
Abstract
Background
The rarity of juvenile psammomatoid ossifying fibroma (JPOF) and lack of cytogenetic studies prompted us to report a novel SETD2 gene mutation in a benign odontogenic tumour.
Case presentation
A 21-year-old man presented with a hard, expanded mandibular cortex. Computed tomography revealed multilocular radiopacity in the mandible; this was reconstructed via segmental mandibulectomy using a vascularised iliac crest flap. Based on the clinical and histological findings, we diagnosed JPOF associated with an aneurysmal bone cyst. Microscopically, the solid area was characterised by many rounded or angular ossicles in a cellular fibrous stroma. The stromal cells were spindle-like or stellate. Next-generation sequencing detected a frame shift mutation of the SETD2 gene, while the copy number was normal.
Conclusions
Our findings suggest further genetic studies should be performed to assess whether this mutation is related to tumour genesis.
Similar content being viewed by others

Background
According to the World Health Organization (WHO) classification, there are three subtypes of ossifying fibromas of the craniofacial skeleton and jaw, differing in clinical presentation and histopathological appearance: cemento-ossifying fibroma (COF), juvenile trabecular ossifying fibroma (JTOF), and juvenile psammomatoid ossifying fibroma (JPOF) [1]. JPOFs show a slight male predominance and, in one study, the mean age of the patients was 18.9 ± 12.0 years [2]. JPOFs in the mandible are rare [2,3,4]. An association with aneurysmal bone cysts or cortical bone perforation with local aggressive growth has been reported in some cases [2]. Approximately one-third of JPOFs of the jaws recur; this is mainly dependent on the extent of the surgical excision [2]. The rarity of JPOF in the mandible associated with an aneurysmal bone cyst, and the lack of cytogenetic studies, prompted us to report the clinicopathological features and mutational status of such a case.
Case presentation
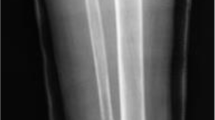
A 21-year-old man was transferred to the Department of Oral and Maxillofacial Surgery, Medical University of Graz when a slow-growing tumour in the left mandible, which had been present for 10 years and could not be diagnosed or treated adequately in his home country. On admission, he had facial asymmetry with adequate mouth opening. The intraoral examination revealed left-sided, hard, expanded buccal and lingual mandibular cortices. Computed tomography showed an 8.5-cm multilocular radiopacity in the left mandibular neck extending to the midline. Despite occupying the soft tissues, the mainly cystic, focal, solid lesion (arrow) was completely covered by thin, non-perforated, pre-existing cortical bone (Fig. 1). Laboratory work-up was normal, including endocrine parameters. Of note, the somatomedin C, calcium, and phosphate levels were within their normal ranges. The medical and family histories were non-contributory. Following a preoperative biopsy, he underwent segmental mandibulectomy for reconstruction with a vascularised iliac crest flap. The osseous flap was inserted into the defect and fixed with a plate. The postoperative healing was uneventful. After removing the plate, osseointegrated dental implants were used for dental rehabilitation. No further tumours were seen in the remaining jaw bone during a 7-year follow-up period.

Computed tomography shows a mainly cystic lesion; the tumour proper is the solid area in the labial aspect of the lower part (arrow)
Pathology and cytogenetics
The surgical specimen was 12 cm in its greatest longitudinal diameter and dominated by an 8-cm-diameter, nodular, bone-distending tumour. On cross-sections, the tumour consisted mainly of cystic spaces varying in size and sometimes filled with blood clots; a solid, nodular area measuring 3 cm was seen only at the lingual aspect of the lower part (Fig. 2). Microscopically, the cysts were mostly lined with granulation tissue and scar-like fibrous tissue (Fig. 3). The actual tumour, corresponding to the solid area, was characterised by many ossicles of different sizes in cellular fibrous stroma (Fig. 4). The ossicles were rounded or angular and comprised woven-bone deposits with distinct, sometimes laminated calcifications resembling psammoma bodies (Fig. 5). The stromal cells were spindle-shaped or stellate (Fig. 6). The tumour was covered peripherally by a thin shell of cortical bone, with no evidence of perforation (Fig. 7). The histopathological diagnosis of the preoperative biopsy was JPOF with an associated aneurysmal bone cyst; this was confirmed by examination of the surgical specimen, which had identical microscopic features. Paraffin-embedded tumour tissue was analysed by next-generation sequencing (NGS) using a PCR-based gene panel to detect mutations in the coding regions of 409 genes relevant to tumours, and low-density whole genome sequencing was performed to assess copy number variation. NGS showed a frame-shift mutation of the SETD2 gene, while the copy number was normal (Fig. 8).

