
Abstract
Background
During the COVID-19 pandemic, antigen diagnostic tests were frequently used for screening, triage, and diagnosis. Novel instrument-based antigen tests (iAg tests) hold the promise of outperforming their instrument-free, visually-read counterparts. Here, we provide a systematic review and meta-analysis of the SARS-CoV-2 iAg tests’ clinical accuracy.
Methods
We systematically searched MEDLINE (via PubMed), Web of Science, medRxiv, and bioRxiv for articles published before November 7th, 2022, evaluating the accuracy of iAg tests for SARS-CoV-2 detection. We performed a random effects meta-analysis to estimate sensitivity and specificity and used the QUADAS-2 tool to assess study quality and risk of bias. Sub-group analysis was conducted based on Ct value range, IFU-conformity, age, symptom presence and duration, and the variant of concern.
Results
We screened the titles and abstracts of 20,431 articles and included 114 publications that fulfilled the inclusion criteria. Additionally, we incorporated three articles sourced from the FIND website, totaling 117 studies encompassing 95,181 individuals, which evaluated the clinical accuracy of 24 commercial COVID-19 iAg tests. The studies varied in risk of bias but showed high applicability. Of 24 iAg tests from 99 studies assessed in the meta-analysis, the pooled sensitivity and specificity compared to molecular testing of a paired NP swab sample were 76.7% (95% CI 73.5 to 79.7) and 98.4% (95% CI 98.0 to 98.7), respectively. Higher sensitivity was noted in individuals with high viral load (99.6% [95% CI 96.8 to 100] at Ct-level ≤ 20) and within the first week of symptom onset (84.6% [95% CI 78.2 to 89.3]), but did not differ between tests conducted as per manufacturer’s instructions and those conducted differently, or between point-of-care and lab-based testing.
Conclusion
Overall, iAg tests have a high pooled specificity but a moderate pooled sensitivity, according to our analysis. The pooled sensitivity increases with lower Ct-values (a proxy for viral load), or within the first week of symptom onset, enabling reliable identification of most COVID-19 cases and highlighting the importance of context in test selection. The study underscores the need for careful evaluation considering performance variations and operational features of iAg tests.
Similar content being viewed by others

Background and rationale
Antigen diagnostic tests have been a key component of the COVID-19 response [1], as they allow early and prompt identification of SARS-CoV-2-positive individuals, especially when the viral load is high [2,3,4,5,6]. Antigen diagnostic tests are immunoassays designed to detect SARS-CoV-2 antigen targets, primarily the nucleocapsid protein. A wide variety of antigen diagnostic tests have been commercialized during the pandemic, enabling use in different settings from self-testing to specialized laboratories [7, 8]. Among those, the instrument-free lateral flow assays (LFAs) are the most widely used antigen detection tests, especially in circumstances where fast results are needed and only limited resources are available (including a lack of highly skilled personnel or specialized laboratory equipment). Their clinical performance has been extensively reviewed and falls short in comparison to the gold standard, reverse transcription-polymerase chain reaction (RT-PCR) [9,10,11]. On the other hand, instrument-based antigen diagnostic tests (iAg tests) leverage a variety of antigen-detection technologies with varying levels of automation and infrastructure requirements. For instance, iAg tests include high-throughput, fully automated, laboratory-based instruments as well as LFAs with results amplification (e.g., fluorescence) and dedicated readers that enable standardized result interpretation and connectivity. Their diversity encompasses a broad range of potential benefits for streamlining clinical procedures. For instance, some enable the concurrent detection of multiple pathogens [12] and/or expedite and automate testing processes. Consequently, these methods have garnered significant attention, not only for their aforementioned advantages, but also due to their reported higher sensitivity compared to conventional lateral flow tests [9, 13, 14].
There have been a number of systematic evidence syntheses on the diagnostic performance of instrument-free antigen detection tests for SARS-CoV-2 [9, 11, 14,15,16]; however, the performance of iAg tests has received relatively little attention. Previous review articles combined data from instrument-free and instrument-based antigen tests regardless of their development stage [10, 14]. Only digital and fluorescence immunoassays, which are often more appropriate for point-of-care (POC) use, were considered in the review by Keskin et al., specifically focused on iAg tests [17]. Here, we conducted a systematic review and meta-analysis to assess the clinical performance of commercially available iAg tests for detecting current SARS-CoV-2 infection compared to RT-qPCR and/or culture as the reference standard.
Methods
Overview
This study expands on systematic reviews previously published by our group, which assessed the clinical accuracy of commercially available instrument-free and instrument-based antigen-detection rapid diagnostic tests (Ag-RDTs) for SARS-CoV-2 [9, 18, 19]. The methodology of our most recent analysis was applied in the present review as appropriate [19], following the standards of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) checklist [20] (Supplementary Material, File S1). The study protocol was registered on PROSPERO (ID CRD42021276232) [21].
The following is a summary of the study protocol deviations: Analytical studies were excluded because it was not possible to conduct the analysis due to inconsistent and inadequate reporting and disparate methodologies. For similar reasons, we were unable to include in the analysis all the protocol-specified variables—viral load and antigen levels, for example—that could affect test performance.
Searched databases
We systematically searched MEDLINE (PubMed), Web of Science, medRxiv, and bioRxiv using search terms and strategy on the basis of the earlier reviews cited above [9, 18]. The full list of search terms was adapted from our previous reviews and is available in Supplementary Material (File S1) [9, 19]. No language or geographic restrictions were applied. Searches were carried out until November 7th, 2022. Additional relevant papers were identified by manually searching the website of FIND, the global alliance for diagnostics, on February 19th, 2023 (https://www.finddx.org/sarscov2-eval-antigen/).
Eligibility criteria
Clinical studies evaluating the clinical accuracy of commercially available iAg tests for SARS-CoV-2 detection against RT-PCR or viral culture as a reference standard were considered eligible. Retrospective and prospective clinical diagnostic accuracy studies with a cohort, case-control, cross-sectional, or randomized design were eligible. We included both peer-reviewed publications and preprints to present the most recent data. We excluded studies on monitoring or analytical studies and studies with a sample size of less than ten, as smaller sample sizes are prone to yielding erroneous estimations of accuracy.
Index test
Fully automated, high-throughput laboratory-based iAg tests as well as POC iAg devices requiring a specialized reader (and if the use of the appropriate reader was stated) were included in the review.
In order to further categorize the technologies under investigation, we divided the diverse group of iAg tests based on whether they were applicable to POC (henceforth called ‘POC’) or laboratory-based testing (henceforth called ‘lab-based’). A detailed description of the included tests is provided in Table S1 and File S2.
Assessment of methodological quality
The QUADAS-2 tool, which had been adjusted for a prior review [9], was used to evaluate the quality of clinical accuracy studies. The relevant publication has comprehensive details on the adjustments that were made [9]. To assess whether studies performed iAg tests as per manufacturers’ instructions for use (IFU), given that it was not possible to trace back version differences across studies due to software and hardware changes, we exclusively relied on the unambiguous statement of IFU compliance, as described in each study.
Study selection and data extraction
The selection of studies, data extraction, and quality assessment were carried out as described previously [9, 19]. When needed, we reached out to the study authors to request any missing information or clarification. The final data set is available in the Supplementary Material (File S2).
Statistical analysis and data synthesis
Statistical analysis and data synthesis were conducted as previously established [19]. In summary, we calculated the performance estimates when these outcomes were assessed in at least four studies (with ≥ 20 positive samples). If there were a minimum of four data sets, each including at least 20 RT-PCR-positive samples, a bivariate meta-analysis or a univariate random effects inverse variance meta-analysis was conducted. Otherwise, only a descriptive analysis was carried out. We predefined subgroups for meta-analysis based on the following criteria: Ct value range, testing in accordance with IFU, age, presence of symptoms, duration of symptoms, and viral variant of concern. R 4.2.1 (R Foundation for Statistical Computing, Vienna, Austria) was used for the analyses.
We used the Deeks test for funnel-plot asymmetry to investigate small study effects (the “midas” command in Stata, version 15) [22, 23]. A significant asymmetry is indicated by a p-value for the slope coefficient less than 0.10.
For more information on the methodology, please refer to the original publications [9, 18, 19].
Results
Summary of studies
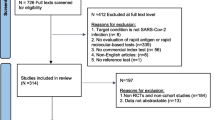
The systematic search resulted in 40,595 records. After removing duplicates, 20,431 articles were left for title and abstract screening. Out of these, 807 were considered eligible for full-text screening, of which a total of 114 were finally included. Three additional articles were included after screening the articles found on the FIND website. Thus, a total of 117 articles incorporating 159 data sets reporting on 24 unique iAg tests were included in the review (Fig. 1).

