to interfere with ATM function [45] and functional TP53 might be necessary for PARPi effectiveness [46], there is currently no approved targeted therapy for tumors harboring TP53 mutations.
In the present study, two CDK12 mutations and one deleterious variant in RAD54L were detected in patients with PC. Those genes are used as predictive biomarkers in the PROfound phase 3 study, which the FDAs approval for olaparib was based on, with PARPi in castration-resistant PC [11]. The PROfound study compared olaparib as monotherapy to physician’s choice (enzalutamide or abiraterone). Overall, 27.9% of patients were identified as HR deficient and significant beneficial effects of PARPi concerning progression free survival were reported. Whereby patients with mutations in BRCA1/2 and ATM showed the best response [47]. Patients with variants in CDK12 or RAD54L had a better calculated survival, suggesting a beneficial effect of PARPi [17]. Another study in primary PC used HRD score to define genomic instability and reported a significantly higher score in tumors with germline BRCA2 mutations than in ATM and CHEK2 altered cases [48]. However, the evidence of a beneficial effect of PARPi in patients with CDK12 mutations is more ambiguous. A recent study investigated the responsiveness of patients with loss-of-function CDK12 mutations regarding PARPi and PD-1 inhibitor (programmed cell death receptor-1) and detected no beneficial effect with PARPi on PSA response or objective response, but patients might be sensitive to immunotherapy treatment with PD-1 [49].
In the present cohort, patients with at least one mutation in ARID1A, ATM and ATR represent a substantial subgroup and account for 13.3% of all patients. The signaling of ARID1A, ATM and ATR interact and affect each other [50,51,52,53] and implicate that defects in those genes are targetable for a novel lethal synthetic approach in patient treatment [50, 51]. In many cancer types, mutations in ARID1A are the most common among genes associated with HR and were reported to account for 6.4% in OvCa, 3.7% in BC, 5.5% in PaC and 0% in PC of mutations found in a retrospective study of 21 cancer lineages [1]. In the present study, two mutations of ARID1A each were detected in PaC and OvCa. Several studies have shown that loss of ARID1A sensitized tumors to PARPi therapy. This effect was enhanced when tumors presented additional defects in ATR function [51] or were pre-treated with ionizing radiation [53]. The ongoing phase 2 study ATARI is investigating the impact of olaparib (PARPi) monotherapy and in combination with an ATR inhibitor drug (AZD6738) in gynecological cancers and compares tumors with and without ARID1A loss (NCT04065269). An additional study of AZD6738 investigates solid tumors (including pancreatic cancer) and determines response rates to therapy with ATR kinase inhibitor monotherapy and in combination with olaparib in an ATM loss and an ARID1A mutated subgroup (NCT03682289). Another ATR inhibitor (M4344) in combination with PARPi (niraparib) is investigated in PARPi resistant ovarian cancer in a clinical trial (NCT04149145). However, the outcomes of those studies are yet to be determined. In cells harboring ATM mutations, PARPi or ATR inhibitors enhanced the apoptosis rate of pancreatic cancer and subsequently increased synthetic lethality [54]. In this study, damaging ATR variants were found in pancreatic, breast and ovarian cancer in one patient each. Furthermore, pathogen variants in ATM were the third leading mutation after TP53 and BRCA1 and were detected in 9% of all patients. Contradictory to the reported frequencies of ATM mutations in other studies (OvCa = 1.5% [1, 13], BC = 4% [14], PaC = 5% [16] and PC = 4% [15]), the present one found that ATM variants were most common in OvCa (n = 7) and absent in PC and BC, which can be explained by the limited overall sample size. In ATM deficient cell lines, monotherapy of PARPi (olaparib) showed proficient results and lead to reduction of viability but the apoptosis rate was significantly increased in combination with ATR inhibitors [54,55,56,57].