Sagittal cross-section of the tumour reveals many cystic spaces in the solid part, mirroring the computed tomography findings

Cystic spaces (asterisks) are part of the multilocular aneurysmal bone cyst. A peripheral extension of the solid tumour is seen in the centre

An area of the solid tumour consisting of many ossicles set in cellular stroma

The ossicles are often rounded and show laminated calcification, reminiscent of psammoma bodies

Chromotrope aniline blue staining highlights the fibrous stroma

Tumour tissue infiltrating the mandibular bone, which leads to distinct osseous thinning. The bone is covered by the mucosal membrane

Genetic aberrations. NGS sequencing revealed a somatic deletion of two nucleotides in exon 3 of SETD2
Discussion
Only a few JPOFs located in the mandible associated with an aneurysmal bone cyst have been reported [3,4,5,6,7,8,9,10,11]. The rarity of this incidental lesion is underlined by the fact that aneurysmal bone cysts comprise only 0.15–0.4% of all jaw cysts [12], with two-thirds located in the mandible [12]. To our knowledge, this is the first report to compare whole-mount sections with their imaging equivalents, highlighting the dominating cystic character of the lesion.
On searching the literature, we found no cytogenetic studies of a mandibular JPOF with an associated aneurysmal bone cyst. However, we found cases that were diagnosed retrospectively as psammomatoid ossifying fibromas on re-evaluation their histopathological appearance. One of these was located in the mandible and had an interstitial deletion on chromosome 2 between q31–32 and q35–36 [13]; three cases were in the orbit and had a chromosomal translocation t(X;2) (q26;q33) [14, 15]. Tabareau-Delalande et al. [16] presented three JPOFs of non-mandibular or unknown location, with a chromosome 12 long arm rearrangement and amplification of the MDM2 and RASR1 genes. Although not subtyping ossifying fibromas despite the introduction of JPOF as a distinct clinicopathological entity by the WHO in 2005 [1, 17], several authors have published cytogenetic studies on fibro-osseous tumours, calling them simple ossifying fibromas, COFs, or juvenile ossifying fibromas [18,19,20,21,22,23,24,25,26,27,28]. We speculate that some of these tumours were JPOFs. Those tumours contained mutations of the HRPT2/CDC73 gene [18, 22, 25], showed upregulated expression of Notch receptors and ligands [21], down- or upregulation of miRNA [26], or deregulation of the Wnt/ß–catenin pathway [27]. However, no GNAS [19, 20, 23, 24, 28] or HRPT2, a component of PAF1 complex, [24] gene mutations were observed. In this context, it is interesting that the first cytogenetic analysis of a gnathic ossifying fibroma was probably performed in 1992, on a cemento-ossifying fibroma of the maxilla with three chromosomal translocations [29].
Knocking out Hrpt2 in the mouse results in preimplantation lethality as well as when conditionally deleted in adult animals [30]. De Mesquita et al. found a loss of heterozygosity at the HRPT2 gene locus in ossifying fibromas, fibrous dysplasia and osteosarcomas; nonetheless, only a limited contribution to the pathogenesis was detected [31]. On the other hand, PAF1 complex is required for mammalian development, likely through regulation of H3K36me3. Knockdown of either SETD2 or RTF1 results in similar phenotypes [32]. The SETD2 gene is located on chromosome 3 p21.31 and encodes a histone