PRISMA flow diagram [20]. Abbreviations Ag-RDT = antigen rapid diagnostic test; RT-PCR = reverse transcription polymerase chain reaction; sens = sensitivity; spec = specificity; iAg test = instrument-based antigen test
Study description
A total of 109 of the 117 studies included in the review were conducted in high-income countries (HICs) and only seven studies were conducted in low- and middle-income countries (LMICs) [24,25,26,27,28,29,30]. Two studies were multi-country studies conducted in the USA/India and the UK/USA [31, 32].
A case-control design was used in 32 of the studies (27.4%) [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64], while the remaining 85 (72.6%) were cohort studies. RT-PCR was the reference method for all but one study that used viral culture [65]. Out of a total of 159 data sets, 44 (27.7%) reported on adult populations, seven data sets (4.4%) on children, and 32 data sets (20.1%) on mixed populations. In less than half of the data sets (n = 76; 47.8%), the age group of the target population was not reported. Across all the studies, the main reasons for testing were screening regardless of symptom status (70/159 data sets, 44.0%), contact investigations (67/159 data sets, 42.1%), and/or presence of symptoms (117/159 data sets; 73.5%). In 36 data sets (22.6%), the reasons for testing were not reported by the authors.
The most common specimen used for iAg testing was nasopharyngeal (‘NP’; 107 data sets, 67.3%). Other studies used combined anterior nasal/mid-turbinate (AN/MT) specimens (35 data sets, 22.0%), saliva (3 data sets, 1.9%), or oropharyngeal (‘OP’; 1 data set, 0.6%) specimens. The specimen type used was unclear in seven studies (13 data sets, 8.1%). Two of the studies pooled nasopharyngeal samples from multiple patients for testing ([51, 52]; also see Supplementary File S2).
Of the 24 unique iAg tests evaluated across all studies, 15 were suitable for POC use and nine were lab-based immunoassays. The most frequently used iAg test was the Sofia SARS Antigen FIA test by Quidel (US; henceforth called Sofia) with 22 data sets (13.8%) and 20,970 (21.8%) tests. The STANDARD F COVID-19 Ag FIA (SD Biosensor Inc., South Korea; henceforth called STANDARD F) was assessed in 18 data sets (11.3%) with 19,617 (20.4%) tests and the BD Veritor System for Rapid Detection of SARS-CoV-2 (Becton, Dickinson and Company [BD], MD, US; henceforth called BD Veritor) in 17 data sets (10.7%) with 11,878 (12.4%) tests, followed by the LumiraDx SARS-CoV-2 Ag test (LumiraDx UK Ltd., UK; henceforth called LumiraDx) with 24 data sets (15.1%) and 10,136 (10.5%) tests. Additional details on each of the iAg tests included in the review are provided in the supplements (Table S1 and File S1).
Methodological quality of included studies
The included studies were found to have a variable risk of bias, but high applicability (Fig. 2). Of the data sets evaluated, only 37 (23.3%) data sets included a representative study population by avoiding inappropriate exclusions or a case-control design, resulting in a low risk of bias. A majority of studies were carried out in a routine practice setting, resulting in a high applicability of the included study population to the review in terms of patient selection in a majority of data sets (n = 145; 91.2%), while the applicability of the study population was unclear in the remaining data sets (n = 14; 8.8%).
The interpretation of the index test results was of low concern for 59 (37.1%) data sets because it was carried out without knowledge of the results of the reference standard; however, the majority of the data sets (n = 96; 60.4%) failed to report on the blinded interpretation of the index test results. A predefined threshold was used (n = 138; 86.8%) or tests were conducted in accordance with IFU in a majority of the studies (n = 120; 75.5%). Index test applicability was judged to be of low concern in 120 (75.5%) data sets, which explicitly mentioned IFU compliance, but high in the remaining 39 (24.5%).
In 104 data sets (65.4%), the reference standard selection, its conduct, or its interpretation was insufficiently described and thus resulted in an unclear risk of bias, which was primarily caused by inadequate reporting of the results' blinded interpretation. The risk of bias in this aspect was low for the remaining data sets (n = 55; 34.6%) since the reference standard was administered prior to the iAg tests, and/or the operator administering the reference standard was blinded to the iAg test results, thereby minimizing the potential for bias. The applicability of the reference test was determined to be of low concern for all data sets, because the target condition for this review was defined by viral culture or RT-PCR.
Samples taken simultaneously were used for index and reference testing in 140 (88.1%) of the data sets. In 100 (62.9%) data sets, a single assay was consistently used as the reference, whereas multiple RT-PCR assays were used as the reference in 43 (27.0%) of the data sets (specified in S1). As a result, while also accounting for the possibility that not all patients were included in the analysis, the risk of bias related to flow and timing was assessed to be low in 54.7% of the data sets, intermediate in 27.0%, high in 5.7% and unclear in 12.6%.

(A) QUADAS assessment for risk of bias and (B) applicability
The test manufacturers provided financial support for 41 (35.0%) of the studies. In addition, they coauthored 15 of these and two additional studies, accounting for 14.5% of all studies. Moreover, a conflict of interest due to receiving funding from or employment with the test manufacturer was disclosed in 34 studies (29.1%) (File S3).
Analysis of small study effects, which may indicate publication bias, yielded no significant evidence for such effects (p = 0.39) (Figure S1).
Performance of iAg tests in comparison to RT-PCR and/or viral culture
The pooled estimates of sensitivity and specificity for all iAg tests were 76.0% (95% CI 72.7 to 79.0) and 98.5% (95% CI 98.1 to 98.8), respectively, based on the bivariate analysis of the 127 data sets from a total of 99 studies that evaluated 83,993 tests (Fig. 3A, Figure S2). This was slightly higher than a pooled sensitivity of 74.6% (95% CI 71.7 to 77.6) obtained from the univariate analysis of 144 data sets (Fig. 3B, Figure S3). The point estimate of pooled specificity was the same in a univariate analysis of 134 data sets (98.5%; 95% CI 98.0 to 98.9) (Fig. 3C, Figure S4).

Pooled accuracy of (A) bivariant analysis and (B) + (C) univariant analysis. Abbreviations CI = confidence interval
Lumipulse G had the highest pooled sensitivity (86.5% [95% CI 79.9 to 91.2]) but the lowest pooled specificity (96.4% [95% CI 94.2 to 97.8]) among the eight tests that were eligible for test-specific meta-analysis (Fig. 4A). LIAISON had the lowest pooled sensitivity (62.5% [95% CI 47.1 to 75.8]). VITROS had the highest pooled specificity at 99.7% (95% CI 99.1 to 99.9). The POC-applicable digital immunoassay BD Veritor had a pooled sensitivity of 73.9% (95% CI 63.2 to 82.3) and a pooled specificity of 99.4% (95% CI 98.9 to 99.7). Among the fluorescence immunoassays (FIAs) with sufficient numbers of data sets (> 4), LumiraDx had the highest pooled sensitivity at 81.1% (95% CI 73.2 to 87.0) but the lowest specificity at 97.3% (95% CI 95.7 to 98.3).
The pooled sensitivity and specificity for IFU-conforming data sets (n = 95) were estimated to be 75.8% (95% CI 71.9 to 79.4) and 98.5% (95% CI 98.1 to 98.9), respectively (Fig. 4B). The pooled performance for data sets without reported IFU conformity showed slightly higher sensitivity (76.5%; 95% CI 70.0 to 82.0) and similar specificity (98.4%; 95% CI 97.4 to 99.0).
The highest pooled sensitivity, 78.2% (95% CI 74.7 to 85.5), was observed when the wild-type SARS CoV-2 was predominant (64 data sets, 50.4%) (Fig. 4C). The pooled sensitivity across all studies conducted during a wave of the SARS CoV-2 Alpha variant (11 data sets, 8.7%) was 54.8% (95% CI 37.3 to 71.2), which was the lowest. Based on only six data sets, the pooled sensitivity during the Delta variant was determined to be 74.5% (95% CI 48.8 to 90) with the highest specificity (99.2%; 95% CI 96.6 to 99.8). Only two studies were conducted during the wave of the Omicron variant (2 data sets, 1.6%), with sensitivities ranging from 76.5 to 88.5% [62, 66].
After analyzing the pooled accuracy per intended use setting, the tests intended for lab-based use achieved a sensitivity of 75.9% (95% CI 69.9 to 80.9) and therefore performed similarly to the POC tests, with 76.1% (95% CI 72.1 to 79.7) sensitivity; specificity was almost identical (Fig. 4D).
When only NP samples (88 data sets) were considered, the pooled sensitivity and specificity were estimated to be 76.5% (95% CI 73.0 to 79.7) and 98.4% (95% CI 97.8 to 98.8), respectively (Fig. 4E). Analysis of AN and/or MT samples resulted in a pooled sensitivity of 80.0% with a wide confidence interval (95% CI 73.5 to 85.2) and a pooled specificity of 98.5% (95% CI 97.7 to 99.0).

