
Abstract
Background
The co-occurrence of chronic hepatitis B (CHB) and metabolic dysfunction-associated fatty liver disease (MAFLD) has drawn considerable attention due to its impact on disease outcomes. This study aimed to investigate the association between hepatic steatosis and hepatitis B virus (HBV) and analyzed the influence of hepatic steatosis on hepatitis B virology in patients with CHB.
Methods
In this cross-sectional study, 272 patients infected with HBV who were treatment-naïve or had ceased antiviral treatment for > 6 months were categorized into the CHB group (n = 128) and CHB + MAFLD group (n = 144). Furthermore, based on whether HBV DNA was higher than 2000 IU/mL, patients were categorized into the high-level HBV DNA group (n = 129) and the low-level HBV DNA group (n = 143). The impact of hepatic steatosis on hepatitis B virology was analyzed within the CHB cohort. Multivariate logistic regression analysis was employed to identify independent factors influencing pre-genomic RNA (pgRNA) levels below the lower limit of detection (LLD) in patients with CHB.
Results
Among the 272 patients, compared with CHB group, HBV DNA levels (4.11 vs. 3.62 log10 IU/mL, P = 0.045), hepatitis B surface antigen (HBsAg) levels (3.52 vs. 3.20 log10 IU/mL, P = 0.008) and the hepatitis B e antigen (HBeAg) positive rate (33.6% vs. 22.2%, P = 0.036) were significantly decreased in the CHB + MAFLD group; In 143 low-level HBV DNA patients, the CHB + MAFLD group exhibited decreased levels of pgRNA and HBsAg compared to the CHB group. However, in 129 high-level HBV DNA patients, a more significant decrease was observed in pgRNA (3.85 vs 3.35 log10 copies/mL, P = 0.044) and HBsAg (3.85 vs 3.59 log10 IU/mL, P = 0.033); Spearman correlation analysis revealed a negative correlation between hepatic steatosis and pgRNA (r = − 0.529, P < 0.001), HBV DNA (r = − 0.456, P < 0.001), HBsAg (r = − 0.465, P < 0.001) and HBeAg (r = − 0.339, P < 0.001) levels; Multivariate logistic regression analysis identified HBV DNA (odds ratio [OR] = 0.283, P < 0.001), HBsAg (OR = 0.300, P < 0.001), and controlled attenuation parameter (CAP) values (OR = 1.013, P = 0.038) as independent factors influencing pgRNA levels below the LLD in patients with CHB.
Conclusions
This study establishes a negative correlation between hepatic steatosis and hepatitis B virology, demonstrating decreased HBV expression in patients with CHB + MAFLD.
Similar content being viewed by others


Introduction
Hepatitis B virus (HBV) and metabolic dysfunction-associated fatty liver disease (MAFLD) are prevalent chronic liver diseases with the potential to progress to liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC), significantly impacting patient prognosis. The coexistence of these conditions in many patients can further influence disease evolution and prognosis.
The pathogenesis of the coexistence of chronic hepatitis B (CHB) and MAFLD is intricate. Numerous studies have aimed to elucidate their interaction, marking a challenging and prominent research focus in chronic liver disease [1, 2]. An animal study indicated that hepatic steatosis attenuated the expression of HBV DNA, hepatitis B surface antigen (HBsAg), and pre-genomic RNA (pgRNA) in immunocompetent mice, inhibiting HBV replication [3]. This inhibition could potentially benefit achieving clinical cure in patients with CHB. However, some scholars argue that MAFLD-related lipid metabolism disorders and lipotoxicity may induce inflammation, disrupting hepatocyte homeostasis, resulting in elevated alanine aminotransferase (ALT) levels in patients with CHB, with potential progression to cirrhosis and HCC [4]. Consequently, further investigation into the role of hepatic steatosis in HBV infection is warranted. Recent research suggests that the true virological response requires HBV DNA below detection limits, considering the recognized replication cycle of HBV infection. HBV DNA disappearance indicates the inhibition of reverse transcription and not that of covalently closed circular DNA (cccDNA) transcription. A comprehensive virological response necessitates both HBV DNA and HBV RNA to be below the lower limit of detection (LLD) [5]. As a precursor of HBV RNA, pgRNA authentically reflects cccDNA transcriptional activity in hepatocytes. Therefore, serum pgRNA levels can reflect HBV replication in the hepatocytes of patients. In summary, this study investigated hepatic steatosis and hepatitis B virology, explored the influencing factors of pgRNA below LLD in patients infected with HBV, and delved into the potential impact of liver steatosis on HBV replication ability.
