
Abstract
Influenza A viruses cause severe respiratory illnesses in humans and animals. Overreaction of the innate immune response to influenza virus infection results in hypercytokinemia, which is responsible for mortality and morbidity. The influenza A virus surface glycoprotein neuraminidase (NA) plays a vital role in viral attachment, entry, and virion release from infected cells. NA acts as a sialidase, which cleaves sialic acids from cell surface proteins and carbohydrate side chains on nascent virions. Here, we review progress in understanding the role of NA in modulating host immune response to influenza virus infection. We also discuss recent exciting findings targeting NA protein to interrupt influenza-induced immune injury.
Similar content being viewed by others


Introduction
Influenza is highly contagious, and acute viral respiratory infections may occur as pandemics, epidemics, and outbreaks [1,2,3]. The susceptibility to infection involvement in all age stages, prominent individuals with chronic comorbid diseases, immunosuppression, pregnant and postpartum women, and frail older adults; most commonly occurs in spring and winter, and the virus causes significant mortality and morbidity worldwide [2, 4, 5]. Influenza viruses belong to the Orthomyxoviridae RNA virus and classify into four distinct types based on their antigenic differences: influenza A, influenza B, influenza C, and influenza D. Influenza A viruses infect a broad range of host species. However, the main hosts are humans for influenza B and C, and influenza D viruses have not infected humans. Influenza A is the type most responsible for causing pandemics because of its high susceptibility to antigenic variation [6,7,8], so we will mainly discuss understanding influenza A viruses (IAV) infection.
IAV virions are constructed from a host cell-derived membrane and various viral proteins [9, 10]. Eight single-stranded viral RNA segments for 10 structural and at least 9 nonstructural/regulatory proteins. PB1, PB2, PA, NP, M1, NS1, and NEP are inside the lipid envelope. At the same time, M2, Hemagglutinin (HA), and Neuraminidase (NA) are embedded in the envelope and available for antibody binding [10,11,12,13,14,15]. IAV infects the upper respiratory tract at first, enters epithelial cells through endocytosis, and infects the lower respiratory tract with the disease developed [16, 17]. The HA protein binds to sialic acid residues expressed in the airway or alveolar epithelium, triggering the endocytosis of viral particles [18]. The virus completes the shedding, assembly, and release on the membrane. In the procession of viral shedding, NA cuts the connection between the HA of newly formed viral particles and sialic acid receptors on the cell surface, releasing progeny viruses, which promotes viral replication, transcription, and translation, infecting neighboring cells or leaving the individual through respiratory droplets [19, 20]. For the past few years, the widespread use of NA inhibitors has raised the concern about drug resistance. Here, we review the role of NA in modulating the host immune response to influenza virus infection.
Neuraminidase: structure, mutation, and function
NA structure
Neuraminidase, located on the virus's surface and belongs to glycoproteins, next to HA and matrix protein2(M2), is also called sialidase [21]. NA is a tetramer of four identical polypeptides, presenting the mushroom-like structure consisting of four domains: an N-terminal cytoplasmic sequence, followed by a membrane-anchoring hydrophobic transmembrane domain (TMD) and a thin stalk of variable length, ending in a globular head domain, the binding site for sialic acid is located in the head domain [21,22,23,24]. Each protomer comprises approximately 470 amino acid residues [23]. NA tetramer dimensions about 10 nm × 15 nm, cleaves sialic acid for virion release 20–50 tetramers per spherical virion. Space for about 13 bound Fabs per tetramer, containing 1880 aa/tetramer, weight 220 kDa/tetramer for prototypical lab-adapted strains of the influenza A virus [25].
The N-terminal cytoplasmic domain has six amino acid residues (MNPNQK); this sequence is nearly 100% conserved across all influenza A subtypes. The arrangement has been highly preserved in influenza A and B viruses [26, 27]. The cytoplasmic domain is also essential for attaching NA with lipid rafts [19].
The TMD can transport newly expressed NA to the apical plasma membrane with the N-terminal cytoplasmic tail [28]. The TMD that follows the short cytoplasmic sequence is variable in sequence among subtypes. Still, all subtypes form a transmembrane helix encompassing amino acids 7–29 when analyzed by the highly reliable program TMHMM [29,30,31].
The stalk domain connects the TMD with the catalytic head domain. The NA stalk varies in length within and across NA subtypes and contains multiple predicted N-linked glycosylation sites. And, with few exceptions, it has at least one cysteine residue that can form an intermolecular disulfide bond with a neighboring NA molecule. Glycosylation of the stalk region may contribute to NA stability; the disulfide bond needs to format tetramer [31]. A discovery indicated that specificity and affinity to sialic acids by the HA is highly dependent on sugar conformation and extension, and the stalk length of NA can impact combining HA with sialic acids [31,32,33,34,35].
The NA head domain is characterized by a six-bladed propeller folded around the catalytic site and is typical for all known sialidases [36]. Each blade comprises four antiparallel beta sheets stabilized by disulfide bonds and is connected by variable-length loops [31]. In of to classify with glycoproteins, NA possess nine different classes, N1–N9, crystal structures of the head domain of at least one representative NA from N1 to N9 and from influenza B NA have been analyzed, and crystal structures of NA encompass the catalytically active heads [36,37,38,39] (Fig. 1).