Pooled accuracy of (A) per test assessed, (B) per IFU-conformity, (C) per predominant variant of concern, (D) per intended setting, and (E) per sample type. Abbreviations CI = confidence interval; IFU = instructions for use
Subgroup analyses
By age
Thirty data sets with 14,451 samples from adults (age ≥ 18 years) were available for a meta-analysis, and the results showed a pooled sensitivity and specificity of 72.9% (95% CI 63.2 to 80.9) and 98.8% (95% CI 98.0 to 99.3), respectively (Fig. 5A). Only five datasets with 1,655 samples were available for the pediatric group (age < 18 years) with, compared to adults, a higher pooled sensitivity (81.9%, 95% CI 63.5 to 92.2) and a comparable pooled specificity (98.3%, 95% CI 95.9 to 99.3).
By presence of symptoms
Compared to that in the symptomatic group (sensitivity 79.9%; 95% CI 76.5 to 83.0), the pooled sensitivity in the asymptomatic group was substantially lower at 50.3% (95% CI 33.5 to 67.0) (Fig. 5B). Both subgroups had comparably high specificity. As the analysis was repeated per intended use setting, POC tests showed higher sensitivity than the tests intended for lab use in the symptomatic group (81.1% [95% CI 77.6 to 84.1] vs. 69.1% [95% CI 60.8 to 76.4]) (Figure S5A). There were not enough data sets available for the lab tests in the asymptomatic group for the analysis to be repeated (Figure S5B).
By duration of symptoms
Data from 1,724 people who were tested within 7 days of the onset of their symptoms were available for the analysis, compared to a very small number of patients (177) who were tested ≥ 7 days after the onset of symptoms (Fig. 5B). In comparison to 84.6% (95% CI 78.2–89.3%) sensitivity for people tested within 7 days of the onset of symptoms, the pooled sensitivity for people tested ≥ 7 days was much lower with only 57.8% (95% CI 48.5–66.6%). The pooled specificity estimates were 98.4% (95% CI 97.3 to 99.1) in the < 7 days group and 97.0% (95% CI 86.2 to 99.4) in the ≥ 7 days group.
By Ct values
Fifty-five studies (255 data sets) reported on the performance values based on various Ct value groups, allowing for univariate meta-analysis, which showed that higher Ct values were associated with decreased pooled sensitivity (Fig. 5C). For the Ct value groups < 20 and ≥ 20, the pooled sensitivities were 99.6% (95% CI 98.8 to 100.0) and 94.8% (95% CI 91.0 to 98.6), respectively. For the Ct value group < 25, the pooled sensitivity was 97.8% (95% CI 96.7 to 98.5) but decreased to 85.3% (95% CI 81.7 to 89.0) for the CT value group < 30. The pooled sensitivity for the Ct value group ≥ 30 was estimated to be very low at 26.4% (95% CI 15.8 to 37.1).

Pooled accuracy of the subgroups (A) aged < 18 years and ≥ 18 years, (B) symptomatic and asymptomatic persons and (C) Ct-values. Abbreviations CI = confidence interval; Ct = cycle threshold
Sensitivity analyses
When case-control studies were excluded the sensitivity and specificity remained similar to the overall pooled sensitivity and specificity estimates with 76.6% (95% CI 72.4 to 80.3) and 98.5% (95% CI 98.0 to 98.9), respectively (Figure S6). Exclusion of preprints did not change the sensitivity or specificity significantly (75.8% [95% CI 72.3 to 79.0] and 98.4% [95% CI 98.0 to 98.8]) (Figure S7). Data from manufacturer-independent studies (68 data sets) produced results with a similar specificity of 98.4% (95% CI 97.8 to 98.8) and a slightly lower sensitivity of 74.4% (95% CI 69.5 to 78.7) (Figure S8).
The studies were also categorized by country income level of the country where participants were enrolled. No significant differences were found between HICs and LMICs for pooled sensitivity (HICs: 75.1%; 95% CI 71.5 to 78.4; LMICs: 76.6%; 95% CI 73.4 to 79.5) or specificity (HICs: 98.6; 95% CI 98.2–98.9; LMICs: 97.1; 95% CI 93.7 to 98.7), with overlapping confidence intervals (Figure S9).
Discussion
Our systematic review summarized the data from 117 studies that evaluated the clinical accuracy of 24 commercial COVID-19 iAg tests and included a total of 95,181 individuals. The meta-analysis estimated a pooled sensitivity and specificity of 76.0% (95% CI 72.7 to 79.0) and 98.5% (95% CI 98.1 to 98.8), respectively. In a mixed population of symptomatic and asymptomatic individuals, the sensitivity estimate falls short of the WHO’s minimal acceptable sensitivity requirement (≥ 80%), while the pooled specificity exceeded the acceptable specificity requirement (≥ 97%) [4]. One test (LumiraDx) met the requirements for both sensitivity and specificity at 81.1% (95% CI 73.2 to 87.0%) and 97.3% (95% CI 95.7 to 98.3), respectively, aligning with earlier reports [11, 17, 18].
However, when assessing symptomatic individuals within the first week of symptom onset, the pooled performance estimates for all iAg tests satisfied the WHO requirements, indicating the high utility of these tests in this particular population. The lower performance estimates in asymptomatic populations are consistent with previous reports that also considered instrument-free antigen tests [10, 11]. Similarly, subgroup analysis based on Ct values yielded results that were in line with previous studies, indicating that the primary factor influencing test sensitivity is viral load [9, 11, 13, 67]. While the sensitivity estimate for the pediatric group was found to be higher than that of the adult group, we are unable to draw any firm conclusions about how age affects test performance due to the small number of data sets available for the prior age group.
When the wild type was predominant (79.3% [95% CI 75.6 to 82.5]), our subgroup analysis on VoC revealed greater sensitivity than when the Alpha variant was predominant (54.8% [95% CI 37.3 to 71.2]). Notably, out of 11 studies where Alpha predominated, six had unknown symptom statuses, and four had unclear testing timing. Of the 64 studies carried out during the period when the wild type was primarily circulating, 31 reported that testing was conducted regardless of symptoms, and nine did not report on the symptom status. Overall, the substantial interstudy heterogeneity makes it difficult to draw conclusions about performance differences between VoCs. Moreover, due to small sample sizes, the CIs largely overlapped in all variants except the wild type, although this does not rule out a difference between groups.
Overall, we found that the clinical accuracy of the POC and lab-based iAg tests included in the review was comparable. Of note, we estimated the pooled sensitivity of POC-applicable iAg tests to be 76.1% (95% CI 72.1 to 79.7), higher than the 67.1% sensitivity that was previously reported by Keskin et al. [17]. Although there were notably more studies in our review, the overall sample size was smaller. It is also important to note that the analysis of a wide range of lab-based tests with varying analytical sensitivity and potential differences in populations studied may have obscured variations in clinical performance between these platforms.
While our findings indicate that iAg tests alone have no discernible advantages in terms of accuracy over their instrument-free counterparts overall and across all groups as reported in prior systematic reviews [9, 11, 15, 68], iAg tests are still likely to offer benefits from an operational standpoint. Available evidence suggests that errors in the reading and interpretation of instrument-free rapid antigen tests are common and primarily stem from a lack of training and a failure to follow test instructions [69]. Instrument-based POC antigen tests can help decrease human error and subjectivity and, thereby, improve the interpretability of test results. Through their connectivity features, they can also enable automated reporting, which facilitates real-time surveillance; some iAg tests can also potentially increase accessibility, for example for those who are visually impaired. In addition, some lab-based iAg tests have the distinct advantage of offering quick, high-throughput results, making them appropriate for large-scale testing in hospitals and reference laboratories. However, there are infrastructural factors that need to be considered before implementation, such as the availability of trained personnel and laboratory facilities.
To our knowledge, this is the first systematic review that provides a comprehensive summary of the clinical performance of iAg tests for COVID-19 in their intended use settings (laboratory versus POC). The main strengths of our study are the broad search terms we used and the rigorous methodology applied. Additionally, we used an interpretation guide developed a priori to assess the methodological quality of the included studies. Moreover, we reported on all literature that was accessible throughout the search period, including preprints and peer-reviewed publications, regardless of language restrictions. There is currently only one other systematic review and meta-analysis that focuses on COVID-19 iAg tests; however, this review is not as comprehensive, does not include a quality assessment, and does not include subgroup analysis [17].
