
Abstract
Idiopathic normal pressure hydrocephalus (iNPH) is considered an age-dependent chronic communicating hydrocephalus associated with cerebrospinal fluid (CSF) malabsorption; however, the aetiology of ventricular enlargement in iNPH has not yet been elucidated. There is accumulating evidence that support the hypothesis that various alterations in CSF dynamics contribute to ventricle dilatation in iNPH. This review focuses on CSF dynamics associated with ventriculomegaly and summarises the current literature based on three potential aetiology factors: genetic, environmental and hydrodynamic. The majority of gene mutations that cause communicating hydrocephalus were associated with an abnormal structure or dysfunction of motile cilia on the ventricular ependymal cells. Aging, alcohol consumption, sleep apnoea, diabetes and hypertension are candidates for the risk of developing iNPH, although there is no prospective cohort study to investigate the risk factors for iNPH. Alcohol intake may be associated with the dysfunction of ependymal cilia and sustained high CSF sugar concentration due to uncontrolled diabetes increases the fluid viscosity which in turn increases the shear stress on the ventricular wall surface. Sleep apnoea, diabetes and hypertension are known to be associated with the impairment of CSF and interstitial fluid exchange. Oscillatory shear stress to the ventricle wall surfaces is considerably increased by reciprocating bidirectional CSF movements in iNPH. Increased oscillatory shear stress impedes normal cilia beating, leading to motile cilia shedding from the ependymal cells. At the lack of ciliary protection, the ventricular wall is directly exposed to increased oscillatory shear stress. Additionally, increased oscillatory shear stress may be involved in activating the flow-mediated dilation signalling of the ventricular wall. In conclusion, as the CSF stroke volume at the cerebral aqueduct increases, the oscillatory shear stress increases, promoting motor cilia shedding and loss of ependymal cell coverage. These are considered to be the leading causes of ventricular enlargement in iNPH.
Similar content being viewed by others
Introduction
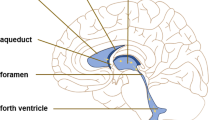
Hydrocephalus is characterised by pathological enlargement of the ventricles. Communicating chronic hydrocephalus in adult is synonymous with normal pressure hydrocephalus (NPH) in which the ventricles expand while the intracranial pressure remains within normal range [1]. Patients with NPH present the typical triad of cognitive decline, gait imbalance and urinary incontinence. NPH has been categorised into congenital/developmental aetiologies, idiopathic (iNPH) and secondary NPH (sNPH) developing after haemorrhagic stroke, trauma, infection and tumour [2–6]. The pathophysiology of iNPH has not yet been elucidated, although advanced age and alterations in cerebrospinal fluid (CSF) dynamics have been recognised as potential contributory factors. Initially, iNPH was considered as a disease associated with CSF malabsorption, due to the factor that the intracranial CSF volume in iNPH was significantly larger than that in age-matched controls or patients with other brain diseases [7]. Typical iNPH neuroimaging findings were expressed as disproportionally enlarged subarachnoid space hydrocephalus (DESH), characterised by ventricular enlargement, high convexity/midline tight sulci, and enlarged Sylvian fissures and basal cistern [5–9]. As an aetiology of DESH-pattern CSF distribution in iNPH, we previously reported that the inferior horn of the lateral ventricles directly communicates with the basal cistern via the choroidal fissure, serving as an overflow device that works with increased volume of intracranial CSF [5]. However, the mechanisms contributing to the enlargement of ventricles rather than subarachnoid spaces in iNPH remain to be determined. The development of imaging technology has enabled the observation of pulsatile and bidirectional CSF movements that are mainly driven by blood circulation and respiration under normal conditions [10,11,12,13,14,15,16,17,18]. CSF normally flows outward from the cranium during systole due to the expansion of the brain and intracranial vasculature, and flows inward during diastole. The CSF stroke, corresponding to one reciprocating motion during a cardiac cycle, is known to be increased at the cerebral aqueduct in most NPH patients [10,11,12,13,14, 16–18]. Similarly, the flow void sign, determined by signal loss on T2-weighted fast or turbo spin echo sequence that indicates a fast flow velocity, as depicted in Fig. 1, was also reported to be prominent at the cerebral aqueduct in NPH patients [11, 12, 19]. Previously, we found that an increased CSF stroke volume at the lower end of the cerebral aqueduct in iNPH was significantly associated with the dilation of the foramen of Magendie [17]. Therefore, we hypothesised that the foramen of Magendie serves as the first water gate to reduce backward CSF inflow into the ventricular system, and its subsequent dilation leads to an increase reverse flow into the ventricle systems. Furthermore, an increase in CSF stroke volume at the foramen of Monro, and not at the cerebral aqueduct, was significantly associated with the dilation of the lateral ventricles toward the vertex in iNPH [17]. Recently, CSF flow produced by the directional beating of the motile cilia on the ventricle surfaces was reported to be crucial in the regulation of the CSF distribution within and across brain ventricles, and the dysfunction of ciliary beat leads to reduced hydrodynamic coupling between the ventricles [20]. Additionally, coordinated directional beating of ependymal cilia is essential for appropriate CSF flow [21–23]. Therefore, motile cilia lining ventricular ependymal surfaces can be considered as a mechanism of ventricular enlargement in the association with CSF stroke volume. Thus, we speculated that the coordinated directional movements of the motile cilia on the apical surface of ependymal cells may be impaired by an increase in CSF stroke volume. To investigate a possible mechanism for the relationship between the development of ventricular enlargement and degeneration of ependymal cilia in iNPH, we reviewed the current literature on genetic and environmental factors for ventricular enlargement in communicating hydrocephalus.