Materials and methods
Study population
This cross-sectional study involved 272 patients with chronic HBV infection who visited the Liver Disease Center of Integrated Traditional Chinese and Western Medicine at Dalian Medical University between July 2021 and November 2023. All patients were either treatment-naïve or had ceased antiviral treatment for > 6 months. Based on the presence of fatty liver, patients were categorized into the CHB group (CHB) with 128 cases, and the CHB combined with the MAFLD group (CHB + MAFLD) with 144 patients. Further classification into high-level HBV DNA (HBV DNA ≥ 2000 IU/mL) (n = 129) and low-level HBV DNA (HBV DNA < 2000 IU/mL) (n = 143) groups was done based on whether HBV DNA was higher than 2000 IU/mL. Patient data encompassing general conditions were collected, and various examinations, including serum biochemistry, HBV serology, HBV DNA quantification, pgRNA quantification, and FibroScan examination, were conducted. Serum samples were collected and stored at − 80°C.
The inclusion criteria were as follows: (1) age ≥ 18 years; (2) Positive HBsAg, or HBV DNA, or both for > 6 months; (3) Diagnosis of MAFLD met the criteria outlined in “A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement” [6]. Exclusion criteria included: (1) Concurrent presence of other viral hepatitis; (2) Coexistence with other liver diseases such as autoimmune liver disease or drug-induced liver injury; (3) Incomplete clinical data collection.
Serum pgRNA and HBV DNA detection
Serum pgRNA was assessed using RNA simultaneous amplification testing (SAT) with the SAT isothermal amplification real-time fluorescence detection kit (Rendu Biotechnology, Shanghai, China). The Auto SAT system (Rendu Biotechnology) facilitated RNA extraction, amplification, and detection. The linear concentration range covered 2 log copies/mL to 8 log copies/mL, with a 50 copies/mL LLD.
Serum HBV DNA levels were determined through real-time polymerase chain reaction utilizing the HBV nucleic acid quantitative detection kit (Shengxiang Biotechnology, Hunan, China), with a reference range of HBV DNA < 20 IU/mL.
HBV serology detection
Serum HBsAg was detected by Abbott Microparticle chemiluminescence i2000 (Abbott, Chicago, USA), with a reference range set at HBsAg > 0.05 IU/mL. HBeAg levels were assessed using a Roche e801 automatic electrochemiluminescence immunoassay analyzer (Roche, Basel, Switzerland), and the reference range was < 1.0 signal-to-cutoff ratio [S/Co].
FibroScan assessment
For the evaluation of the controlled attenuation parameter (CAP) and liver stiffness measurement (LSM), the FibroScan®502 and M-type probe (Echosens, Paris, France) were utilized. The procedure required successful execution ten times at the same position, and the median of the effective measurement results was considered the outcome. Following the meta-analysis published in 2017 [7], CAP values were categorized as follows: no steatosis (grade 0 [S0]) with CAP < 248 dB/M; mild steatosis (grade 1 [S1]) with 248 dB/M ≤ CAP < 268 dB/M; moderate steatosis (grade 2 [S2]) with 268 dB/M ≤ CAP < 280 dB/M; and severe hepatic steatosis (grade 3 [S3]) with CAP ≥ 280 dB/M.
Statistical analysis
Continuous variables were expressed as median (interquartile range [IQR]), and group comparisons were conducted using the Mann‒Whitney U test or Kruskal–Wallis test. Categorical variables were presented as percentages (%), and inter-group differences were assessed using the Chi-squared and Fisher’s exact tests. One-way analysis of variance detected variations in HBV levels among groups with varying hepatic steatosis. Spearman correlation coefficient was employed to assess the correlation between hepatic steatosis and hepatitis B virology. Univariate and multivariate logistic regression analyses were performed to identify factors influencing pgRNA below the LLD, and odds ratio (OR) with 95% confidence intervals (95% CI) were calculated. Statistical significance was set at P < 0.05. Statistical analysis was conducted using IBM SPSS (Version 25.0).