The limitations of this study include the following: (i) Not all commercially available iAg tests were included in our evidence synthesis. As of November 6th, 2023, the FIND COVID-19 test directory lists a total of 85 iAg tests, while we captured only 24 in our study, suggesting that the majority of commercial COVID-19 iAg tests have not been evaluated in published studies [7]. (ii) For the most part, studies that are part of the main analysis either include a mixed population of individuals who are symptomatic and asymptomatic, or they fail to report on the population specifics. As expected, our study confirmed that when symptom status data were available, the sensitivity of iAg tests in symptomatic individuals was substantially higher than that in asymptomatic individuals. Therefore, we cannot completely rule out the possibility that variations in the study populations can account for some of the findings. Similarly, it is unclear whether study participants have received any vaccinations, which may have an effect on their antigen levels and the outcomes of the tests. Additionally, nine studies allowed the use of banked samples, which may have reduced sensitivity estimates in those studies. (iii) In one review study, the reference standard was solely culture [66]. The performance of the index antigen test in this study may have been overestimated due to the lower sensitivity of culture compared to PCR. (iv) The assessment of whether studies followed the manufacturer’s instructions proved to be challenging due to reasons such as poor reporting and version updates. As a result, we relied on whether or not IFU compliance was explicitly stated (in 75% of the studies), which may not be as precise as a comprehensive assessment. (v) As the quality assessment indicated, the shortcomings of the included studies and the fact that there were fewer eligible studies for most individual products limited the scope of our systematic review. By conducting a sensitivity analysis, we aimed to address any data quality constraints caused by the inclusion of preprints in the study. (vi) As previously indicated, the meta-analysis excluded several tests due to the relatively small overall sample size, which limits the significance of the overall results.
Conclusion
Our systematic review and meta-analysis indicate that commercially available instrument-based antigen diagnostic tests can accurately detect SARS-CoV-2 infections in both laboratory and point-of-care settings, with similar performance estimates to instrument-free antigen tests. As a result, they can have high utility for diagnosing COVID-19 in the early stages of the disease, enabling standardized result interpretation, automated reporting, upscaling test runs and additional advantages such as the simultaneous identification of different pathogens. Choosing which tests to use in clinical settings necessitates a careful evaluation of each product’s performance, as confirmed by independent studies and operational features.
Data availability
Data is provided within the manuscript or supplementary information files.
Abbreviations
- Ag-RDT:
-
Antigen detection rapid diagnostic test
- AN:
-
Anterior nasal
- CI:
-
Confidence interval
- CLEIA:
-
Chemiluminescence enzyme immunoassay
- CLIA:
-
Chemiluminescence immunoassay
- Ct:
-
Cycle threshold
- DIA:
-
Diffusion immunoassay
- DOS:
-
Duration of symptoms
- ECLIA:
-
Electrochemiluminescence immunoassay
- ELISA:
-
Enzyme-linked immunosorbent assay
- FIA:
-
Fluorescence immunoassay
- FN:
-
False negative
- FP:
-
False positive
- HIC:
-
High-income countries
- iAg test:
-
Instrument-based antigen test
- IFU:
-
Instructions for use
- MIC:
-
Middle-income countries
- NAATs:
-
Nucleic acid amplification tests
- NMT:
-
Nasal mid-turbinate
- NP:
-
Nasopharyngeal
- NPA:
-
Negative percentage agreement
- OP:
-
Oropharyngeal
- POC:
-
Point of care
- PRISMA:
-
Preferred Items for Systematic Reviews and Meta-analysis
- RCT:
-
Randomized controlled trial
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- TN:
-
True negative
- TP:
-
True positive
- VoC:
-
Variant of Concern
- WHO:
-
World Health Organization
References
FDA. In Vitro Diagnostics EUAs 2022 [updated 14.4.2022. https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas.
Mina MJ, Parker R, Larremore DB. Rethinking Covid-19 test sensitivity - A strategy for Containment. N Engl J Med. 2020;383(22):e120.
Peto T, Affron D, Afrough B, Agasu A, Ainsworth M, Allanson A et al. COVID-19: Rapid Antigen detection for SARS-CoV-2 by lateral flow assay: a national systematic evaluation of sensitivity and specificity for mass-testing. eClinicalMedicine. 2021;36.
WHO. Antigen-detection in the diagnosis of SARS-CoV-2 infection 2021 [updated 6.10.2021. https://www.who.int/publications/i/item/antigen-detection-in-the-diagnosis-of-sars-cov-2infection-using-rapid-immunoassays.
Parvu V, Gary DS, Mann J, Lin Y-C, Mills D, Cooper L et al. Factors that influence the reported sensitivity of Rapid Antigen Testing for SARS-CoV-2. Front Microbiol. 2021;12.
He X, Lau EHY, Wu P, Deng X, Wang J, Hao X, et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat Med. 2020;26(5):672–5.
FINDDX. Test Directory 2022 [ https://www.finddx.org/covid-19/test-directory/.
Johns Hopkins CfHS. Antigen and Molecular Tests for COVID-19 2022 [ https://www.centerforhealthsecurity.org/covid-19TestingToolkit/molecular-based-tests/current-molecular-and-antigen-tests.html.
Brümmer LE, Katzenschlager S, McGrath S, Schmitz S, Gaeddert M, Erdmann C, et al. Accuracy of rapid point-of-care antigen-based diagnostics for SARS-CoV-2: an updated systematic review and meta-analysis with meta-regression analyzing influencing factors. PLoS Med. 2022;19(5):e1004011.
Fragkou PC, Moschopoulos CD, Dimopoulou D, Ong DSY, Dimopoulou K, Nelson PP, et al. Performance of point-of care molecular and antigen-based tests for SARS-CoV-2: a living systematic review and meta-analysis. Clin Microbiol Infect. 2023;29(3):291–301.
Dinnes J, Sharma P, Berhane S, van Wyk SS, Nyaaba N, Domen J et al. Rapid, point-of‐care antigen tests for diagnosis of SARS‐CoV‐2 infection. Cochrane Database Syst Reviews. 2022(7).
Ltd AIO. mariPOC tests [cited 2023 20.12.]. https://www.arcdia.com/maripoc/tests/.
Pollock Nira R, Savage Timothy J, Wardell H, Lee Rose A, Mathew A, Stengelin M, et al. Correlation of SARS-CoV-2 Nucleocapsid Antigen and RNA concentrations in nasopharyngeal samples from children and adults using an ultrasensitive and Quantitative Antigen Assay. J Clin Microbiol. 2021;59(4). https://doi.org/10.1128/jcm.03077-20.
Khalid MF, Selvam K, Jeffry AJN, Salmi MF, Najib MA, Norhayati MN, et al. Performance of Rapid Antigen tests for COVID-19 diagnosis: a systematic review and Meta-analysis. Diagnostics (Basel). 2022;12(1):110.
Khandker SS, Nik Hashim NHH, Deris ZZ, Shueb RH, Islam MA. Diagnostic accuracy of Rapid Antigen Test kits for detecting SARS-CoV-2: a systematic review and Meta-analysis of 17,171 suspected COVID-19 patients. J Clin Med. 2021;10(16):3493.
Fragkou PC, De Angelis G, Menchinelli G, Can F, Garcia F, Morfin-Sherpa F, et al. Update of European Society of Clinical Microbiology and Infectious diseases coronavirus disease 2019 guidelines: diagnostic testing for severe acute respiratory syndrome coronavirus 2. Clin Microbiol Infect. 2023;29(7):876–86.
Keskin AU, Ciragil P, Topkaya AE. Clinical accuracy of Instrument-Read SARS-CoV-2 Antigen Rapid Diagnostic tests (Ag-IRRDTs). Int J Microbiol. 2022;2022:9489067.
Brümmer L, Katzenschlager S, Gaeddert M, Erdmann C, Schmitz S, Bota M, et al. The accuracy of novel antigen rapid diagnostics for SARS-CoV-2: a living systematic review and meta-analysis. PLoS Med. 2021;18(8):e1003735.
Katzenschlager S, Brümmer LE, Schmitz S, Tolle H, Manten K, Gaeddert M, et al. Comparing SARS-CoV-2 antigen-detection rapid diagnostic tests for COVID-19 self-testing/self-sampling with molecular and professional-use tests: a systematic review and meta-analysis. Sci Rep. 2023;13(1):21913.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. PLoS Med. 2021;18(3):e1003583.
Katharina Manten SK, Lukas Brümmer, Claudia M, Denkinger. Feasibility and accuracy of novel instrument-based antigen testing against SARS-CoV-2: a systematic review and meta-analysis [Protocol]. NIHR, PROSPERO. International prospective register of systematic reviews2021 [updated 13 September 2021. https://www.crd.york.ac.uk/prospero/display_record.php?RecordID=276232.
van Enst WA, Ochodo E, Scholten RJPM, Hooft L, Leeflang MM. Investigation of publication bias in meta-analyses of diagnostic test accuracy: a meta-epidemiological study. BMC Med Res Methodol. 2014;14(1):70.