Flow void signs and flow vectors in normal elderly and three types of normal-pressure hydrocephalus (NPH). The left figures (a, c, e, g) depict the flow void sign at the top of the cerebral aqueduct (red arrow head) and dorsal region of the foramen magnum (yellow arrow head) from the T2-weighted fast spin-echo sequence, and the right figures (b, d, f, h) show the flow vectors from 4D flow MRI in normal elderly (a and b) and patients diagnosed with idiopathic NPH (c and d), secondary NPH (e and f) and late-onset congenital or developmental hydrocephalus (g and h). The colour of the vector represents flow velocity; pink indicates highest velocity and blue indicates lowest velocity. C: corpus callosum, P: pons, II: lateral ventricle, III: third ventricle, IV: fourth ventricle
Current concept of CSF dynamics
In the early 1900s, Cushing proposed the third circulation theory that CSF is secreted by the choroid plexus in the ventricles, flows from the lateral ventricles to the third and fourth ventricles via the foramina of Monro and cerebral aqueduct, moves to the subarachnoid spaces through the foramina of Magendie and Luschka, circulates from the basal cistern to the subarachnoid spaces in the convexity part of the brain, and finally be absorbed into the blood through the arachnoid villi [24]. Although many researchers continue to believe third circulation by the CSF bulk flow, remarkable research findings of the last decade have questioned this traditional understanding of CSF physiology [25–33]. The current concept of CSF dynamics was summarized in Fig. 2. First, magnetic resonance flow images visualized complex pulsatile CSF motion (red double-headed arrows in Fig. 2), not bulk flow [10–18]. Second, in the glymphatic pathway, interstitial fluid flows from the arterial perivascular space to the venous perivascular space in the brain parenchyma and exchanges to the CSF through the water channel aquaporin-4 (purple double-headed arrows) [26, 27, 30, 31]. Therefore, the efflux of interstitial fluid can be the main CSF production, rather than the choroid plexus. Finally, CSF drains from the subarachnoid spaces to the nasal lymphatics via the cribriform plate (yellow arrows) [33] and to the meningeal lymphatics (green arrows) [26, 28, 29, 32] These lymphatic drainages were considered as the major outflow pathway rather than the dural venous sinuses through arachnoid villi. Based on these new concepts of CSF dynamics, we considered the mechanisms of ventricular enlargement in communicating hydrocephalus.

Current concept of CSF dynamics. This figure was drawn by using the Paint 3D application (Microsoft Corporation) and SYNAPSE 3D (FUJIFILM Corporation). The colour arrows indicate direction of CSF movement
Genetic factors
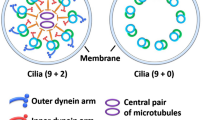
Genetic-based pathways offer valuable insights into understanding the underlying mechanisms of communicating hydrocephalus. There has been accumulating evidence which supports the association of ciliary dysfunction lining the ventricle surfaces with congenital hydrocephalus [22, 34–36]. Several mutations in Ccdc39, Celsr2, Celsr3, Cetn2, Dvls, FoxJ1, Hydin, Mdnah5, Pkd1, Tg737, Daple, Dnah14, Cfap43 and Cwh43 genes, which were related to the structure or function of motile cilia on ependymal cells, have been reported to develop hydrocephalus in animal models [21, 22, 34, 37–51]. For example, the transcription factor FoxJ1 plays a crucial role in the constant maintenance of motile cilia, and mutation in FoxJ1 causes ciliopathy characterised by foetal hydrocephalus [47]. Hydrocephalus can also be induced through dysfunction of motile cilia on the ependymal cells by experimentally degrading the Foxj1 protein with IκB kinase-2 inhibitors [52]. Furthermore, Hydin, a gene that encodes a central pair protein within the axon of motile cilia and expresses ependymal cell layer lining the ventricles, also may induce hydrocephalus when mutated [39, 40, 44]. Mdnah5 is specifically expressed in ependymal cells, and is essential for ultra-structural and functional integrity of ependymal cilia [41]. Both the polycystin-1 encoded by the Pkd1 gene, required for cilia function, and polaris encoded by the Tg737 gene, required for cilia structure, are reported to be crucial mechanosensitive molecules to control fluid flow through primary cilia in endothelial cells [22, 53]. Daple regulates the direction and coordination of motile cilia beating in the ependymal cells, and determines the direction of CSF flow [54]. Daple-deficient mice present communicating hydrocephalus [54], and mutation in human DAPLE causes autosomal recessive congenital hydrocephalus [55]. Mutation in DNAH14 results in motile cilia dysfunction was identified in familial cases diagnosed with panventriculomegaly with a wide foramen of Magendie and large cisterna magna (PaVM) that belongs to a subtype of late-onset congenital communicating hydrocephalus [43]. Mutation in CFAP43, encoding a protein associated with ependymal cilia, was also identified as a cause of familial NPH, and CFAP43 knock-out mice exhibited hydrocephalus due to morphologic abnormality of motile cilia [42]. Two deletions in CWH43 which regulates the membrane targeting of glycosylphosphatidylinositol-anchored proteins were identified in 8 of 53 iNPH patients (15 %), and CWH43 mutant mice exhibited communicating hydrocephalus with gait and balance dysfunction [50]. Because CWH43 mutant mice appear normal at birth and show no major deficits through middle age, they are considered to be the animal model close to iNPH or late-onset congenital or developmental hydrocephalus. The number of ependymal cilia of CWH43 mutant mice was significantly decreased compared to that of wild type mice, although CWH43 has not been reported to be a gene associated with dysfunction or degeneration of motile cilia and ependymal cells. This review excluded genes that were reported to be associated with cerebral aqueduct stenosis and non-communicating hydrocephalus. These genetic studies provide robust support regarding the importance of ciliary function and offer a commonality among the varied causes of communicating hydrocephalus. However, less is understood on how these gene mutations associated with ciliary dysfunction result in ventricle dilation. Other genes which were reported to cause hydrocephalus in rodents are associated with several inflammatory cytokines. Transgenic mice that overexpressed the transforming growth factor β1 (TGF-β1) die within 3 weeks after developing a severe form of hydrocephalus [56]. In the hyh mouse, one-third survive longer and exhibit congenital hydrocephalus [45, 57]. Here the cytokine tumour necrosis factor alpha (TNFα) and its receptor TNFαR1 were upregulated in the brain, particularly in the periventricular white matter lesion [57]. The production of TNFα in glial cells could mediate the permeability of the endothelium of the brain blood barrier [58]. Therefore, these inflammatory cytokines might affect CSF malabsorption due to dysfunction of the glymphatic pathway in the aetiology of iNPH.