Moreover, 14% of the patients included in this study harbored deleterious mutations in Fanconi anemia genes (FANCA, FANCD2, FANCF, FANCG FANCI and FANCL) and in WRN, which have been reported to interact with BRCA1 [58,59,60]. Furthermore, substantial crosstalk between WRN and PARP1 is described [61]. In patients with triple negative BC, a selection of Fanconi anemia genes, amongst other genes, are used as biomarkers in an ongoing phase 2 clinical trial with PARPi (Talazoparib) (NCT02401347). Fanconi anemia genes and also WRN are subject to another phase 2 trial in germline mutated metastatic BC, where the response of patients with mutations in those genes is compared to the response of patients with mutations in BRCA1/2, PALB2 and CHEK2 in regard to combination therapy of immunotherapeutic agent PD-1 (HX008) and PARPi (niraparib) (NCT04508803). All genes included in this study were reported to impact HR function but the relevance of some genes has recently been challenged.
Contradictory to the theoretical assumption, about 40% of patients with BRCA1/2 deficient tumors fail to respond do PARPi monotherapy [62, 63] or acquire resistance over the course of application time [64, 65] revealing that HRD status is a very dynamic phenotype. Mechanisms of PARPi resistance include reversion or secondary mutations which restore protein expression, enhanced stabilization and protection of the replication fork (upregulation of PARG) and inhibiting NHEJ factors (downregulation of the SHIELDIN complex, 53BP1 and REV7). Those mechanisms lead to restoration of HR and subsequently promoting PARPi resistance [66,67,68,69,70]. Secondary or reversion mutations restoring HR competence, and therefore PARPi resistance, are most commonly described in BRCA1/2 but are also reported in RAD51C and RAD51D [70, 71]. Therefore especially for patients with PARPi resistance, combination therapy with agents inhibiting ATM, ATR, PARG or immune checkpoints offer a substantial benefit in treatment response to overcome resistance mechanisms and reverse sensitivity to PARPi [51, 55, 56, 72]. The new 42 HRD gene panel allows for screening a large number of HR associated genes for the detection of primary, reversion and secondary mutations in genes besides BRCA1/2. Those analyses allow for targeting acquired resistance mutations, guide therapy decisions and may explain reverse sensitivity to PARPi resistance mechanisms on a genetical level. By monitoring a broad number of genes involved in HR, these findings may be particularly relevant in generating new insights into PAPRi resistance mechanisms and may help to guide treatment decisions in order to adapt personalized therapy of patients.
In summary, the results of the present study show the feasibility of the new 42 HRD gene panel and reflect the prevalence of HR-associated genes in routine conditions. The NGS approach detected variants with complete concordance resulting in reliable panel performance. Characteristic HR phenotype was present in each cancer entity and was heterogeneous in that way. BRCAm was shown to account only for a subgroup of HRD tumors. Although BRCA1/2 HR-phenotype claims to achieve most proficient effects with PARPi therapy, other genes manifest to be promising in some (pre-) clinical settings; especially in combination with other agents like ATR inhibitors or immunotherapy. However, the conclusive clinical relevance and impact in treatment strategy needs to be determined by further research. In particular, in tumors harboring mutations in uncommon HR-associated genes or VUS, the contribution to synthetic lethality is still uncertain. Lastly, our data highlight the importance of further evaluating HRD genes and addressing PARPi resistance mechanisms, beyond BRCA1/2, in clinical settings to expand the scope of therapeutic approaches with PARPi or combination therapies to refine treatment selection for optimal personalized therapy in cancer patients.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- BC:
-
Breast cancer
- BER:
-
Base Excision Repair
- BRCAm:
-
BRCA mutations
- DSB:
-
Double strand breaks
- EMA:
-
European Medicines Agency
- FDA:
-
U.S. Food and Drug Administration
- FFPE:
-
Formalin-fixed paraffin-embedded
- HR:
-
Homologous recombination
- HRD:
-
Homologous recombination deficiency
- LOH:
-
Loss of heterozygosity
- LST:
-
Large-scale state transitions
- NGS:
-
Next-generation sequencing
- NHEJ:
-
Non-homologous end joining
- OvCa:
-
Ovarian cancer
- PaC:
-
Pancreatic cancer
- PARP:
-
Poly (ADP-ribose) polymerase
- PARPi:
-
PARP inhibitors
- PC:
-
Prostate cancer
- TAI:
-
Telomeric allelic imbalance
- VUS:
-
Variants of uncertain significance
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
References
Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen W-J, et al. Prevalence of homologous recombination–related gene mutations across multiple Cancer types. JCO Precis Oncol. 2018;2(2):1–13.