Deeks JJ, Macaskill P, Irwig L. The performance of tests of publication bias and other sample size effects in systematic reviews of diagnostic test accuracy was assessed. J Clin Epidemiol. 2005;58(9):882–93.
Neukam K, Lucero A, Gutierrez-Valencia A, Amaya L, Echegoyen N, Martelli A, et al. Point-of-care detection of SARS-CoV-2 antigen among symptomatic vs. asymptomatic persons: testing for COVID-19 vs. infectivity. Front Public Health. 2022;10:995249.
Sitoe N, Sambo J, Mabunda N, Nguenha N, Chilaule J, Rafael J et al. Clinical performance of self-collected nasal swabs and Antigen Rapid tests for SARS-CoV-2 detection in resource-poor settings. Biomedicines. 2022;10(9).
FINDDx. FIND Evaluation of LumiraDx. SARS-CoV-2 Ag Test. External Report. [updated Version 2.0, 8 October 2021. https://www.finddx.org/wp-content/uploads/2023/01/20211008_lumiradx_lumiradx_FV_EN.pdf.
Paul D, Gupta A, Rooge S, Gupta E. Performance evaluation of automated chemiluminescence immunoassay based antigen detection - moving towards more reliable ways to predict SARS-CoV-2 infection. J Virol Methods. 2021;298:114299.
Montalvo Villalba MC, Sosa Glaria E, Rodriguez Lay LLA, Valdes Ramirez O, Vallina Garcia D, Arencibia Garcia A, et al. Performance evaluation of Elecsys SARS-CoV-2 Antigen immunoassay for diagnostic of COVID-19. J Med Virol. 2022;94(3):1001–8.
Iqbal B, Khan M, Shah N, Dawood MM, Jehanzeb V, Shafi M. Comparison of SARS-CoV-2 antigen electrochemiluminescence immunoassay to RT-PCR assay for laboratory diagnosis of COVID-19 in Peshawar. Diagnosis (Berl). 2022;9(3):364–8.
FINDDx FIND. Evaluation of SD Biosensor, Inc. STANDARDTM F COVID-19 Ag FIA. External Report Site Specific Report [updated Version 1.1 3 June 2022. https://www.finddx.org/wp-content/uploads/2023/01/20221004_sd_biosensor_standard_F_india_FV_EN.pdf.
Drain PK, Ampajwala M, Chappel C, Gvozden AB, Hoppers M, Wang M, et al. A Rapid, High-Sensitivity SARS-CoV-2 Nucleocapsid Immunoassay to Aid diagnosis of Acute COVID-19 at the point of care: a clinical performance study. Infect Dis Ther. 2021;10(2):753–61.
Amrish M, Bri S, Ashok G, Pradeep K, Raúl AOG, Kate T et al. Performance of Gazelle COVID-19 point-of-care test for detection of nucleocapsid antigen from SARS-CoV-2. medRxiv. 2022:2022.03.23.22272094.
Pekosz A, Parvu V, Li M, Andrews JC, Manabe YC, Kodsi S, et al. Antigen-based testing but not real-time polymerase chain reaction correlates with severe Acute Respiratory Syndrome Coronavirus 2 viral culture. Clin Infect Dis. 2021;73(9):e2861–6.
Porte L, Legarraga P, Vollrath V, Aguilera X, Munita JM, Araos R, et al. Evaluation of a novel antigen-based rapid detection test for the diagnosis of SARS-CoV-2 in respiratory samples. Int J Infect Dis. 2020;99:328–33.
Porte L, Legarraga P, Iruretagoyena M, Vollrath V, Pizarro G, Munita J, et al. Evaluation of two fluorescence immunoassays for the rapid detection of SARS-CoV-2 antigen-new tool to detect infective COVID-19 patients. PeerJ. 2021;9:e10801.
Weitzel T, Legarraga P, Iruretagoyena M, Pizarro G, Vollrath V, Araos R, et al. Comparative evaluation of four rapid SARS-CoV-2 antigen detection tests using universal transport medium. Travel Med Infect Dis. 2021;39:101942.
Jaaskelainen AE, Ahava MJ, Jokela P, Szirovicza L, Pohjala S, Vapalahti O, et al. Evaluation of three rapid lateral flow antigen detection tests for the diagnosis of SARS-CoV-2 infection. J Clin Virol. 2021;137:104785.
Gili A, Paggi R, Russo C, Cenci E, Pietrella D, Graziani A, et al. Evaluation of Lumipulse® G SARS-CoV-2 antigen assay automated test for detecting SARS-CoV-2 nucleocapsid protein (NP) in nasopharyngeal swabs for community and population screening. Int J Infect Dis. 2021;105:391–6.
Lefever S, Indevuyst C, Cuypers L, Dewaele K, Yin N, Cotton F, et al. Comparison of the quantitative DiaSorin Liaison Antigen Test to reverse Transcription-PCR for the diagnosis of COVID-19 in symptomatic and asymptomatic outpatients. J Clin Microbiol. 2021;59(7):e0037421.
Koeleman JGM, Brand H, de Man SJ, Ong DSY. Clinical evaluation of rapid point-of-care antigen tests for diagnosis of SARS-CoV-2 infection. Eur J Clin Microbiol Infect Dis. 2021;40(9):1975–81.
Blairon L, Cupaiolo R, Thomas I, Piteüs S, Wilmet A, Beukinga I, et al. Efficacy comparison of three rapid antigen tests for SARS-CoV-2 and how viral load impact their performance. J Med Virol. 2021;93(10):5783–8.
Frediani JK, Levy JM, Rao A, Bassit L, Figueroa J, Vos MB, et al. Multidisciplinary assessment of the Abbott BinaxNOW SARS-CoV-2 point-of-care antigen test in the context of emerging viral variants and self-administration. Sci Rep. 2021;11(1):14604.
Van der Moeren N, Zwart VF, Goderski G, Rijkers GT, van den Bijllaardt W, Veenemans J, et al. Performance of the Diasorin SARS-CoV-2 antigen detection assay on the LIAISON XL. J Clin Virol. 2021;141:104909.
Karon BS, Donato LJ, Bridgeman AR, Blommel JH, Kipp B, Maus A, et al. Analytical Sensitivity and specificity of Four Point of Care Rapid Antigen Diagnostic tests for SARS-CoV-2 using real-time quantitative PCR, quantitative Droplet Digital PCR, and a Mass Spectrometric Antigen Assay as Comparator methods. Clin Chem. 2021;67(11):1545–53.
Levett PN, Cheung B, Kustra J, Pidduck T, Mak A, Tsang F, et al. Evaluation of a high volume antigen test for detection of SARS-CoV-2. J Clin Virol. 2021;142:104938.
Audigé A, Böni J, Schreiber PW, Scheier T, Buonomano R, Rudiger A et al. Reduced relative sensitivity of the Elecsys SARS-CoV-2 Antigen Assay in Saliva compared to nasopharyngeal swabs. Microorganisms. 2021;9(8).
Koskinen JM, Antikainen P, Hotakainen K, Haveri A, Ikonen N, Savolainen-Kopra C, et al. Clinical validation of automated and rapid mariPOC SARS-CoV-2 antigen test. Sci Rep. 2021;11(1):20363.
Tanimoto Y, Mori A, Miyamoto S, Ito E, Arikawa K, Iwamoto T. Comparison of RT-PCR, RT-LAMP, and Antigen quantification assays for the detection of SARS-CoV-2. Jpn J Infect Dis. 2022;75(3):249–53.
Wang H, Hogan CA, Verghese M, Solis D, Sibai M, Huang C, et al. Ultra-sensitive severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Antigen Detection for the diagnosis of Coronavirus Disease 2019 (COVID-19) in Upper respiratory samples. Clin Infect Dis. 2021;73(12):2326–8.
Andreani J, Lupo J, Germi R, Laugier C, Roccon M, Larrat S, et al. Evaluation of six commercial SARS-CoV-2 rapid antigen tests in nasopharyngeal swabs: better knowledge for better patient management? J Clin Virol. 2021;143:104947.
Gourgeon A, Soulier A, Audureau É, Khouider S, Galbin A, Langlois C, et al. Performance of 22 Rapid lateral Flow tests for SARS-CoV-2 Antigen Detection and Influence of variants of concern: implications for clinical use. Microbiol Spectr. 2022;10(4):e0115722.
Greub G, Caruana G, Schweitzer M, Imperiali M, Muigg V, Risch M et al. Multicenter Technical Validation of 30 Rapid Antigen tests for the detection of SARS-CoV-2 (VALIDATE). Microorganisms. 2021;9(12).
Hirotsu Y, Maejima M, Shibusawa M, Natori Y, Nagakubo Y, Hosaka K, et al. Direct comparison of Xpert Xpress, FilmArray Respiratory Panel, Lumipulse antigen test, and RT-qPCR in 165 nasopharyngeal swabs. BMC Infect Dis. 2022;22(1):221.