Results
Clinical characteristics of 272 patients
Throughout the study, 272 patients were enrolled, and Table 1 summarizes their clinical profiles. The cohort comprised 156 men (57.4%) and 116 women (42.6%), with a median age of 44 years. Among them, 138 cases (50.7%) had a family history of HBV, 31 (11.4%) had diabetes mellitus, 34 (12.5%) had cirrhosis, and 14 (5.1%) had HCC. The distribution of hepatic steatosis was as follows: 128 cases without hepatic steatosis, 51 with S1, 41 with S2, and 52 with S3.
Significant differences were observed between the CHB + MAFLD and CHB groups in terms of male proportion, history of diabetes mellitus, history of cirrhosis, body mass index (BMI), ratio of overweight/obesity individuals, CAP values, and proportion of elevated ALT (P < 0.05). However, the two groups showed no significant differences in age, family history of HBV, history of HCC, waist-to-hip ratio, LSM values, and ALT levels.
Association between hepatic steatosis and hepatitis B virology
Comparative analysis of CHB and CHB + MAFLD in 272 patients infected with HBV
In the cohort of 272 cases with HBV infection, CHB + MAFLD exhibited an increased proportion of HBV DNA and pgRNA below the LLD compared to CHB (HBV DNA: 39.6% vs. 29.7%; pgRNA: 27.8% vs. 18.8%, respectively). Moreover, the levels of HBV DNA (4.11 vs. 3.62 log10 IU/mL, P = 0.045) and HBsAg (3.53 vs. 3.15 log10 IU/mL, P = 0.008) were significantly decreased in CHB + MAFLD, along with a lower positive rate of HBeAg (33.6% vs. 22.2%, P = 0.036) (Table 2).
Comparative analysis of hepatitis B virology in the high-level HBV DNA cohort with CHB and CHB + MAFLD
In 129 high-level HBV DNA patients, compared with CHB group, the levels of pgRNA (3.85 vs. 3.35 log10 copies/mL, P = 0.044) and HBsAg (3.85 vs. 3.59 log10 IU/mL, P = 0.033) were significantly decreased in CHB + MAFLD group. Furthermore, the positive rate of HBeAg was lower in CHB + MAFLD group (Table 3).
Comparative analysis of hepatitis B virology in the low-level HBV DNA cohort with CHB and CHB + MAFLD
In 143 low-level HBV DNA patients, CHB + MAFLD showed an increase in the proportion of pgRNA below the LLD (36.7% vs. 44.6%, respectively). Furthermore, the levels of pgRNA (3.09 vs. 2.96 log10 copies/ml, respectively), HBsAg (2.74 vs. 2.69 log10 IU/ml, respectively) and the positive rate of HBeAg (15.0% vs. 7.2%, respectively) were decreased in CHB + MAFLD (Table 4).
Correlation analysis between hepatic steatosis and hepatitis B virology
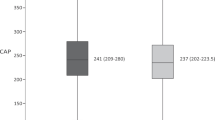
A correlation analysis investigating the relationship between the severity of liver steatosis and hepatitis B virology was performed on 144 patients with CHB + MAFLD. The results revealed negative correlations between liver steatosis and pgRNA, HBV DNA, HBsAg, and HBeAg (pgRNA: r = − 0.529, P < 0.001; HBV DNA: r = − 0.456, P < 0.001; HBsAg: r = − 0.465; P < 0.001; HBeAg: r = − 0.339, P < 0.001) (Fig. 1). As the severity of liver steatosis increased, there was a corresponding decrease in the levels of pgRNA, HBV DNA, HBsAg, and HBeAg.

Correlation between hepatic steatosis and hepatitis B virology in 144 patients with CHB + MAFLD. A pgRNA, B HBV DNA, C HBsAg, and D HBeAg were negatively correlated with the degree of hepatic steatosis
In liver steatosis grades S1, S2, and S3, a consistent descending trend was observed for the levels of pgRNA, HBV DNA, HBsAg, and the positive rate of HBeAg (pgRNA: 3.51 vs. 2.93 vs. 2.72 log10 copies/mL, P < 0.001; HBV DNA: 4.04 vs. 3.53 vs. 2.73 log10 IU/mL, P < 0.001; HBsAg: 3.47 vs. 3.09 vs. 2.52 log10 IU/mL, P < 0.001; positive rate of HBeAg: 41.2% vs. 19.5% vs. 5.8%, P < 0.001) (Table 5).