Environmental factors
Alcohol abuse during pregnancy is a known risk factor for foetal hydrocephalus [59, 60]. Furthermore, oral intake of ethanol was reported to decrease the beating frequency of motile cilia on the ependymal cells of lateral and third ventricles in a rat model [61]. Recently, two case-control studies provided evidence indicating that alcohol consumption showed a higher frequency in iNPH patients compared with an age-matched general population [62, 63]. Therefore, alcohol consumption is a potential risk factor for developing iNPH due to motile cilia dysfunction on the ependymal cells.
Patients with iNPH have a higher comorbidity rate of diabetes, which is more than double, compared with an age-adjusted general population [64–66]; however, the causal relationship between diabetes and iNPH has not yet been determined. Studies from patients with vestibular schwannoma have reported to have higher occurrence of communicating-type hydrocephalus, rather than non-communicating hydrocephalus due to the fourth ventricular compression [67–72]. Furthermore, sNPH related to vestibular schwannoma is frequently observed as focal dilation of the Sylvian fissure concurrent with ventricular enlargement, similar to DESH in iNPH [68, 72]. Following the complete removal of the schwannoma, 78–100% of sNPH patients were improved without persistent CSF shunt surgery [68–70]. Preoperative protein concentration in the lumber CSF was higher, [67] but significantly decreased after tumour removal [69]. Therefore, a higher protein concentration in CSF produced by schwannomas is a potential biomarker for communicating-type hydrocephalus pathogenesis associated with vestibular schwannoma. If sNPH associated with vestibular schwannoma is a reversible disease without CSF shunt surgery by tumor removal, permanent impaired absorption of CSF is unlikely to be the cause of hydrocephalus. Instead, dysfunction of CSF dynamics should be considered as the main cause of communicating-type hydrocephalus associated with vestibular schwannoma. A high concentration of sugars and proteins in CSF increases the fluid viscosity which in turn increases the shear stress on the ventricular wall surface, as shear stress is dependent on the flow velocity and fluid viscosity. In vivo experiments have shown that the increase in fluid viscosity enhances the dilatation of the vasculature, in parallel with changes in blood viscosity, through the sensitivity of the endothelium to shear stress [73, 74].
A higher frequency of sleep apnoea was reported in iNPH patients in a case-control study [75]. Indeed, there has been growing interest in the relationship between sleep and the glymphatic system [76–78]. The clearance activity of the glymphatic system is stimulated during sleep by increased CSF and interstitial fluid exchange. The amount of interstitial fluid increased by 60 % during sleep compared with being awake [78]. In addition, disorders of the sleep-wake cycle are closely associated with the accumulation of brain waste products, including amyloid-β and τ, and optimising the sleep-wake cycle would be important for the prevention of Alzheimer’s disease and other tauopathy [77, 79, 80]. Recently, deep non-rapid eye movements sleep has been reported to amplify reciprocating CSF movements synchronised with cerebral blood flow in conjunction with slow-delta electrophysiologic oscillations [81]. During deep sleep, the large slow waves of cerebral blood circulation and neural activity were interlinked to the CSF inflow into the ventricles through the foramen of Magendie.
In a nested case-control study, hypertension was reported to be a potential risk factor for hydrocephalic ventricular enlargement, which was distinct from ventricular enlargement in iNPH [65]. In rodents, angiotensin-II induced hypertension and decreased the speed of CSF net flow in the periarterial space by less than 40% [30], suggesting a decline in glymphatic function.
Aging effect on ventricular enlargement in iNPH
Current CSF dynamic results suggest that age-related impairment of meningeal lymphatic CSF drainage and glymphatic fluid exchange between CSF and interstitial fluid may contribute to the pathogenesis of iNPH [32, 82–85]. Indeed, the meningeal lymphatic outflow of CSF is reduced in aged mice compared with young mice [86]. The reduction of cerebral blood flow, arterial pulsations and arterial elasticity could contribute to age-related glymphatic decline, and furthermore the age-related decline in sleep duration and efficiency may also impair the overall sleep-active glymphatic function. Sleep quality and quantity decline rapidly over the age of 65 and is characterised by the significant decrease of deep sleep with non-rapid eye movements-slow wave [80, 87 ]. In general, older individuals (> 65-years) sleep lighter and cannot stay asleep, resulting in awakening early in the morning. Because aging is considered to be the most relevant to the development of iNPH, the pathogenic mechanism of ventricular enlargement in iNPH may be associated with age-related sleep disorders. In addition, it is important to note the phenomenon of the aging brain, i.e. as the brain volume decreases, the intracranial CSF volume increases. Age-related declines in the function and number of motile cilia and ependymal cells also lead to ventricular enlargement in iNPH, as detailed in the next chapter.
Oscillation of shear stress on the ventricular wall surface
Under normal conditions, the movement of CSF in the cerebral aqueduct is much less than those in the foramen magnum and premedullary cistern [10, 17] The coordinated beating of motile cilia on the ependymal cells covering ventricular surface generates a directional steady laminar flow of CSF, which maintains the ventricles in a calm environment [20, 88]. Conversely, subarachnoid space is covered by the meningeal cells forming the leptomeninges, arachnoid and pia mater, and forms a transparent membrane composed of fibrous tissue with no motile cilia. The arachnoid membrane is connected to the pia mater by many trabeculae, just like a cobweb, and structurally continuous with the pia mater. In iNPH, the CSF stroke volume at the cerebral aqueduct is significantly increased, while unchanged at the foramen magnum [13, 17, 18]. Inevitably, the oscillatory shear stress produced by the reciprocating CSF flow acting tangentially on the wall surface is also increased in the cerebral aqueduct [18]. Increase in the CSF oscillation directly impedes normal cilia beating. The impaired beating of ependymal cilia is compounded by decreased apical actin filaments, which leads to motile cilia shedding from the ependymal cells, since the coordinated directional beating of motile cilia contributes to centriole stabilisation [88]. Oscillatory shear stress produced by the complex turbulent flow is known to be a detrimental cellular stress and be associated with autophagy in endothelial cells [89, 90]. The shedding of motor cilia can cause glial scarring on the surface of the ventricles by direct exposure to strong oscillatory shear stresses, as illustrated in Fig. 3. It has been demonstrated that the lateral ventricles expand due to the loss or degeneration of ependymal cell coverage on the ventricular surface [91].