Melinda LT, Kirsten MT, Julia R, Bryan H, Gordon BM, Kristin CJ, et al. Homologous recombination Deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative Breast Cancer. Clin Cancer Res. 2016;22(15):3764–73.
Gulhan DC, Lee JJK, Melloni GEM, Cortés-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet [Internet]. 2019;51(5):912–9. https://doi.org/10.1038/s41588-019-0390-2.
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20.
Patel KJ, Yu VPCC, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, et al. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1(3):347–57.
Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404(6773):37–41.
Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Sci (80-). 2017;355(3):1152–8.
Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020;34(5):360–94.
Pilié PG, Gay CM, Byers LA, O’Connor MJ, Yap TA. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res. 2019;25(13):3759–71.
Hu Y, Guo M. Synthetic lethality strategies: beyond BRCA1/2 mutations in Pancreatic cancer. Cancer Sci. 2020;111(9):3111–21.
Antonarakis ES, Gomella LG, Petrylak DP. When and How to Use PARP Inhibitors in Prostate Cancer: A Systematic Review of the Literature with an Update on On-Going Trials. Eur Urol Oncol [Internet]. 2020;3(5):594–611. https://doi.org/10.1016/j.euo.2020.07.005.
Sokol ES, Pavlick D, Khiabanian H, Frampton GM, Ross JS, Gregg JP, et al. Pan-cancer Analysis of BRCA1 and BRCA2 genomic alterations and their Association with genomic instability as measured by genome-wide loss of Heterozygosity. JCO Precis Oncol. 2020;4(4):442–65.
Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15.
Heeke AL, Xiu J, Elliott A, Korn WM, Lynce F, Pohlmann PR et al. Actionable co-alterations in breast tumors with pathogenic mutations in the homologous recombination DNA damage repair pathway. Breast Cancer Res Treat [Internet]. 2020;184(2):265–75. https://doi.org/10.1007/s10549-020-05849-2.
Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, et al. The Molecular Taxonomy of primary Prostate Cancer. Cell. 2015;163(4):1011–25.
Jensen DM. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32(2):185–203.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant Prostate Cancer. N Engl J Med. 2020;382(22):2091–102.
Lang H, Baumgart J, Heinrich S, Tripke V, Passalaqua M, Maderer A et al. Extended molecular profiling improves stratification and prediction of Survival after Resection of Colorectal Liver metastases. Ann Surg. 2019;270(5).
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagnostics [Internet]. 2017;19(1):4–23. https://doi.org/10.1016/j.jmoldx.2016.10.002.
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–74.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94.
Tripathi A, Balakrishna P, Agarwal N. PARP inhibitors in castration-resistant prostate cancer. Cancer Treat Res Commun [Internet]. 2020;24:100199. https://doi.org/10.1016/j.ctarc.2020.100199.
Jiang Y, Dai H, Li Y, Yin J, Guo S, Lin SY, et al. PARP inhibitors synergize with gemcitabine by potentiating DNA damage in non-small-cell Lung cancer. Int J Cancer. 2019;144(5):1092–103.
Lin PH, Chen M, Tsai LW, Lo C, Yen TC, Huang TY, et al. Using next-generation sequencing to redefine BRCAness in triple-negative Breast cancer. Cancer Sci. 2020;111(4):1375–84.
Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol [Internet]. 2020;31(12):1606–22. https://doi.org/10.1016/j.annonc.2020.08.2102.
Schram AM, Aghajanian CA, Hyman DM. Niraparib in recurrent Ovarian Cancer. N Engl J Med. 2017;376(8):801–2.
Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23(4):517–25.
Castroviejo-Bermejo M, Cruz C, Llop‐Guevara A, Gutiérrez‐Enríquez S, Ducy M, Ibrahim YH, et al. A RAD 51 assay feasible in routine Tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol Med. 2018;10(12):1–16.
Cruz C, Castroviejo-Bermejo M, Gutiérrez-Enríquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated Breast cancer. Ann Oncol. 2018;29(5):1203–10.