Logistic regression analysis of pgRNA below the LLD in patients infected with HBV
In 272 patients with HBV infection, univariate logistic regression analysis revealed significant correlations between pgRNA below the LLD and HBV DNA, HBsAg, triacylglycerol (TG), and CAP values (P < 0.05). Subsequently, a multivariable logistic regression incorporating these indicators was conducted. The results indicated that HBV DNA (OR: 0.283, 95% CI 0.146–0.550, P < 0.001), HBsAg (OR: 0.300, 95% CI 0.148–0.610, P < 0.001), and CAP values (OR: 1.013, 95% CI 1.001–1.026, P = 0.038) independently influenced the presence of pgRNA below the LLD in chronic HBV infection (Table 6).
Discussion
Globally, the coexistence of CHB and MAFLD poses a significant public health challenge, with biopsy-proven MAFLD prevalence ranging from 13.5 to 30% in patients with CHB [8,9,10]. Despite the extensive prevalence, the causal relationship between these conditions remains elusive. The intricate interplay of metabolic disorders and immune dysregulation in MAFLD adds complexity to the HBV-MAFLD interaction, potentially accelerating the progression of severe liver disease. This study investigated the association between liver steatosis and hepatitis B virology to delve deeper into the impact of MAFLD on HBV infection, investigating factors influencing pgRNA levels in patients infected with HBV. The key findings are summarized below: (1) The expression of HBV in patients with CHB and liver steatosis exhibited a significant decrease, particularly in the high-level HBV DNA cohort, with significant reductions in HBsAg and pgRNA levels; (2) Liver steatosis demonstrated a negative correlation with hepatitis B virology, with higher degrees of liver steatosis associated with lower expression of HBV DNA, pgRNA, HBsAg, and HBeAg; (3) Independent influencing factors for pgRNA below the LLD in patients with HBV infection including HBV DNA, HBsAg, and CAP values.
A 3-year longitudinal cohort study reported that patients with CHB and liver steatosis had a higher chance of achieving HBsAg loss, with the HBsAg clearance rate after the 3-year follow-up being three times greater than that of those with CHB and without liver steatosis. This finding suggests an increased potential for clinical cure in patients with CHB and liver steatosis [11]. In this cross-sectional study of 272 patients with CHB, the association between liver steatosis and hepatitis B virology was sought to be investigated. Our findings revealed diminished HBV DNA, pgRNA and HBsAg levels in the CHB + MAFLD group, and a significant decrease in HBeAg positivity rates. These results suggest that hepatic steatosis inhibits HBV replication. Hu et al. [3] corroborated this perspective through mice model experiments, asserting that the decline in HBV DNA levels in mice with hepatic steatosis was linked to hepatocyte metabolism, indicating a direct impact of hepatic steatosis on HBV replication. Furthermore, our study unveiled a significant negative correlation between liver steatosis and hepatitis B virology. Spearman correlation analysis revealed a negative association between the degree of hepatic steatosis and HBV DNA, pgRNA, HBsAg, and HBeAg. Patients with severe liver steatosis exhibited significantly lower levels of HBV DNA, pgRNA, and HBsAg compared to those with mild or moderate liver steatosis, indicating a marked decrease in HBV levels with increasing severity of hepatic steatosis. Consistent with our findings, a retrospective study of 3212 patients with CHB demonstrated a negative correlation between the degree of hepatic steatosis and the level of HBsAg in hepatocytes. In patients with hepatic steatosis > 5%, HBsAg levels in hepatocytes were significantly lower than those in patients with hepatic steatosis < 5% and without hepatic steatosis [12]. According to the present study, MAFLD may inhibit HBV through various mechanisms. Firstly, MAFLD inhibits HBV replication by modulating immune responses, activating NK cells and CD8 + T cells through the Toll-like receptor (TLR) pathway to accelerate HBV clearance [13, 14]. TLR4/myeloid differentiation factor 88 (MyD88), a common signaling pathway in MAFLD and CHB, is critical in inhibiting HBV replication. TLR4/MyD88 downregulates the expression of two transcription factors (hepatocyte nuclear factor 1-alpha and hepatocyte nuclear factor 4-alpha) during HBV replication, potentially explaining the reduction in HBV replication in patients with CHB and MAFLD [15]. Some scholars propose that hepatic steatosis inhibits HBV replication by enhancing T-cell responses [16]. Secondly, MAFLD may accelerate the induction of apoptosis and inhibit autophagy in HBV-infected cells, ultimately leading to the clearance of HBsAg and HBV DNA [17, 18]. In addition, it has been confirmed that miR-122 inhibits HBV transcription by reducing the expression of cyclin G1 [19], while the expression level of miR-122 was increased in patients with MAFLD [20], which ultimately leads to the reduction of HBV expression in patients with CHB combined with MAFLD. Finally, MAFLD-related metabolic disorders, such as insulin resistance, diminish HBV replication by suppressing the activity of peroxisome proliferator-activated receptor-gamma (PPAR-γ) coactivator one alpha [21, 22].