Illustration of cilia loss from the cerebral aqueduct wall surface due to oscillatory shear stress produced by the reciprocating CSF flow. This figure was drawn by our imagination using the Paint 3D application (Microsoft Corporation) and SYNAPSE 3D (FUJIFILM Corporation). The two upper figures depict the cerebral aqueduct in which the coordinated beating of motile cilia on the ependymal cells generate a directional steady flow of CSF under normal conditions. The lower figures illustrate the shedding of the motile cilia from the degenerated ependymal cells due to increased CSF backflow into the ventricles in iNPH
Oscillation of the shear stress on the vascular endothelial cells has been noted in the pathogenesis of vascular dilation through activation of inflammatory signals [74, 92–97]. Compared with physiological steady shear stress, oscillatory shear stress induces higher activation of TGF-β1 which drives pathological vascular remodelling [94]. Endothelial cilia dysfunction contributes to aberrant fluid-sensing and thus results in vascular disorders, such as aneurysm formation and atherosclerosis [98, 99] Therefore, increased oscillatory shear stress due to reciprocating CSF movements may directly activate flow-mediated dilation at the cerebral aqueduct, and it may generate a synergistic effect of increased stroke volumes at the nearby upstream CSF pathways in the ventricles. Finally, the lateral ventricles might gradually expand due to an increase in oscillatory shear stress at the ependymal surface and loss of motile cilia and ependymal cells.
Potential biomarker of cerebrospinal fluid indicating degeneration of motile cilia or ventriculomegaly
Recently, Nakajima et al. reported that iNPH patients had significantly higher concentration of the protein tyrosine phosphatase receptor type Q (PTPRQ) in the lumbar CSF (mean: 619 pg/mL), compared with either healthy control individuals (296 pg/mL), patients with Alzheimer’s disease (365 pg/mL) or Parkinson’s disease (338 pg/mL) [100]. Furthermore, the mean levels of PTPRQ for sNPH (1697 pg/mL) and late-onset congenital or developmental hydrocephalus (1720 pg/mL) were double that of iNPH levels. Patients with sNPH and late-onset congenital or developmental hydrocephalus usually had larger ventricles and higher CSF stroke volume at the cerebral aqueduct, compared with patients with iNPH, as depicted in Fig. 2. PTPRQ is a membrane protein involved in actin polymerisation and ciliary movement and structure. In addition, PTPRQ are expressed in the ependymal cells of the ventricles and choroid plexus indicated by immunostaining and in situ hybridisation. Therefore, PTPRQ concentration in the CSF is a good candidate as a CSF biomarker for ventricular enlargement in NPH due to the degeneration of motile cilia on the ependymal cells.
Conclusions
This review summarises the current literature related to the mechanism of ventricular enlargement in iNPH. Although the aetiology of ventricular enlargement in iNPH has not yet been demonstrated, several reports suggest that the increase oscillatory shear stress on the ventricular wall, shedding of motor cilia and the subsequent loss of ependymal cell coverage may be crucially involved for ventricular enlargement in iNPH (Fig. 4). The cumulative evidence from these studies support the notion of CSF malabsorption as the combined impairment of glymphatic and meningeal lymphatic function is located upstream of ciliary dysfunction in the mechanism of ventricular enlargement in iNPH (Fig. 4). In summary, we hope that this review will be valuable for future basic and clinical research focusing on the mechanism of ventricular enlargement in iNPH.

Potential mechanisms of ventricular enlargement in iNPH. The schematic diagram provides an overview of possible aetiological factors for ventricular dilatation in iNPH
Availability of data and materials
Not applicable.
Abbreviations
- CSF:
-
Cerebrospinal fluid
- DESH:
-
Disproportionally enlarged subarachnoid space hydrocephalus
- iNPH:
-
Idiopathic normal pressure hydrocephalus
- NPH:
-
Normal pressure hydrocephalus
- PaVM:
-
Panventriculomegaly with a wide foramen of Magendie and large cisterna magna
- PTPRQ:
-
Protein tyrosine phosphatase receptor type Q
- sNPH:
-
Secondary normal pressure hydrocephalus
References
Adams RD, Fisher CM, Hakim S, et al. Symptomatic occult hydrocephalus with normal cerebrospinal-fluid pressure. A treatable syndrome. N Eng J Med. 1965;273:117–26. https://doi.org/10.1056/NEJM196507152730301 (published Online First: Epub Date).
Marmarou A, Bergsneider M, Relkin N, et al. Development of guidelines for idiopathic normal-pressure hydrocephalus: introduction. Neurosurgery. 2005;57(3_Suppl):S1–3.
Mori E, Ishikawa M, Kato T, et al. Guidelines for management of idiopathic normal pressure hydrocephalus: second edition. Neurologia medico-chirurgica. 2012;52(11):775–809.
Nakajima M, Yamada S, Miyajima M, et al.: Guidelines for Management of Idiopathic Normal Pressure Hydrocephalus (Third Edition): Endorsed by the Japanese Society of Normal Pressure Hydrocephalus. Neurologia medico-chirurgica 2021:(online first) doi: https://doi.org/10.2176/nmc.st.2020-0292. (published Online First: Epub Date).
Yamada S, Ishikawa M, Iwamuro Y, et al. Choroidal fissure acts as an overflow device in cerebrospinal fluid drainage: morphological comparison between idiopathic and secondary normal-pressure hydrocephalus. Sci Rep. 2016;6:39070. https://doi.org/10.1038/srep39070 (published Online First: Epub Date).
Yamada S, Ishikawa M, Yamamoto K. Fluid distribution pattern in adult-onset congenital, idiopathic and secondary normal-pressure hydrocephalus: implications for clinical care. Front Neurol. 2017;8:583. https://doi.org/10.3389/fneur.2017.00583 (published Online First: Epub Date).