Horban A, Kowalska JD, Dyda T, Ząbek P, Czeszko-Paprocka H. The utility of rapid FIA antigen test to detect SARS-CoV-2 infection - preliminary experiences of the hospital for infectious diseases in Warsaw. Postepy Dermatol Alergol. 2022;39(3):613–4.
Kirby JE, Riedel S, Dutta S, Arnaout R, Cheng A, Ditelberg S, et al. Sars-Cov-2 antigen tests predict infectivity based on viral culture: comparison of antigen, PCR viral load, and viral culture testing on a large sample cohort. Clin Microbiol Infect. 2023;29(1):94–100.
Kliegr T, Jarkovský J, Jiřincová H, Kuchař J, Karel T, Tachezy R. Role of population and test characteristics in antigen-based SARS-CoV-2 diagnosis, Czechia, August to November 2021. Euro Surveill. 2022;27(33).
Fernández-Rivas G, Barallat J, Gonzalez V, Martinez S, Bordoy AE, Jimenez L, et al. Analytical Performance of quantitative DiaSorin Liaison SARS-COV-2 Antigen Test for the Asymptomatic Population. Front Public Health. 2021;9:788581.
Lau CS, Phua SK, Hoo SP, Jiang B, Aw TC. Evaluation and validation of the Roche Elecsys SARS-CoV-2 Antigen Electro-Chemiluminescent Immunoassay in a southeast Asian Region. Vaccines (Basel). 2022;10(2).
Navero-Castillejos J, Casals-Pascual C, Narváez S, Cuesta G, Hurtado JC, Fernandez M, et al. Diagnostic performance of six Rapid Antigen tests for SARS-CoV-2. Microbiol Spectr. 2022;10(2):e0235121.
Peck Palmer O, Hasskamp JH, La HS, Pramod Patwardhan P, Ghumman S, Baloda V, et al. Performance of high Throughput SARS-CoV-2 Antigen Testing compared to nucleic acid testing. Lab Med. 2023;54(2):e54–7.
Petonnet D, Marot S, Leroy I, Cohier J, Ramahefasolo C, Mansaly S et al. Comparison of Rapid and Automated Antigen Detection tests for the diagnosis of SARS-CoV-2 infection. Diagnostics (Basel). 2022;12(1).
Uster S, Topalli Z, Sasse T, Suter-Riniker F, Barbani MT. Evaluation of the DiaSorin LIAISON SARS-CoV-2 antigen assay on nasopharyngeal swabs in two different SARS-CoV-2 pandemic waves in Switzerland: the impact of the Omicron variant on its performance. J Clin Virol Plus. 2022;2(3):100095.
Yu K, Song J, Kim D, Park Y, Jeong SH. Clinical evaluation of two Rapid Antigen tests for severe Acute Respiratory Syndrome Coronavirus 2 detection. Ann Lab Med. 2023;43(1):120–3.
Saito K, Ai T, Kawai A, Matsui J, Fukushima Y, Kikukawa N, et al. Performance and usefulness of a novel automated immunoassay HISCL SARS-CoV-2 Antigen assay kit for the diagnosis of COVID-19. Sci Rep. 2021;11(1):23196.
Horber S, Drees C, Ganzenmueller T, Schmauder K, Peter S, Biskup D, et al. Evaluation of a laboratory-based high-throughput SARS-CoV-2 antigen assay. Clin Chem Lab Med. 2022;60(9):1478–85.
Rabaan AA, Tirupathi R, Sule AA, Aldali J, Mutair AA, Alhumaid S et al. Viral dynamics and Real-Time RT-PCR ct values correlation with Disease Severity in COVID-19. Diagnostics (Basel). 2021;11(6).
Tapari A, Braliou GG, Papaefthimiou M, Mavriki H, Kontou PI, Nikolopoulos GK et al. Performance of Antigen Detection tests for SARS-CoV-2: a systematic review and Meta-analysis. Diagnostics (Basel). 2022;12(6).
Mukadi P, Gillet P, Lukuka A, Mbatshi J, Otshudiema J, Muyembe JJ, et al. External quality assessment of reading and interpretation of malaria rapid diagnostic tests among 1849 end-users in the Democratic Republic of the Congo through Short Message Service (SMS). PLoS ONE. 2013;8(8):e71442.
Baccani I, Morecchiato F, Chilleri C, Cervini C, Gori E, Matarrese D, et al. Evaluation of three immunoassays for the Rapid detection of SARS-CoV-2 antigens. Diagn Microbiol Infect Dis. 2021;101(2):115434.
Lanari M, Biserni GB, Pavoni M, Borgatti EC, Leone M, Corsini I et al. Feasibility and Effectiveness Assessment of SARS-CoV-2 antigenic tests in Mass Screening of a Pediatric Population and correlation with the kinetics of viral loads. Viruses. 2021;13(10).
Van der Moeren N, Zwart VF, Lodder EB, Van den Bijllaardt W, Van Esch H, Stohr J, et al. Evaluation of the test accuracy of a SARS-CoV-2 rapid antigen test in symptomatic community dwelling individuals in the Netherlands. PLoS ONE. 2021;16(5):e0250886.
Caruana G, Croxatto A, Kampouri E, Kritikos A, Opota O, Foerster M et al. Implementing SARS-CoV-2 Rapid Antigen Testing in the Emergency Ward of a Swiss University Hospital: the INCREASE Study. Microorganisms. 2021;9(4).
Gomez Marti JL, Gribschaw J, McCullough M, Mallon A, Acero J, Kinzler A et al. Differences in detected viral loads guide use of SARS-CoV-2 antigen-detection assays towards symptomatic college students and children. medRxiv. 2021:2021.01.28.21250365.
Kilic A, Hiestand B, Palavecino E. Evaluation of performance of the BD Veritor SARS-CoV-2 Chromatographic Immunoassay Test in patients with symptoms of COVID-19. J Clin Microbiol. 2021;59(5).
Yin N, Debuysschere C, Decroly M, Bouazza F-Z, Collot V, Martin C, et al. SARS-CoV-2 diagnostic tests: algorithm and field evaluation from the Near patient testing to the Automated Diagnostic platform. Front Med (Lausanne). 2021;8:581–650.
Christensen K, Ren H, Chen S, Cooper CK, Young S. Clinical evaluation of BD Veritor SARS-CoV-2 and Flu A + B assay for Point-Of-Care system. Microbiol Spectr. 2022;10(2):e0180721.
Wee LE, Conceicao EP, Sim JX, Venkatachalam I, Wan PW, Zakaria ND, et al. Utilization of rapid antigen assays for detection of severe acute respiratory coronavirus virus 2 (SARS-CoV-2) in a low-incidence setting in emergency department triage: does risk-stratification still matter? Infect Control Hosp Epidemiol. 2022;43(12):1974–6.
Robinson ML, Mirza A, Gallagher N, Boudreau A, Garcia L, Yu T et al. Limitations of molecular and antigen test performance for SARS-CoV-2 in symptomatic and asymptomatic COVID-19 contacts. medRxiv. 2022.
Wells CR, Pandey A, Moghadas SM, Singer BH, Krieger G, Heron RJL, et al. Comparative analyses of eighteen rapid antigen tests and RT-PCR for COVID-19 quarantine and surveillance-based isolation. Commun Med (Lond). 2022;2:84.
Parada-Ricart E, Gomez-Bertomeu F, Picó-Plana E, Olona-Cabases M. Usefulness of the antigen test for diagnosing SARS-CoV-2 infection in patients with and without symptoms. Enferm Infecc Microbiol Clin (Engl Ed). 2021;39(7):357–8.
Krüger LJ, Tanuri A, Lindner AK, Gaeddert M, Köppel L, Tobian F, et al. Accuracy and ease-of-use of seven point-of-care SARS-CoV-2 antigen-detecting tests: a multi-centre clinical evaluation. eBioMedicine. 2022;75:103774.
Nörz D, Olearo F, Perisic S, Bauer MF, Riester E, Schneider T, et al. Multicenter evaluation of a fully automated high-throughput SARS-CoV-2 Antigen Immunoassay. Infect Dis Therapy. 2021;10(4):2371–9.
Osterman A, Iglhaut M, Lehner A, Spath P, Stern M, Autenrieth H, et al. Comparison of four commercial, automated antigen tests to detect SARS-CoV-2 variants of concern. Med Microbiol Immunol. 2021;210(5–6):263–75.
Ben Abdelhanin M, Mvumbi DM, Agathine A, Nanos N, Gidenne S. Evaluation of the Elecsys SARS-CoV-2 antigen assay for the detection of SARS-CoV-2 in nasopharyngeal swabs. J Clin Virol. 2021;144:104991.