To further investigate the relationship between liver steatosis and HBsAg, HBeAg, and pgRNA in patients with similar viral loads, our study encompassed both high-level HBV DNA and low-level HBV DNA populations. The findings indicated that, in both groups, the levels of pgRNA and HBsAg in CHB + MAFLD were lower compared to the CHB group. However, in high-level HBV DNA patients, the reduction was more substantial for pgRNA (3.85 vs. 3.35 log10 copies/mL, P = 0.044) and HBsAg (3.85 vs. 3.59 log10 IU/mL, P = 0.033). These results confirmed that liver steatosis led to varying HBV replication reduction in high-level HBV DNA and low-level HBV DNA populations. Adiponectin, a downstream target gene of PPAR-γ known to upregulate HBV replication, experiences reduced levels in patients with hepatic steatosis [23, 24]. Mohamadkhani et al. [25] demonstrated a negative correlation between HBV DNA levels and adiponectin. These results showed that in the CHB + MAFLD group with higher viral loads, adiponectin levels were lower, ultimately contributing to the decline in HBV replication ability. Given the relatively high viral load in high-level HBV DNA patients in this study, the presence of hepatic steatosis aggravated HBV clearance, suppressing HBV replication.
In recent years, accumulating evidence suggests that achieving HBV DNA below the LLD is not a sufficient endpoint for discontinuing antiviral therapy [26, 27]. As a precursor of HBV RNA, pgRNA provides insight into the transcriptional activity of cccDNA in hepatocytes [28]. Distinguished from HBsAg and HBV DNA, serum pgRNA exclusively originates from cccDNA, rendering it a valuable criterion for determining a complete virological response to antiviral therapy and a predictive indicator for virological recurrence post-discontinuation [29, 30]. The conversion to negative pgRNA, synonymous with cccDNA depletion or transcriptional silencing [31], signifies increased accessibility to clinical cures for CHB. This study employed multivariate logistic regression analysis to explore further the relevant influencing factors of pgRNA below the LLD in patients infected with HBV. The results revealed that HBV DNA, HBsAg, and CAP values emerged as independent influencing factors for pgRNA negativity in CHB. Therefore, lower HBsAg and HBV DNA levels, coupled with higher CAP values, indicated a higher likelihood for patients with CHB to achieve pgRNA below the LLD, reaffirming the supportive role of liver steatosis in HBV clearance.
This study carries certain limitations. Firstly, a cross-sectional analysis was conducted, lacking a dynamic observation of hepatic steatosis’s influence on HBV through longitudinal follow-up. Future studies will address this by implementing longitudinal follow-up to gauge the impact of liver steatosis on antiviral therapy for CHB. Secondly, this study lacked liver pathological biopsy, relying solely on the serological analysis of HBV expression. To provide a more comprehensive understanding of hepatic steatosis’s impact on hepatitis B virology, disease progression, and prognosis, a multicenter, large-sample prospective cohort study is warranted.