Yamada S, Ishikawa M, Yamamoto K. Optimal diagnostic indices for idiopathic normal pressure hydrocephalus based on the 3D quantitative volumetric analysis for the cerebral ventricle and subarachnoid space. AJNR Am J Neuroradiol. 2015;36(12):2262–9. https://doi.org/10.3174/ajnr.A4440 (published Online First: Epub Date).
Yamada S, Ishikawa M, Yamamoto K. Comparison of CSF distribution between idiopathic normal pressure hydrocephalus and Alzheimer disease. AJNR Am J Neuroradiol. 2016;37(7):1249–55. https://doi.org/10.3174/ajnr.A4695 (published Online First: Epub Date).
Hashimoto M, Ishikawa M, Mori E, et al. Diagnosis of idiopathic normal pressure hydrocephalus is supported by MRI-based scheme: a prospective cohort study. Cerebrospinal Fluid Res. 2010;7:18. https://doi.org/10.1186/1743-8454-7-18 (published Online First: Epub Date).
Baledent O, Gondry-Jouet C, Meyer ME, et al. Relationship between cerebrospinal fluid and blood dynamics in healthy volunteers and patients with communicating hydrocephalus. Investigative Radiol. 2004;39(1):45–55. https://doi.org/10.1097/01.rli.0000100892.87214.49 (published Online First: Epub Date).
Bradley WG Jr, Queralt SD, et al. Normal-pressure hydrocephalus: evaluation with cerebrospinal fluid flow measurements at MR imaging. Radiology. 1996;198(2):523–9. https://doi.org/10.1148/radiology.198.2.8596861 (published Online First: Epub Date).
Bradley WG Jr. CSF flow in the brain in the context of normal pressure hydrocephalus. AJNR Am J Neuroradiol. 2014;36(5):831–8. https://doi.org/10.3174/ajnr.A4124 (published Online First: Epub Date).
Lindstrom EK, Ringstad G, Mardal KA, et al. Cerebrospinal fluid volumetric net flow rate and direction in idiopathic normal pressure hydrocephalus. NeuroImage Clin. 2018;20:731–41. https://doi.org/10.1016/j.nicl.2018.09.006 (published Online First: Epub Date).
Shanks J, Markenroth Bloch K, Laurell K, et al. Aqueductal CSF stroke volume is increased in patients with idiopathic normal pressure hydrocephalus and decreases after shunt surgery. AJNR Am J Neuroradiol. 2019;40(3):453–9. https://doi.org/10.3174/ajnr.A5972 (published Online First: Epub Date).
Yamada S, Miyazaki M, Kanazawa H, et al. Visualization of cerebrospinal fluid movement with spin labeling at MR imaging: preliminary results in normal and pathophysiologic conditions. Radiology. 2008;249(2):644–52. https://doi.org/10.1148/radiol.2492071985 (published Online First: Epub Date).
Yamada S, Tsuchiya K, Bradley WG, et al. Current and emerging MR imaging techniques for the diagnosis and management of CSF flow disorders: a review of phase-contrast and time-spatial labeling inversion pulse. AJNR Am J Neuroradiol. 2015;36(4):623–30. https://doi.org/10.3174/ajnr.A4030 (published Online First: Epub Date).
Yamada S, Ishikawa M, Ito H, et al. Cerebrospinal fluid dynamics in idiopathic normal pressure hydrocephalus on four-dimensional flow imaging. Eur Radiol. 2020;30(8):4454–65. https://doi.org/10.1007/s00330-020-06825-6 (published Online First: Epub Date).
Yamada S, Ito H, Ishikawa M, et al. Quantification of oscillatory shear stress from reciprocating CSF motion on 4D flow imaging. AJNR Am J Neuroradiol. 2021. https://doi.org/10.3174/ajnr.A6941 (published Online First: Epub Date).
Bradley WG Jr, Whittemore AR, Kortman KE, et al. Marked cerebrospinal fluid void: indicator of successful shunt in patients with suspected normal-pressure hydrocephalus. Radiology. 1991;178(2):459–66. https://doi.org/10.1148/radiology.178.2.1987609 (published Online First: Epub Date).
Olstad EW, Ringers C, Hansen JN, et al. Ciliary beating compartmentalizes cerebrospinal fluid flow in the brain and regulates ventricular development. Curr Biol. 2019;29(2):229-41 e6. https://doi.org/10.1016/j.cub.2018.11.059 (published Online First: Epub Date).
Ohata S, Nakatani J, Herranz-Perez V, et al. Loss of dishevelleds disrupts planar polarity in ependymal motile cilia and results in hydrocephalus. Neuron. 2014;83(3):558–71. https://doi.org/10.1016/j.neuron.2014.06.022 (published Online First: Epub Date).
Ohata S, Herranz-Perez V, Nakatani J, et al. Mechanosensory genes Pkd1 and Pkd2 contribute to the planar polarization of brain ventricular epithelium. J Neurosci. 2015;35(31):11153–68. https://doi.org/10.1523/JNEUROSCI.0686-1520152015.2015 (published Online First: Epub Date).
Siyahhan B, Knobloch V, De Zelicourt D, et al. Flow induced by ependymal cilia dominates near-wall cerebrospinal fluid dynamics in the lateral ventricles. J R Soc Interface. 2014;11(94):20131189. https://doi.org/10.1098/rsif.2013.1189 (published Online First: Epub Date).
Cushing H. The third circulation and its channels. Lancet. 1925;2:851–7.
Da Mesquita S, Louveau A, Vaccari A, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560(7717):185–91. https://doi.org/10.1038/s41586-018-0368-8 (published Online First: Epub Date).
Da Mesquita S, Fu Z, Kipnis J. The meningeal lymphatic system: a new player in neurophysiology. Neuron. 2018;100(2):375–88. https://doi.org/10.1016/j.neuron.2018.09.022 (published Online First: Epub Date).
Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra11. https://doi.org/10.1126/scitranslmed.3003748 (published Online First: Epub Date).
Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–41. https://doi.org/10.1038/nature14432 (published Online First: Epub Date).