methyltransferase, which is responsible for the trimethylation of lysine 36 of histone H3 to H3K36me. This is the first report of a mutation of the SETD2 gene in a JPOF for an odontogenic tumour. Furthermore, no SETD2 mutation has been detected previously in a benign tumour. Proteins listed by H3K36me readers are involved in various consecutive events, such as transcription elongation, RNA processing, and DNA repair; therefore, SETD2 is considered a tumour-suppressor gene [33, 34]. Mutations of the SETD2 gene have been found in a variety of malignant tumours, especially renal cell carcinoma [33]. SETD2 mutations have also been reported in patients with Sotos syndrome and Sotos-like syndromes [35].
Conclusions
In our case, the SETD2 gene mutation likely played an important role in tumourigenesis. Therefore, it would be interesting to determine whether this mutation is typical of JPOF in general. To this end, further genetic studies on a JPOF series are required.
Abbreviations
- JPOF:
-
juvenile psammomatoid ossifying fibroma
- WHO:
-
World Health Organization
- CPO:
-
cemento-ossifying fibroma
- JTOF:
-
juvenile trabecular ossifying fibroma
- NGS:
-
next-generation sequencing
References
El Mofty SK, Nelson B, Toyosawa S: Ossifying fibroma. In: Barnes L, Eveson JW, Reichart P, Sidransky D, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Head and Neck Tumours. Lyon: IARC Press; 2017: 251–252.
Chrcanovic BR, Gomez RS. Juvenile ossifying fibroma of the jaws and paranasal sinuses: a systematic review of the cases reported in the literature. Int J Oral Maxillofac Surg. 2020;49(1):28–37. https://doi.org/10.1016/j.ijom.2019.06.029.
El Mofty S. Psammomatoid and trabecular juvenile ossifying fibroma of the craniofacial skeleton: two distinct clinicopathologic entities. Oral Pathol Oral Radiol Endod. 2002;93(3):296–304. https://doi.org/10.1067/moe.2002.121545.
Tolentino ES, Centurion BS, Tjioe KC, Casaroto AR, Tobouti PL, Junior UF, et al. Sant’ana E, Gonçales ES. Psammomatoid juvenile ossifying fibroma: an analysis of 2 cases affecting the mandible with review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113(6):e40–5. https://doi.org/10.1016/j.oooo.2011.08.005.
Smith SF, Newman L, Walker DM, Papadopoulos H. Juvenile aggressive psammomatoid ossifying fibroma: an interesting, challenging, and unusual case report and review of the literature. J Oral Maxillofac Surg. 2009;67(1):200–6. https://doi.org/10.1016/j.joms.2007.12.009.
Sarode SC, Sarode GS, Waknis P, Patil A, Jashika M. Juvenile psammomatoid ossifying fibroma: a review. Oral Oncol. 2011;47(12):1110–6. https://doi.org/10.1016/j.oraloncology.2011.06.513.
Waknis P, Sarode SC, Dolas R. Psammomatoid juvenile ossifying fibroma of the mandible with secondary aneurysmal bone cyst: a case report. Asian J Oral Maxillofac Surg. 2011;23(2):83–6. https://doi.org/10.1016/j.ajoms.2010.12.001.
Deshingkar SA, Barpande SR, Bhavthankar JD. Juvenile psammomatoid ossifying fibroma with secondary aneurysmal bone cyst of mandible. Saudi J Dent Res. 2014;5(2):5135–8. https://doi.org/10.1016/j.sjdr.2013.12.002.
Tamgadge S, Avinash T, Bhalerao S, Rajhans S. Juvenile psammomatoid ossifying fibroma with aneursmal bone cyst in the posterior mandible. Ecancermedicalscience. 2014;8:471.
Shruti SK, Kamath W, Hegde S, Sreevidya B. Psammous desmo-osteoblastoma with concomitant aneurysmal bone cyst of mandible. Ann Maxillofac Surg. 2015;5(1):130–4. https://doi.org/10.4103/2231-0746.161136.