Conclusion
Our study revealed reduced HBV expression among patients with CHB and hepatic steatosis, particularly significant in high-level HBV DNA patients with a higher viral load. Furthermore, a negative correlation was identified between hepatic steatosis and hepatitis B virology, observing a gradual decline in the levels of HBsAg, HBV DNA, and pgRNA and the positive rate of HBeAg with worsening hepatic steatosis. Finally, our findings highlighted HBV DNA, HBsAg, and CAP values as independent factors influencing pgRNA levels below the LLD in patients infected with HBV. These insights could hold clinical significance for the future monitoring and treatment of patients with CHB and MAFLD. Subsequent research, encompassing primary and clinical studies, is needed to explore further the impact of hepatic steatosis on the natural course of HBV and patient prognosis.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available but are available from the corresponding author upon reasonable request.
Abbreviations
- CHB:
-
Chronic hepatitis B
- MAFLD:
-
Metabolic dysfunction-associated fatty liver disease
- HBV:
-
Hepatitis B virus
- HBsAg:
-
Hepatitis B surface antigen
- pgRNA:
-
Pregenomic RNA
- HBeAg:
-
Hepatitis B e antigen
- OR:
-
Odds ratio
- ALT:
-
Alanine aminotransferase
- cccDNA:
-
Covalently closed circular DNA
- SAT:
-
Simultaneous amplification testing
- S/Co:
-
Signal-to-cutoff ratio
- LLD:
-
Lower limit of detection
- CAP:
-
Controlled attenuation parameter
- LSM:
-
Liver stiffness measurement
- IQR:
-
Interquartile range
- 95% CI:
-
95% Confidence interval
- BMI:
-
Body mass index
- AST:
-
Aspartate aminotransferase
- ALP:
-
Alkaline phosphatase
- GGT:
-
Gamma-glutamyltransferase
- TC:
-
Total cholesterol
- TG:
-
Triacylglycerol
- TLR4:
-
Toll-like receptor 4
- MyD88:
-
Myeloid differentiation factor 88
- PPAR-γ:
-
Peroxisome proliferator-activated receptor-gamma
References
Zhang J, Lin S, Jiang D, et al. Chronic hepatitis B and non-alcoholic fatty liver disease: Conspirators or competitors? Liver Int. 2020;40(3):496–508.
Yang M, Wei L. Impact of NAFLD on the outcome of patients with chronic hepatitis B in Asia. Liver Int. 2022;42(9):1981–90.
Hu D, Wang H, Wang H, et al. Non-alcoholic hepatic steatosis attenuates hepatitis B virus replication in an HBV-immunocompetent mouse model. Hepatol Int. 2018;12(5):438–46.
Tourkochristou E, Assimakopoulos SF, Thomopoulos K, Marangos M, Triantos C. NAFLD and HBV interplay-related mechanisms underlying liver disease progression. Front Immunol. 2022;13:965548.
Lu F, Wang J, Chen X, Xu D, Xia N. Potential use of serum HBV RNA in antiviral therapy for chronic hepatitis B in the era of nucleos(t)ide analogs. Front Med. 2017;11(4):502–8.
Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73(1):202–9.
Karlas T, Petroff D, Sasso M, et al. Individual patient data meta-analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J Hepatol. 2017;66(5):1022–30.
Shi YW, Yang RX, Fan JG. Chronic hepatitis B infection with concomitant hepatic steatosis: current evidence and opinion. World J Gastroenterol. 2021;27(26):3971–83.
Machado MV, Oliveira AG, Cortez-Pinto H. Hepatic steatosis in hepatitis B virus infected patients: meta-analysis of risk factors and comparison with hepatitis C infected patients. J Gastroenterol Hepatol. 2011;26(9):1361–7.
Wong VW, Wong GL, Chu WC, et al. Hepatitis B virus infection and fatty liver in the general population. J Hepatol. 2012;56(3):533–40.
Mak LY, Hui RW, Fung J, et al. Diverse effects of hepatic steatosis on fibrosis progression and functional cure in virologically quiescent chronic hepatitis B. J Hepatol. 2020;73(4):800–6.
Wang MM, Wang GS, Shen F, Chen GY, Pan Q, Fan JG. Hepatic steatosis is highly prevalent in hepatitis B patients and negatively associated with virological factors. Dig Dis Sci. 2014;59(10):2571–9.