Louveau A, Herz J, Alme MN, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nature Neurosci. 2018;21(10):1380–91. https://doi.org/10.1038/s41593-018-0227-9 (published Online First: Epub Date).
Mestre H, Tithof J, Du T, et al. Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun. 2018;9(1):4878. https://doi.org/10.1038/s41467-018-07318-3 (published Online First: Epub Date).
Nedergaard M. Neuroscience. Garbage truck of the brain. Science. 2013;340(6140):1529–30. https://doi.org/10.1126/science.1240514 (published Online First: Epub Date).
Ringstad G, Eide PK. Cerebrospinal fluid tracer efflux to parasagittal dura in humans. Nat Commun. 2020;11(1):354. https://doi.org/10.1038/s41467-019-14195-x (published Online First: Epub Date).
Kida S, Pantazis A, Weller RO. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol. 1993;19(6):480–8.
Badano JL, Mitsuma N, Beales PL, et al. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genom Hum Genet. 2006;7:125–48. https://doi.org/10.1146/annurev.genom.7.080505.115610 (published Online First: Epub Date).
Furey CG, Zeng X, Dong W, et al. Human genetics and molecular mechanisms of congenital hydrocephalus. World Neurosurg. 2018;119:441–3. https://doi.org/10.1016/j.wneu.2018.09.018 (published Online First: Epub Date).
Zhang J, Williams MA, Rigamonti D. Genetics of human hydrocephalus. J Neurol. 2006;253(10):1255–66. https://doi.org/10.1007/s00415-006-0245-5 (published Online First: Epub Date).
Abdelhamed Z, Vuong SM, Hill L, et al. A mutation in Ccdc39 causes neonatal hydrocephalus with abnormal motile cilia development in mice. Development. 2018. https://doi.org/10.1242/dev.154500 (published Online First: Epub Date).
Banizs B, Pike MM, Millican CL, et al. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development. 2005;132(23):5329–39. https://doi.org/10.1242/dev.02153 (published Online First: Epub Date).
Dawe HR, Shaw MK, Farr H, et al. The hydrocephalus inducing gene product, Hydin, positions axonemal central pair microtubules. BMC Biol. 2007;5:33. https://doi.org/10.1186/1741-7007-5-33 (published Online First: Epub Date).
Doggett NA, Xie G, Meincke LJ, et al. A 360-kb interchromosomal duplication of the human HYDIN locus. Genomics. 2006;88(6):762–71. https://doi.org/10.1016/j.ygeno.2006.07.012 (published Online First: Epub Date).
Ibanez-Tallon I, Pagenstecher A, Fliegauf M, et al. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum Mol Genetics. 2004;13(18):2133–41. https://doi.org/10.1093/hmg/ddh219 (published Online First: Epub Date).
Morimoto Y, Yoshida S, Kinoshita A, et al. Nonsense mutation in CFAP43 causes normal-pressure hydrocephalus with ciliary abnormalities. Neurology. 2019;92(20):e2364–74. https://doi.org/10.1212/WNL.0000000000007505 (published Online First: Epub Date).
Kageyama H, Miyajima M, Ogino I, et al. Panventriculomegaly with a wide foramen of Magendie and large cisterna magna. J Neurosurg. 2016;124(6):1858–66. https://doi.org/10.3171/2015.6.JNS.15162 (published Online First: Epub Date).
Lechtreck KF, Delmotte P, Robinson ML, et al. Mutations in Hydin impair ciliary motility in mice. J Cell Biol. 2008;180(3):633–43. https://doi.org/10.1083/jcb.200710162 (published Online First: Epub Date).
Roales-Bujan R, Paez P, Guerra M, et al. Astrocytes acquire morphological and functional characteristics of ependymal cells following disruption of ependyma in hydrocephalus. Acta Neuropathol. 2012;124(4):531–46. https://doi.org/10.1007/s00401-012-0992-6 (published Online First: Epub Date).
Tissir F, Qu Y, Montcouquiol M, et al. Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci. 2010;13(6):700–7. https://doi.org/10.1038/nn.2555 (published Online First: Epub Date).
Wallmeier J, Frank D, Shoemark A, et al. De novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. Am J Hum Genet. 2019;105(5):1030–9. https://doi.org/10.1016/j.ajhg.2019.09.022 (published Online First: Epub Date).
Wang X, Zhou Y, Wang J, et al. SNX27 deletion causes hydrocephalus by impairing ependymal cell differentiation and ciliogenesis. J Neurosci. 2016;36(50):12586–97. https://doi.org/10.1523/JNEUROSCI.1620-16.2016 (published Online First: Epub Date).
Wodarczyk C, Rowe I, Chiaravalli M, et al. A novel mouse model reveals that polycystin-1 deficiency in ependyma and choroid plexus results in dysfunctional cilia and hydrocephalus. PloS One. 2009;4(9):e7137. https://doi.org/10.1371/journal.pone.0007137 (published Online First: Epub Date).
Yang HW, Lee S, Yang D, et al. Deletions in CWH43 cause idiopathic normal pressure hydrocephalus. EMBO Mol Med. 2021. https://doi.org/10.15252/emmm.202013249 (published Online First: Epub Date).
Ying G, Avasthi P, Irwin M, et al. Centrin 2 is required for mouse olfactory ciliary trafficking and development of ependymal cilia planar polarity. J Neurosci. 2014;34(18):6377–88. https://doi.org/10.1523/JNEUROSCI.0067-14.2014 (published Online First: Epub Date).
Abdi K, Lai CH, Paez-Gonzalez P, et al. Uncovering inherent cellular plasticity of multiciliated ependyma leading to ventricular wall transformation and hydrocephalus. Nat Commun. 2018;9(1):1655. https://doi.org/10.1038/s41467-018-03812-w (published Online First: Epub Date).
Nauli SM, Kawanabe Y, Kaminski JJ, et al. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117(9):1161–71. https://doi.org/10.1161/CIRCULATIONAHA.107.710111 (published Online First: Epub Date).
Takagishi M, Sawada M, Ohata S, et al. Daple coordinates planar polarized microtubule dynamics in ependymal cells and contributes to hydrocephalus. Cell Rep. 2017;20(4):960–72. https://doi.org/10.1016/j.celrep.2017.06.089 (published Online First: Epub Date).