Kolesova O, Tomassetti F, Cerini P, Finucci D, Turchetti G, Capogreco F, et al. Evaluation of ECLIA antigen detection tests as screening methods for COVID-19 in comparison with molecular analysis. Ir J Med Sci. 2022;191(5):2213–7.
Salvagno GL, Pighi L, De Nitto S, Lippi G. Clinical performance of the Roche Elecsys SARS-CoV-2 antigen fully automated electrochemiluminescence immunoassay. Pract Lab Med. 2022;29:e00265.
Miłosz M, Słota M, Jakubowski J, Kwiatkowski P, Kasperczyk S, Kozłowska M, et al. Validation of the test for detecting SARS–CoV-2 antigens in the Polish population in patients with suspected SARS-CoV-2 infection. Cent Eur J Immunol. 2022;47(1):58–62.
Bruzzone B, De Pace V, Caligiuri P, Ricucci V, Guarona G, Pennati BM, et al. Comparative diagnostic performance of rapid antigen detection tests for COVID-19 in a hospital setting. Int J Infect Dis. 2021;107:215–8.
Orsi A, Pennati BM, Bruzzone B, Ricucci V, Ferone D, Barbera P, et al. On-field evaluation of a ultra-rapid fluorescence immunoassay as a frontline test for SARS-CoV-2 diagnostic. J Virol Methods. 2021;295:114201.
Mehta A, Spencer B, Garg A, Kalmorge P, Ocasio Gonzalez R, Taussig K, et al. Performance of Gazelle COVID-19 point-of-care test for detection of nucleocapsid antigen from SARS-CoV-2. medRxiv; 2022.
Linssen J, Schapendonk C, Münster M, Daemen P, Rahamat-Langendoen J, Wertheim H. A method comparison study of the high throughput automated HISCL® SARS-CoV-2 antigen assay using nasopharyngeal swab samples from symptomatic and asymptomatic subjects against conventional RT-PCR. J Med Virol. 2022;94(7):3070–80.
FINDDX. FIND Evaluation of Boditech Medical, Inc. iChroma COVID-19 Ag Test [updated Version 1.0, 23 February 2021. https://www.finddx.org/wp-content/uploads/2023/01/20210223_boditech_ichroma_FV_EN.pdf.
Salvagno GL, Gianfilippi G, Fiorio G, Pighi L, De Nitto S, Henry BM, et al. Clinical Assessment of the DiaSorin LIAISON SARS-CoV-2 Ag Chemiluminescence Immunoassay. Ejifcc. 2021;32(2):216–23.
Hartard C, Berger S, Josse T, Schvoerer E, Jeulin H. Performance evaluation of an automated SARS-CoV-2 ag test for the diagnosis of COVID-19 infection on nasopharyngeal swabs. Clin Chem Lab Med. 2021;59(12):2003–9.
Alghounaim M, Bastaki H, Bin Essa F, Motlagh H, Al-Sabah S. The performance of two Rapid Antigen tests during Population-Level Screening for SARS-CoV-2 infection. Front Med (Lausanne). 2021;8:797109.
Hirotsu Y, Maejima M, Shibusawa M, Amemiya K, Nagakubo Y, Hosaka K, et al. Prospective study of 1308 nasopharyngeal swabs from 1033 patients using the LUMIPULSE SARS-CoV-2 antigen test: comparison with RT-qPCR. Int J Infect Dis. 2021;105:7–14.
Asai N, Sakanashi D, Ohashi W, Nakamura A, Kawamoto Y, Miyazaki N, et al. Efficacy and validity of automated quantitative chemiluminescent enzyme immunoassay for SARS-CoV-2 antigen test from saliva specimen in the diagnosis of COVID-19. J Infect Chemother. 2021;27(7):1039–42.
Matsuzaki N, Orihara Y, Kodana M, Kitagawa Y, Matsuoka M, Kawamura R, et al. Evaluation of a chemiluminescent enzyme immunoassay-based high-throughput SARS-CoV-2 antigen assay for the diagnosis of COVID-19: the VITROS® SARS-CoV-2 Antigen Test. J Med Virol. 2021;93(12):6778–81.
Loconsole D, Centrone F, Morcavallo C, Campanella S, Sallustio A, Casulli D, et al. The challenge of using an Antigen Test as a Screening Tool for SARS-CoV-2 infection in an Emergency Department: experience of a Tertiary Care Hospital in Southern Italy. Biomed Res Int. 2021;2021:3893733.
Sberna G, Basile F, Guarino ML, Capobianchi MR, Bordi L, Parisi G. Comparison of Allplex™ SARS-CoV-2 assay, Easy SARS-CoV-2 WE and Lumipulse quantitative SARS-CoV-2 antigen test performance using automated systems for the diagnosis of COVID-19. Int J Infect Dis. 2021;113:113–5.
Nomoto H, Yamamoto K, Isaka E, Miyazato Y, Suzuki T, Maruki T, et al. Potential usage of anterior nasal sampling in clinical practice with three rapid antigen tests for SARS-CoV-2. J Infect Chemother. 2023;29(1):15–9.
Salvetti S, Lavinia F, Rosi N, Vanni S, Masotti L, Tarquini R, et al. A rapid and cost-effective diagnostic algorithm for the detection of SARS-CoV-2 infection in the emergency area by combining highly sensitive antigenic test and RT-PCR. Diagn Microbiol Infect Dis. 2022;103(4):115727.
Kohmer N, Toptan T, Pallas C, Karaca O, Pfeiffer A, Westhaus S et al. The comparative clinical performance of four SARS-CoV-2 Rapid Antigen tests and their correlation to Infectivity in Vitro. J Clin Med. 2021;10(2).
Krüger LJ, Klein JAF, Tobian F, Gaeddert M, Lainati F, Klemm S et al. Evaluation of accuracy, exclusivity, limit-of-detection and ease-of-use of LumiraDx™: An antigen-detecting point-of-care device for SARS-CoV-2. Infection. 2021:1–12.
Micocci M, Buckle P, Hayward G, Allen AJ, Davies K, Kierkegaard P et al. Point of Care Testing using rapid automated Antigen Testing for SARS-COV-2 in Care Homes – an exploratory safety, usability and diagnostic agreement evaluation. medRxiv. 2021:2021.04.22.21255948.
Bianco G, Boattini M, Barbui AM, Scozzari G, Riccardini F, Coggiola M, et al. Evaluation of an antigen-based test for hospital point-of-care diagnosis of SARS-CoV-2 infection. J Clin Virol. 2021;139:104838.
Caramello V, Boccuzzi A, Basile V, Ferraro A, Macciotta A, Catalano A, et al. Are antigenic tests useful for detecting SARS-CoV-2 infections in patients accessing to emergency departments? Results from a North-West Italy hospital. J Infect. 2021;83(2):237–79.
Dierks S, Bader O, Schwanbeck J, Groß U, Weig MS, Mese K et al. Diagnosing SARS-CoV-2 with Antigen Testing, transcription-mediated amplification and real-time PCR. J Clin Med. 2021;10(11).
Fernandez MD, Estevez AS, Alfonsin FL, Arevalo GB. [Usefulness of the Lumiradx Sars-Cov-2 Antigen test in nursing home]. Enferm Infecc Microbiol Clin (Engl Ed; 2021.
Leli C, Di Matteo L, Gotta F, Cornaglia E, Vay D, Megna I, et al. Performance of a SARS-CoV-2 antigen rapid immunoassay in patients admitted to the emergency department. Int J Infect Dis. 2021;110:135–40.
Cento V, Renica S, Matarazzo E, Antonello M, Colagrossi L, Di Ruscio F, et al. Frontline screening for SARS-CoV-2 infection at Emergency Department Admission by Third Generation Rapid Antigen Test. Volume 13. Can We Spare RT-qPCR? Viruses; 2021. 5.
Burdino E, Cerutti F, Panero F, Allice T, Gregori G, Milia MG, et al. SARS-CoV-2 microfluidic antigen point-of-care testing in Emergency Room patients during COVID-19 pandemic. J Virol Methods. 2022;299:114337.
Denina M, Giannone V, Curtoni A, Zanotto E, Garazzino S, Urbino AF, et al. Can we trust in Sars-CoV-2 rapid antigen testing? Preliminary results from a paediatric cohort in the emergency department. Ir J Med Sci. 2022;191(4):1767–70.
Drain P, Sulaiman R, Hoppers M, Lindner NM, Lawson V, Ellis JE. Performance of the LumiraDx microfluidic immunofluorescence point-of-care SARS-CoV-2 Antigen Test in asymptomatic adults and children. Am J Clin Pathol. 2022;157(4):602–7.