Zhang RN, Pan Q, Zhang Z, Cao HX, Shen F, Fan JG. Saturated Fatty Acid inhibits viral replication in chronic hepatitis B virus infection with nonalcoholic Fatty liver disease by toll-like receptor 4-mediated innate immune response. Hepat Mon. 2015;15(5):e27909.
Khanmohammadi S, Kuchay MS. Toll-like receptors and metabolic (dysfunction)-associated fatty liver disease. Pharmacol Res. 2022;185:106507.
Broering R, Lu M, Schlaak JF. Role of Toll-like receptors in liver health and disease. Clin Sci (Lond). 2011;121(10):415–26.
Patel N, Boghici D, Rava M, et al. Enhanced T cell responses in patients and mouse model of hepatitis B and comorbid non-alcoholic fatty liver disease. J Hepatol. 2022;77:S259–60.
Mao Y, Da L, Tang H, et al. Hepatitis B virus X protein reduces starvation-induced cell death through activation of autophagy and inhibition of mitochondrial apoptotic pathway. Biochem Biophys Res Commun. 2011;415(1):68–74.
Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125(2):437–43.
Wang S, Qiu L, Yan X, et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1)-modulated P53 activity. Hepatology. 2012;55(3):730–41.
Long JK, Dai W, Zheng YW, Zhao SP. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol Med. 2019;25(1):26.
Shlomai A, Paran N, Shaul Y. PGC-1alpha controls hepatitis B virus through nutritional signals. Proc Natl Acad Sci U S A. 2006;103(43):16003–8.
Jiang Y, Chen D, Gong Q, et al. Elucidation of SIRT-1/PGC-1α-associated mitochondrial dysfunction and autophagy in nonalcoholic fatty liver disease. Lipids Health Dis. 2021;20(1):40.
Yoon S, Jung J, Kim T, et al. Adiponectin, a downstream target gene of peroxisome proliferator-activated receptor γ, controls hepatitis B virus replication. Virology. 2011;409(2):290–8.
Polyzos SA, Toulis KA, Goulis DG, Zavos C, Kountouras J. Serum total adiponectin in nonalcoholic fatty liver disease: a systematic review and meta-analysis. Metabolism. 2011;60(3):313–26.
Mohamadkhani A, Sayemiri K, Ghanbari R, Elahi E, Poustchi H, Montazeri G. The inverse association of serum HBV DNA level with HDL and adiponectin in chronic hepatitis B infection. Virol J. 2010;7:228.
Terrault NA, Bzowej NH, Chang KM, et al. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63(1):261–83.
Wang J, Shen T, Huang X, et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J Hepatol. 2016;65(4):700–10.
Giersch K, Allweiss L, Volz T, Dandri M, Lütgehetmann M. Serum HBV pgRNA as a clinical marker for cccDNA activity. J Hepatol. 2017;66(2):460–2.
Vachon A, Osiowy C. Novel biomarkers of hepatitis B virus and their use in chronic hepatitis B patient management. Viruses. 2021;13(6):951.
Roncarati G, Galli S, Ferniani T, Moroni A, Lazzarotto T. Evaluation of pregenomic HBV RNA in HBeAg-negative patients. New Microbiol. 2022;45(2):104–10.
Ning Q, Han M, Sun Y, et al. Switching from entecavir to PegIFN alfa-2a in patients with HBeAg-positive chronic hepatitis B: a randomised open-label trial (OSST trial). J Hepatol. 2014;61(4):777–84.
Acknowledgements
We thank all the patients who participated in this research.
Funding
This work was funded by the National Natural Science Foundation of China (Grant Number 82274260).
Author information
Authors and Affiliations
Contributions
Study concept and design: YZ, QC; Collecting data: SY, GR; Data analysis and interpretation: SY, GR; Drafting of the manuscript: SY; Critical revision of the manuscript for important intellectual content: YZ, QC; All the authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
A statement of Helsinki was used in this study and was approved by the Ethics Committee of the First Affiliated Hospital of Dalian Medical University (No. PJ-KS-KY-2022-291) and Clinical trial registration (ChiCTR2200063555). Informed consent was obtained from all subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yi, S., Ren, G., Zhu, Y. et al. Correlation analysis of hepatic steatosis and hepatitis B virus: a cross-sectional study. Virol J 21, 22 (2024). https://doi.org/10.1186/s12985-023-02277-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-023-02277-8