Drielsma A, Jalas C, Simonis N, et al. Two novel CCDC88C mutations confirm the role of DAPLE in autosomal recessive congenital hydrocephalus. J Med Genet. 2012;49(11):708–12. https://doi.org/10.1136/jmedgenet-2012-101190 (published Online First: Epub Date).
Galbreath E, Kim SJ, Park K, et al. Overexpression of TGF-beta 1 in the central nervous system of transgenic mice results in hydrocephalus. J Neuropathol Exp Neurol. 1995;54(3):339–49. https://doi.org/10.1097/00005072-199505000-00007 (published Online First: Epub Date).
Jimenez AJ, Rodriguez-Perez LM, Dominguez-Pinos MD, et al. Increased levels of tumour necrosis factor alpha (TNFalpha) but not transforming growth factor-beta 1 (TGFbeta1) are associated with the severity of congenital hydrocephalus in the hyh mouse. Neuropathol Appl Neurobiol. 2014;40(7):911–32. https://doi.org/10.1111/nan.12115 (published Online First: Epub Date).
Erickson MA, Wilson ML, Banks WA. In vitro modeling of blood-brain barrier and interface functions in neuroimmune communication. Fluids Barriers CNS. 2020;17(1):26. https://doi.org/10.1186/s12987-020-00187-3 (published Online First: Epub Date).
Kahle KT, Kulkarni AV, Limbrick DD Jr, et al. Hydrocephalus in children. Lancet. 2016;387(10020):788–99. https://doi.org/10.1016/S0140-6736(15)60694-8 (published Online First: Epub Date).
Sakata-Haga H, Sawada K, Ohnishi T, et al. Hydrocephalus following prenatal exposure to ethanol. Acta Neuropathol. 2004;108(5):393–8. https://doi.org/10.1007/s00401-004-0901-8 (published Online First: Epub Date).
Omran AJA, Saternos HC, Althobaiti YS, et al. Alcohol consumption impairs the ependymal cilia motility in the brain ventricles. Sci Rep. 2017;7(1):13652. https://doi.org/10.1038/s41598-017-13947-3 (published Online First: Epub Date).
Ghaffari-Rafi A, Gorenflo R, Hu H, et al. Role of psychiatric, cardiovascular, socioeconomic, and demographic risk factors on idiopathic normal pressure hydrocephalus: a retrospective case-control study. Clin Neurol Neurosurg. 2020;193:105836. https://doi.org/10.1016/j.clineuro.2020.105836 (published Online First: Epub Date).
Hickman TT, Shuman ME, Johnson TA, et al. Association between shunt-responsive idiopathic normal pressure hydrocephalus and alcohol. J Neurosurg. 2017;127(2):240–8. https://doi.org/10.3171/2016.6.JNS16496 (published Online First: Epub Date).
Hudson M, Nowak C, Garling RJ, et al. Comorbidity of diabetes mellitus in idiopathic normal pressure hydrocephalus: a systematic literature review. Fluids Barriers CNS. 2019;16(1):5. https://doi.org/10.1186/s12987-019-0125-x (published Online First: Epub Date).
Jaraj D, Agerskov S, Rabiei K, et al. Vascular factors in suspected normal pressure hydrocephalus: a population-based study. Neurology. 2016. https://doi.org/10.1212/WNL.0000000000002369 (published Online First: Epub Date).
Rasanen J, Huovinen J, Korhonen VE, et al. Diabetes is associated with familial idiopathic normal pressure hydrocephalus: a case-control comparison with family members. Fluids Barriers CNS. 2020;17(1):57. https://doi.org/10.1186/s12987-020-00217-0 (published Online First: Epub Date).
Fukuda M, Oishi M, Kawaguchi T, et al. Etiopathological factors related to hydrocephalus associated with vestibular schwannoma. Neurosurgery. 2007;61(6):1186–92. https://doi.org/10.1227/01.neu.0000306096.61012.22 (published Online First: Epub Date. discussion 92 – 3).
Gerganov VM, Pirayesh A, Nouri M, et al. Hydrocephalus associated with vestibular schwannomas: management options and factors predicting the outcome. J Neurosurg. 2011;114(5):1209–15. https://doi.org/10.3171/2010.10.JNS.1029 (published Online First: Epub Date).
Miyakoshi A, Kohno M, Nagata O, et al. Hydrocephalus associated with vestibular schwannomas: perioperative changes in cerebrospinal fluid. Acta Neurochir. 2013;155(7):1271–6. https://doi.org/10.1007/s00701-013-1742-9 (published Online First: Epub Date).
Pirouzmand F, Tator CH, Rutka J. Management of hydrocephalus associated with vestibular schwannoma and other cerebellopontine angle tumors. Neurosurgery. 2001;48(6):1246–53. https://doi.org/10.1097/00006123-200106000-00010 (published Online First: Epub Date. discussion 53 – 4).
Rogg JM, Ahn SH, Tung GA, et al. Prevalence of hydrocephalus in 157 patients with vestibular schwannoma. Neuroradiology. 2005;47(5):344–51. https://doi.org/10.1007/s00234-005-1363-y (published Online First: Epub Date).
Tanaka Y, Kobayashi S, Hongo K, et al. Clinical and neuroimaging characteristics of hydrocephalus associated with vestibular schwannoma. J Neurosurg. 2003;98(6):1188–93. https://doi.org/10.3171/jns.2003.98.6..1188 (published Online First: Epub Date).
Melkumyants AM, Balashov SA. Effect of blood viscocity on arterial flow induced dilator response. Cardiovasc Res. 1990;24(2):165–8. https://doi.org/10.1093/cvr/24.2.165 (published Online First: Epub Date).
Roux E, Bougaran P, Dufourcq P, et al. Fluid shear stress sensing by the endothelial layer. Front Physiol. 2020;11:861. https://doi.org/10.3389/fphys.2020.00861 (published Online First: Epub Date).