Bartow BB, Siegal GP, Yalniz C, Elkhanany AM, Huo L, Ding Q et al. Mutations in homologous recombination genes and loss of Heterozygosity Status in Advanced-Stage breast carcinoma †. Cancers (Basel). 2023;15(9).
Pujade-Lauraine E, Brown J, Barnicle A, Wessen J, Lao-Sirieix P, Criscione SW, et al. Homologous recombination repair gene mutations to Predict Olaparib Plus Bevacizumab Efficacy in the First-Line Ovarian Cancer PAOLA-1/ENGOT-ov25 trial. JCO Precis Oncol. 2023;33(7):1–12.
Wen H, Feng Z, Ma Y, Liu R, Ou Q, Guo Q et al. Homologous recombination deficiency in diverse cancer types and its correlation with platinum chemotherapy efficiency in ovarian cancer. BMC Cancer [Internet]. 2022;22(1):1–11. https://doi.org/10.1186/s12885-022-09602-4.
Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced Ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21(19):4257–61.
FDA. Oncology Drugs approved by the FDA in Early 2018. Am Heal Drug Benefits. 2018;11(4):209–10.
Katayama ES, Hue JJ, Bajor DL, Ocuin LM, Ammori JB, Hardacre JM, et al. A comprehensive analysis of clinical trials in Pancreatic cancer: what is coming down the pike? Oncotarget. 2020;11(38):3489–501.
Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E. PARP inhibitors: clinical relevance, mechanisms of Action and Tumor Resistance. Front Cell Dev Biol. 2020;8(September):1–22.
da Cunha Colombo, Bonadio RR, Fogace RN, Miranda VC, Diz MDPE. Homologous recombination deficiency in Ovarian cancer: a review of its epidemiology and management. Clinics. 2018;73:1–6.
Nguyen L, Martens WM, Van Hoeck J, Cuppen A. Pan-cancer landscape of homologous recombination deficiency. Nat Commun. 2020;11(1):1–12.
Gravells P, Grant E, Smith KM, James DI, Bryant HE. Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA Repair (Amst) [Internet]. 2017;52:81–91. https://doi.org/10.1016/j.dnarep.2017.02.010.
Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34(6):1685–91.
Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74(1):287–97.
Murai J, Huang SYN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99.
Hollstein M, Sidransky D, Vogelstein B, Curtis C. p53 mutations in human cancers. Sci (80-). 1991;253(July):49–54.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–9.
Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9(5):573–80.
Collot T, Niogret J, Carnet M, Chevrier S, Humblin E, Favier L, et al. PARP inhibitor resistance and TP53 mutations in patients treated with olaparib for BRCA-mutated cancer: four case reports. Mol Med Rep. 2021;23(1):1–8.
de Bono JS, Fizazi K, Saad F, Shore N, Sandhu SK, Mehra N, et al. Central, prospective detection of homologous recombination repair gene mutations (HRRm) in tumour tissue from > 4000 men with metastatic castration-resistant Prostate cancer (mCRPC) screened for the PROfound study. Ann Oncol. 2019;30(5):v328–9.
Lotan TL, Kaur HB, Salles DC, Murali S, Schaeffer EM, Lanchbury JS et al. Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod Pathol [Internet]. 2021;https://doi.org/10.1038/s41379-020-00731-4. Available from: http://dx.doi.org/10.1038/s41379-020-00731-4.
Antonarakis ES, Isaacsson Velho P, Fu W, Wang H, Agarwal N, Santos VS, et al. CDK12-Altered Prostate Cancer: clinical features and therapeutic outcomes to standard systemic therapies, poly (ADP-Ribose) polymerase inhibitors, and PD-1 inhibitors. JCO Precis Oncol. 2020;4(4):370–81.
Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A Deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5(7):752–67.
Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun [Internet]. 2016;7:1–13. https://doi.org/10.1038/ncomms13837.
Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5(9):1–18.
Park Y, Chui MH, Rahmanto YS, Yu ZC, Shamanna RA, Bellani MA, et al. Loss of ARID1A in Tumor cells renders selective vulnerability to combined ionizing radiation and PARP inhibitor therapy. Clin Cancer Res. 2019;25(18):5584–93.