Cattelan AM, Sasset L, Zabeo F, Ferrari A, Rossi L, Mazzitelli M et al. Rapid Antigen Test LumiraDx(TM) vs. Real Time Polymerase Chain Reaction for the Diagnosis of SARS-CoV-2 Infection: A Retrospective Cohort Study. Int J Environ Res Public Health. 2022;19(7).
Elli S, Blasi F, Brignolo B, Ceriotti F, Gori A, Piatti A, et al. Diagnostic accuracy of rapid antigen test for COVID-19 in an emergency department. Diagn Microbiol Infect Dis. 2022;102(4):115635.
Gunell M, Rantasärkkä K, Arjonen R, Sandén A, Vuorinen T. Clinical evaluation of an automated, rapid mariPOC antigen test in screening of symptomatics and asymptomatics for SARS-CoV-2 infection. J Med Virol. 2023;95(1):e28189.
Al-Alawi S, Al-Hinai H, Al-Kindi N, Al-Rashidi M, Al-Kindi H, Al-Shukri I, et al. Evaluation of four Rapid Antigen tests for detection of SARS-CoV-2 virus. Oman Med J. 2021;36(5):e297.
Suzuki H, Akashi Y, Ueda A, Kiyasu Y, Takeuchi Y, Maehara Y, et al. Diagnostic performance of a novel digital immunoassay (RapidTesta SARS-CoV-2): a prospective observational study with nasopharyngeal samples. J Infect Chemother. 2022;28(1):78–81.
Bachman CM, Grant BD, Anderson CE, Alonzo LF, Garing S, Byrnes SA, et al. Clinical validation of an open-access SARS-COV-2 antigen detection lateral flow assay, compared to commercially available assays. PLoS ONE. 2021;16(8):e0256352.
Beck ET, Paar W, Fojut L, Serwe J, Jahnke RR. Comparison of the Quidel Sofia SARS FIA Test to the Hologic Aptima SARS-CoV-2 TMA test for diagnosis of COVID-19 in symptomatic outpatients. J Clin Microbiol. 2021;59(2).
Herrera V, Hsu V, Adewale A, Johnson L, Hendrix T, Kuhlman J et al. Testing Healthcare Workers Exposed to COVID19 using Rapid Antigen Detection. medRxiv. 2020:2020.08.12.20172726.
Brihn A, Chang J, K OY, Balter S, Terashita D, Rubin Z, et al. Diagnostic performance of an Antigen Test with RT-PCR for the detection of SARS-CoV-2 in a hospital setting - Los Angeles County, California, June-August 2020. MMWR Morb Mortal Wkly Rep. 2021;70(19):702–6.
Bornemann L, Kaup O, Kleideiter J, Panning M, Ruprecht B, Wehmeier M. Real-life evaluation of the Sofia SARS-CoV-2 antigen assay in a large tertiary care hospital. J Clin Virol. 2021;140:104854.
Harris DT, Badowski M, Jernigan B, Sprissler R, Edwards T, Cohen R et al. SARS-CoV-2 Rapid Antigen Testing of Symptomatic and asymptomatic individuals on the University of Arizona Campus. Biomedicines. 2021;9(5).
Smith RD, Johnson JK, Clay C, Girio-Herrera L, Stevens D, Abraham M, et al. Clinical evaluation of Sofia Rapid Antigen Assay for detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) among emergency department to hospital admissions. Infect Control Hosp Epidemiol. 2022;43(8):968–73.
Mitchell SL, Orris S, Freeman T, Freeman MC, Adam M, Axe M, et al. Performance of SARS-CoV-2 antigen testing in symptomatic and asymptomatic adults: a single-center evaluation. BMC Infect Dis. 2021;21(1):1071.
Bornemann L, Dähne T, Fomenko A, Kaup O, Kleideiter J, Kulis-Horn R, et al. Performance of the Sofia SARS-CoV-2 rapid antigen test as frontline test in a university hospital, Germany. Diagn Microbiol Infect Dis. 2022;103(1):115663.
Černila M, Logar M, Možina H, Osredkar J. Comparison between the Sofia SARS Antigen FIA Test and the PCR test in detection of SARS-CoV-2 infection. Lab Med. 2023;54(2):e44–8.
Freeman MC, Freeman TJ, Iagnemma J, Rasmussen J, Heidenreich K, Wells A, et al. Performance of the Sofia SARS-CoV-2 Rapid Antigen Test in Symptomatic and Asymptomatic Pediatric patients. J Pediatr Infect Dis Soc. 2022;11(9):417–21.
Drevinek P, Hurych J, Kepka Z, Briksi A, Kulich M, Zajac M, et al. The sensitivity of SARS-CoV-2 antigen tests in the view of large-scale testing. Epidemiol Mikrobiol Imunol. 2021;70(3):156–60.
Liotti FM, Menchinelli G, Lalle E, Palucci I, Marchetti S, Colavita F, et al. Performance of a novel diagnostic assay for rapid SARS-CoV-2 antigen detection in nasopharynx samples. Clin Microbiol Infect. 2021;27(3):487–8.
Osterman A, Baldauf HM, Eletreby M, Wettengel JM, Afridi SQ, Fuchs T, et al. Evaluation of two rapid antigen tests to detect SARS-CoV-2 in a hospital setting. Med Microbiol Immunol. 2021;210(1):65–72.
Menchinelli G, De Angelis G, Cacaci M, Liotti FM, Candelli M, Palucci I et al. SARS-CoV-2 Antigen Detection to Expand Testing Capacity for COVID-19: Results from a Hospital Emergency Department Testing Site. Diagnostics (Basel). 2021;11(7).
Kahn M, Schuierer L, Bartenschlager C, Zellmer S, Frey R, Freitag M, et al. Performance of antigen testing for diagnosis of COVID-19: a direct comparison of a lateral flow device to nucleic acid amplification based tests. BMC Infect Dis. 2021;21(1):798.
Weitzel T, Pérez C, Tapia D, Legarraga P, Porte L. SARS-CoV-2 rapid antigen detection tests. Lancet Infect Dis. 2021;21(8):1067–8.
Leiner J, Pellissier V, Nitsche A, König S, Hohenstein S, Nachtigall I, et al. SARS-CoV-2 rapid antigen testing in the healthcare sector: a clinical prediction model for identifying false negative results. Int J Infect Dis. 2021;112:117–23.
García-Fernández S, Pablo-Marcos D, de la Fuente SV, Rodríguez MJR, Gozalo M, Rodríguez-Lozano J, et al. Evaluation of the rapid antigen detection test STANDARD F COVID-19 Ag FIA for diagnosing SARS-CoV-2: experience from an Emergency Department. Diagn Microbiol Infect Dis. 2022;103(2):115683.
Favresse J, Gillot C, Oliveira M, Cadrobbi J, Elsen M, Eucher C, et al. Head-to-Head Comparison of Rapid and Automated Antigen Detection Tests for the diagnosis of SARS-CoV-2 infection. J Clin Med. 2021;10(2):265.
Fourati S, Soulier A, Gourgeon A, Khouider S, Langlois C, Galbin A, et al. Performance of a high-throughput, automated enzyme immunoassay for the detection of SARS-CoV-2 antigen, including in viral variants of concern: implications for clinical use. J Clin Virol. 2022;146:105048.
Routsias JG, Mavrouli M, Tsoplou P, Dioikitopoulou K, Tsakris A. Diagnostic performance of rapid antigen tests (RATs) for SARS-CoV-2 and their efficacy in monitoring the infectiousness of COVID-19 patients. Sci Rep. 2021;11(1):22863.
Acknowledgements
Not applicable.
Funding
This work was supported by the Ministry of Science, Research and Arts of the State of Baden-Wuerttemberg, Germany (no grant number; https://mwk.badenwuerttemberg.de/de/startseite/) and internal funds from the Heidelberg University Hospital (no grant number; https://www.heidelberg-university-hospital.com/de/) to CMD. This publication was supported by the German Federal Ministry of Education and Research (BMBF) as part of the Network University Medicine (NUM): „NaFoUniMedCovid19“, Grant No: 01KX2021, Project: B-FAST. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
KM, SK, LEB, CMD, and SY made substantial contributions to the conception of this work. KM, SK, LEB, CMD and SY designed the work. MGr performed the literature search. SK, LEB, KM and SS performed the data acquisition. Mga, AM, BE, and SY performed the data analy-sis. KM, SK, LEB, JAS, NRP, CMD and SY contributed to the interpretation of the data. KM and SY drafted the manuscript and all authors have substantively revised it. The final version of this manuscript has been approved by all the authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Manten, K., Katzenschlager, S., Brümmer, L.E. et al. Clinical accuracy of instrument-based SARS-CoV-2 antigen diagnostic tests: a systematic review and meta-analysis. Virol J 21, 99 (2024). https://doi.org/10.1186/s12985-024-02371-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-024-02371-5