Gotmare SS, Tamgadge A, Tamgadge S, Kesarkar KS. Recurent psammomatoid juvenile ossifying fibroma with aneurysmal bone cyst: a unusual case presentation. Iran J Med Sci. 2017;42(6):603–6.
Sun ZJ, Sun HL, Yang RL, Zwahlen RA, Zhao YF. Aneurysmal bone cysts of the jaw. Int J Surg Pathol. 2009;17(4):311–22. https://doi.org/10.1177/1066896909332115.
Dal Cin P, Sciot R, Fossion E, Vandamme B, Vandenberghe H. Chromosome abnormalities in cementifying fibroma. Cancer Genet Cytogen. 1993;71(2):170–2. https://doi.org/10.1016/0165-4608(93)90025-H.
Sawyer JR, Tryka AF, Bell JM, Boop FA. Nonrandom chromosome breakpoints at Xq26 and2q33 characterize cemento-ossifying fibromas of the orbit. Cancer. 1995;76(10):1853–9. https://doi.org/10.1002/1097-0142(19951115)76:10<1853::AID-CNCR2820761026>3.0.CO;2-D.
Parham DM, Bridge JA, Lukacs JL, Ding Y, Tryka AF, Sawyer JR. Cytogenetic distinction among benign fibro-osseous lesions of bone in children and adolescents: value of karyotypic findings in differential diagnosis. Pediatr Dev Pathol. 2004;7(2):148–58. https://doi.org/10.1007/s10024-003-6065-z.
Tabareau-Delalande F, Collin C, Gomez-Brouchet A, Bouvier C, Decouvelaere AV, de Muret A, et al. Chromosome 12 long arm rearrange- ment covering MDM2 and RASAL1 is associated with aggressive craniofacial juvenile ossifying fibroma and extracranial psammomatoid fibro-osseous lesions. Mod Pathol. 2015;28(1):48–56. https://doi.org/10.1038/modpathol.2014.80.
Slootweg PJ, El Mofty SK. Ossifying fibroma. In: Barnes L, Eveson JW, Reichart P, Sidransky D, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Head and Neck Tumours. Lyon: IARC Press; 2005: 319–320.
Pimenta FJ, Silveria LFG, Tavares GC, Silva AC, Perdigão PF, Castro WH, et al. HRPT2 gene alterations in ossifying fibroma of the jaws. Oral Oncol. 2006;42(7):735–9. https://doi.org/10.1016/j.oraloncology.2005.11.019.
Toyosawa S, Yuki M, Kishino M, Ogawa Y, Ueda T, Murakami S, et al. Ossifying fibroma vs fibrous dysplasia of the jaw: molecular and immunohistochemical characterization. Mod Pathol. 2007;20(3):389–96. https://doi.org/10.1038/modpathol.3800753.
Patel MM, Wilkey JF, Abdelsayed R. D’Silva NJ, Malchoff C. Mallya SM Analysis of GNAS mutations in cemento-ossifying fibroma and cemento-osseous dysplasia Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109(5):739–43. https://doi.org/10.1016/j.tripleo.2009.12.016.
Zhang GTH, Liu HC, Liao GQ, Liang YJ, Chu M, Wan CQ, et al. Detection of notch signaling molecules in cemento-ossifying fibroma of the jaws. J Oral Pathol Med. 2010;39(3):263–8. https://doi.org/10.1111/j.1600-0714.2009.00842.x.
De Mesquita Netto AC, Gomez RS, Diniz MG, Fonseca-Silva T, Campos K, De Marco L, et al. Assessing the contribution of HRPT2 to the pathogenesis of jaw fibrous dyplasia, ossifying fibroma and osteosarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115(3):359–67. https://doi.org/10.1016/j.oooo.2012.11.015.
Shi RR, Li XF, Zhang R, Chen Y, Li TJ. GNAS mutational analysis in differentiating fibrous dysplasia and ossifying fibroma of the jaw. Mod Pathol. 2013;26(8):1023–31. https://doi.org/10.1038/modpathol.2013.31.
Wang TT, Zhang R, Wang L, Chen Y, Dong Q, Li TJ. Two cases of multiple ossifying fibromas in the jaw. Diagn Pathol. 2014;28(9):75. https://doi.org/10.1186/1746-1596-9-75.
Chen Y, Hu DY, Wang TT, Zhang R, Dong Q, Xu ZX, et al. CDC73 gene mutations in sporadic ossifying fibroma of the jaws. Diagn Pathol. 2016;11(1):91–8. https://doi.org/10.1186/s13000-016-0532-0.