Perkhofer L, Schmitt A, Romero Carrasco MC, Ihle M, Hampp S, Ruess DA, et al. ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy-induced DNA damage. Cancer Res. 2017;77(20):5576–90.
Lloyd RL, Wijnhoven PWG, Ramos-Montoya A, Wilson Z, Illuzzi G, Falenta K, et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene. 2020;39(25):4869–83.
Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, et al. ATM-deficient cancers provide new opportunities for precision oncology. Cancers (Basel). 2020;12(3):1–13.
Xu L, Ma E, Zeng T, Zhao R, Tao Y, Chen X, et al. ATM deficiency promotes progression of CRPC by enhancing Warburg effect. Endocr Relat Cancer. 2019;26(1):59–71.
Swuec P, Renault L, Borg A, Shah F, Murphy VJ, van Twest S et al. The FA Core Complex Contains a Homo-dimeric Catalytic Module for the Symmetric Mono-ubiquitination of FANCI-FANCD2. Cell Rep [Internet]. 2017;18(3):611–23. https://doi.org/10.1016/j.celrep.2016.11.013.
Michl J, Zimmer J, Tarsounas M. Interplay between fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016;35(9):909–23.
Cheng WH, Kusumoto R, Opresko PL, Sui XF, Huang S, Nicolette ML, et al. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006;34(9):2751–60.
von Kobbe C, Harrigan JA, Schreiber V, Stiegler P, Piotrowski J, Dawut L, et al. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res. 2004;32(13):4003–14.
Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier Ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–9.
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent Ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–51.
Janysek DC, Kim J, Duijf PHG, Dray E. Clinical use and mechanisms of resistance for PARP inhibitors in homologous recombination-deficient cancers. Transl Oncol [Internet]. 2021;14(3):101012. https://doi.org/10.1016/j.tranon.2021.101012.
Li H, Liu ZY, Wu N, Chen YC, Cheng Q, Wang J. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer. 2020;19(1):1–16.
Noordermeer SM, van Attikum HPARP, Inhibitor Resistance. A tug-of-war in BRCA-Mutated cells. Trends Cell Biol. 2019;29(10):820–34.
Veneris JT, Matulonis UA, Liu JF, Konstantinopoulos PA. Choosing wisely: Selecting PARP inhibitor combinations to promote anti-tumor immune responses beyond BRCA mutations. Gynecol Oncol [Internet]. 2020;156(2):488–97. https://doi.org/10.1016/j.ygyno.2019.09.021.
Gogola E, Rottenberg S, Jonkers J. Resistance to PARP inhibitors: lessons from preclinical models of BRCA-Associated Cancer. Annu Rev Cancer Biol. 2019;3(1):235–54.
Imyanitov E, Sokolenko A. Mechanisms of acquired resistance of BRCA1/2-driven tumors to platinum compounds and PARP inhibitors. World J Clin Oncol. 2021;12(7):544–56.
Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–9.
Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7(9):984–98.
Wu Z, Cui P, Tao H, Zhang S, Ma J, Liu Z et al. The synergistic effect of PARP inhibitors and Immune Checkpoint inhibitors. Clin Med Insights Oncol. 2021;15.
Acknowledgements
The authors thank Klaus Rehfeldt and Andre Koper from Qiagen for the support in optimizing a custom workflow of the CLC Workbench for the use of variant detection.
Funding
Open Access funding enabled and organized by Projekt DEAL. The authors received no specific funding for this work.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A. V., N. H. and W. R. generated the study concept and design; A. V., A. H. and N. H. performed the experiments; A. V., A. H., M. K., W. R. and N. H. performed analysis and interpretation of data; A. V. and N. H. wrote the paper. All Authors read and approved the final paper.
Corresponding author
Ethics declarations
Ethics approval
The local ethics committee (Ethics Committee of the Rhineland-Palatinate State Medical Association, Mainz, Germany) approved the study design and all samples were handled in compliance with the standards proposed by the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Vogel, A., Haupts, A., Kloth, M. et al. A novel targeted NGS panel identifies numerous homologous recombination deficiency (HRD)-associated gene mutations in addition to known BRCA mutations. Diagn Pathol 19, 9 (2024). https://doi.org/10.1186/s13000-023-01431-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-023-01431-8