Roman GC, Verma AK, Zhang YJ, et al. Idiopathic normal-pressure hydrocephalus and obstructive sleep apnea are frequently associated: a prospective cohort study. J Neurol Sci. 2018;395:164–8. https://doi.org/10.1016/j.jns.2018.10.005 (published Online First: Epub Date).
Hablitz LM, Pla V, Giannetto M, et al. Circadian control of brain glymphatic and lymphatic fluid flow. Nat Commun. 2020;11(1):4411. https://doi.org/10.1038/s41467-020-18115-2 (published Online First: Epub Date).
Holth JK, Fritschi SK, Wang C, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019;363(6429):880–4. https://doi.org/10.1126/science.aav2546 (published Online First: Epub Date).
Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–7. https://doi.org/10.1126/science.1241224 (published Online First: Epub Date).
Brown BM, Rainey-Smith SR, Villemagne VL, et al. The relationship between sleep quality and brain amyloid burden. Sleep. 2016;39(5):1063–8. https://doi.org/10.5665/sleep.5756 (published Online First: Epub Date).
Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–7. https://doi.org/10.1126/science.1180962 (published Online First: Epub Date).
Fultz NE, Bonmassar G, Setsompop K, et al. Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science. 2019;366(6465):628–31. https://doi.org/10.1126/science.aax5440 (published Online First: Epub Date).
Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17(11):1016–24. https://doi.org/10.1016/S1474-4422(18)30318-1 (published Online First: Epub Date).
Ringstad G, Vatnehol SaS, Eide PK. Glymphatic MRI in idiopathic normal pressure hydrocephalus. Brain. 2017;140(10):2691–705. https://doi.org/10.1093/brain/awx191 (published Online First: Epub Date).
Ringstad G, Valnes LM, Dale AM, et al. Brain-wide glymphatic enhancement and clearance in humans assessed with MRI. JCI Insight. 2018. https://doi.org/10.1172/jci.insight.121537 (published Online First: Epub Date).
Taoka T, Naganawa S. Glymphatic imaging using MRI. J Magnetic Resonance Imag. 2020;51(1):11–24. https://doi.org/10.1002/jmri.26892 (published Online First: Epub Date).
Ma Q, Ineichen BV, Detmar M, et al. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. 2017;8(1):1434. https://doi.org/10.1038/s41467-017-01484-6 (published Online First: Epub Date).
Ohayon MM, Carskadon MA, Guilleminault C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep. 2004;27(7):1255–73. https://doi.org/10.1093/sleep/27.7.1255 (published Online First: Epub Date).
Mahuzier A, Shihavuddin A, Fournier C, et al. Ependymal cilia beating induces an actin network to protect centrioles against shear stress. Nat Commun. 2018;9(1):2279. https://doi.org/10.1038/s41467-018-04676-w (Published Online First: Epub Date).
Liu J, Bi X, Chen T, et al. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015;6:e1827. https://doi.org/10.1038/cddis.2015.193 (published Online First: Epub Date).
Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6(1):16–26. https://doi.org/10.1038/ncpcardio1397 (published Online First: Epub Date).
Shook BA, Lennington JB, Acabchuk RL, et al. Ventriculomegaly associated with ependymal gliosis and declines in barrier integrity in the aging human and mouse brain. Aging Cell. 2014;13(2):340–50. https://doi.org/10.1111/acel.12184 (published Online First: Epub Date).
Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330(20):1431–8. https://doi.org/10.1056/NEJM199405193302008 (published Online First: Epub Date).
Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10(1):53–62. https://doi.org/10.1038/nrm2596 (published Online First: Epub Date).
Kouzbari K, Hossan MR, Arrizabalaga JH, et al. Oscillatory shear potentiates latent TGF-beta1 activation more than steady shear as demonstrated by a novel force generator. Sci Rep. 2019;9(1):6065. https://doi.org/10.1038/s41598-019-42302-x (Published online First: Epub Date).
Ku DN, Giddens DP, Zarins CK, et al. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis. 1985;5(3):293–302. (published Online First: Epub Date).
Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. Jama. 1999;282(21):2035–42. https://doi.org/10.1001/jama.282.21.2035. (published Online First: Epub Date).
Meng H, Tutino VM, Xiang J, et al. High WSS or low WSS? Complex interactions of hemodynamics with intracranial aneurysm initiation, growth, and rupture: toward a unifying hypothesis. AJNR Am J Neuroradiol. 2014;35(7):1254–62. https://doi.org/10.3174/ajnr.A3558. (published Online First: Epub Date).
Luu VZ, Chowdhury B, Al-Omran M, et al. Role of endothelial primary cilia as fluid mechanosensors on vascular health. Atherosclerosis. 2018;275:196–204. https://doi.org/10.1016/j.atherosclerosis.2018.06.818. (published Online First: Epub Date).
Pala R, Jamal M, Alshammari Q, et al. The roles of primary cilia in cardiovascular diseases. Cells. 2018. https://doi.org/10.3390/cells7120233 (published Online First: Epub Date).
Nakajima M, Rauramaa T, Makinen PM, et al. Protein tyrosine phosphatase receptor type Q in cerebrospinal fluid reflects ependymal cell dysfunction and is a potential biomarker for adult chronic hydrocephalus. Eur J Neurol. 2020. https://doi.org/10.1111/ene.14575( published Online First: Epub Date).
Acknowledgements
The authors thank Enago (www.enago.jp) for the English language review.
Funding
This research has received the research grants from G-7 Scholarship Foundation for one year in 2020, Taiju Life Social Welfare Foundation for one year in 2020, and FUJIFILM Corporation for two years since 2019. The funding sources for the study had no role in the design and conduct of the study, in the collection, analysis, and interpretation of the data, or in the preparation, review, or approval of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors were involved in conception of the review, and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ethics committees for human research of our institutes approved the study design and protocol (IRB Number: Rakuoto-Rin-17-041 and R2019-227).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yamada, S., Ishikawa, M. & Nozaki, K. Exploring mechanisms of ventricular enlargement in idiopathic normal pressure hydrocephalus: a role of cerebrospinal fluid dynamics and motile cilia. Fluids Barriers CNS 18, 20 (2021). https://doi.org/10.1186/s12987-021-00243-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-021-00243-6