Pereira T, Brito JAR, Guimarães ALS, Gomes CC, de Lacerda JCT, de Castro WH, et al. MicroRNA profiling reveals dysregulated microRNAs and their target gene regulatory networks in cemento-ossifying fibroma. J Oral Pathol Med. 2018;47(1):78–85. https://doi.org/10.1111/jop.12650.
Pereira TDSF, Diniz MG, França JA, Moreira RG, de Menezes GHF, de Sousa SF, et al. The Wnt/beta-catenin pathway is deregulated in cemento-ossifying fibromas. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2018;125(2):172–8. https://doi.org/10.1016/j.oooo.2017.10.004.
Tabareau-Delalande F, Collin C, Gomez-Brouchet A, Decouvelaere AV, Bouvier C, Larousserie F, et al. Diagnostic value of investigating GNAS mutations in fibro-osseous lesions: a retrospective study of 91 cases of fibrous dysplasia and 40 other fibro-osseous lesions. Mod Pathol. 2013;26(7):911–21. https://doi.org/10.1038/modpathol.2012.223.
Gollin SM, Storto PD, Malone PS, Barnes L, Washington JA, Chidambaram A, et al. Cytogenetic abnormalities in an ossifying fibroma from a patient with bilateral retinoblastoma. Genes Chrom Cancer. 1992;4(2):146–52. https://doi.org/10.1002/gcc.2870040208.
Wang P, Bowl MR, Bender S, Peng J, Farber L, Chen J, et al. Parafibromin, a component of the human PAF complex regulates growth factors and is required for embryonic development and survival in adult mice. Mol.Cell.Biol. 2008;8:2930–40.
De Mesquita Netto AC, Santiago Gomez R, Gonçalves Diniz M, Fonseca-Silva T, Campos K, De Marco L, et al. Cavaliéri Gomes C. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115(3):359–67. https://doi.org/10.1016/j.oooo.2012.11.015.
Zhang K, Haversat JM, Mager J. CTR9/PAF1c regulates molecular lineage identity, histone H3K36 Trimethylation and genomic imprinting during preimplantation development. Dev Biol. 2013;383(1):15–27. https://doi.org/10.1016/j.ydbio.2013.09.005.
Carvalho S, Vitor AC, Sridhara SC, Martins FB, Raposo AC, Desterro JMP, et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elifesciences. 2014;02482(002).
Li J, Duns G, Westers H, Sijmons R, van den Berg A, Kok K. SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget. 2016;7(31):50719–34. https://doi.org/10.18632/oncotarget.9368.
Marzin P, Rondeau S, Aldinger KA, Alessandri JL, Isidor B, Heron D, et al. SETD2 related overgrowth syndrome: presentation of four new patients and review of the literature. Am J Med Genet. 2019;181(4):509–18. https://doi.org/10.1002/ajmg.c.31746.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All authors of the manuscript have read and agreed to its content and are accountable for all aspects of the accuracy and integrity of the manuscript in accordance with ICMJE criteria.
The article is original, has not already been published in a journal, and is not currently under consideration by another journal.
All authors agree to the terms of the BioMed Central Copyright and License Agreement.
Competing interests
There is no financial support or any other conflict of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Toferer, A., Truschnegg, A., Kashofer, K. et al. First presentation of a frameshift mutation in the SETD2 gene of a juvenile psammomatoid ossifying fibroma (JPOF) associated with an aneurysmal bone cyst. Diagn Pathol 16, 91 (2021). https://doi.org/10.1186/s13000-021-01160-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-021-01160-w