Neuraminidase structure. Neuraminidase is comprised of N-terminal cytoplasmic tail, transmembrane, NA stalk and NA head. A tetramer dimensions about 10 nm × 15 nm from snapshot
NA mutation
Influenza viruses evolve quickly through frequent antigenic variation. Antigenic drift and shift are terms used to describe how the virus mutates and results in new strains. Drastic changes in the antigenicity of the HA of circulating influenza A viruses is called antigenic shift; the animal strains of the influenza virus can be acquired by human influenza strains through reassortment [40]. We and others established a mice model for H9N2 infection, which revealed multiple amino acid substitutions in NA related to enhanced virulence in mice [41, 42]. There is a significant change in the virus's genome in antigenic shift resulting in new HA and NA protein expression [1, 43], which can cause a medium or small epidemic [44]. Influenza viruses can evade the antibody-mediated immunity induced during infections or vaccinations by gradually accumulating mutations in HA and NA, known as antigenic drift [40]. The antigenic drift of HA has been extensively studied [45, 46]. By using neuraminidase inhibition assays, the antigenic drift of NA mostly matches between vaccines and circulating viruses [40, 47, 48]. The reassortment and evolution of NA and linked HA may result in an antigenic drift of both significant surface glycoproteins, reducing vaccine efficacy and subsequently impacting animal health [49]. K199 and E258 mutations significantly affected Mab binding, NA inhibition, and neutralization. The activity and the modifications help detect antigenically drifted NAs [50]. A less obvious location for functional variation is the fibrous stalk that attacks the globular domain to the membrane, governs the length and the height of the globular domain, and hence its access to substrates and its interactions with HA [51, 52]. However, influenza viruses circulate in different species, such as birds to humans. Subtype antigenic variation is limited, so vaccines can also be selected limited.
NA function
Robust protective human immunity against influenza is primarily provided by antibodies targeting the virus's variable epitopes, those found on portions of its surface glycoproteins. When influenza viruses infect the body, HA mediates binding to sialic acids on host cell glycoproteins or lipids. Then fusion of the host cell and virus membrane through a low pH-induced irreversible conformational changes, primarily as HA2 anchors HA in the envelope and is directly involved in membrane interaction [53]. HA is essential in the entry process. NA is which sialidase catalyzes the removal of the terminal sialic acids. As IAV is essentially reversibly bound in the NA activity, motility enables virion penetration of the mucus layer by cleaving sialic acids as well as attachment to and uptake into the epithelial cells of the respiratory tract, so that infect underlying epithelial cells [54, 55]. Studies show that the enhancement of fusion and infectivity by NA was related to the sialylation of virion-expressed HA, so NA activity plays a critical role in virion infectivity and HA-mediated membrane fusion [56]. On the other hand, the influenza replication cycle needs to release the least newly formed virions from the infected cell and prevent virion aggregation by removing sialic acid from the viral and host cell membrane [57]. Currently, many NA inhibitors are discovered by structure–activity relationship, and these inhibitors fight against the surge in resistance resulting from naturally occurring mutations.
Cytokine storm
The IAV infects epithelial cells, endothelial cells, and alveolar macrophages to produce the first wave of cytokines, especially type I interferons (IFNs), which upregulate the expression of numerous IFN-stimulated genes (ISGs).
Following the type I IFNs released, higher expression of ISGs initiates downstream antiviral responses and subsequent inflammatory cytokine production by innate immune cells. Then the adaptive immune cells (different subsets of T cells and group 2 innate lymphoid cells) are activated and regulated to secrete the second wave of cytokines that promote viral clearance, tissue homeostasis, and lung repair [58]. The innate immune response is regulated by chemokines and cytokines, chemical messengers produced by virus-infected epithelial cells and leukocytes [59], and natural interferon-producing cells, such as plasmacytoid dendritic cells [60]. A study found that the only producers of antiviral cytokines were infecting epithelial cells; plasmacytoid dendritic cells were potent producers of IFNs in the body by using an animal model [61]. After influenza viruses infect the host, the IAV first induces the innate immune system, which can rapidly recruit innate immune cells and cytokines to the site of infection [62]. Cytokines are essential for intercellular communication and viral clearance in the immune system, but excessive cytokines can cause severe immune pathology. Excessive production of pro-inflammatory cytokines leads to aggressive proinflammatory responses. The insufficient control of anti-inflammatory responses is called a cytokine storm or hypercytokinemia, which causes significant immunopathology and serious disease consequences, such as acute respiratory distress syndrome (ARDS) [17, 63,64,65,66,67,68].
From a pathology perspective, the characteristic alveolar changes of influenza-virus pneumonia caused by cytokine storms include capillary thrombosis, focal necrosis, congestion of the alveolar wall, hyaline membrane formation, pulmonary edema, peribronchial hemorrhage, peribronchial pneumonia [69]. The changes characteristic of severe influenza viral pneumonia include capillary and small vessel thromboses, interstitial edema and inflammatory infiltrate, the formation of hyaline membranes in alveoli and alveolar ducts, varying degrees of acute interalveolar edema and hemorrhage, and diffuse alveolar damage in addition to necrotizing bronchitis and bronchiolitis. In later stages of diffuse alveolar damage, fibrosis, epithelial regeneration, and squamous metaplasia [69, 70]. Severe cytokine storms can cause multiple organ dysfunction syndromes, systemic inflammation, and even death [17, 71,72,73]. Cytokine storms can lead to host immune response disorders, primarily the innate immune system, and can cause lung damage after the influenza virus infects the body. Many studies have shown that many factors are related to NA modulating host immune response to influenza virus infection.
Biological factors may affect the host's susceptibility to the influenza virus and its anti-immune response. Activated macrophages were the cellular source of cytokines and chemokines in young and old mice. Macrophages, dendritic cells, and NK cells are activated in younger mice. Dendritic cells did not have this same effect in older mice. The cellular source of many cytokines and chemokines shifted as aging [74]. The different ways and times of vaccine injection also affect immune protection [75]. The risk of influenza-related death increases exponentially after age 65, with over 90% of the annual influenza virus-related mortality from this age stage group in the United States [76]. The severe consequences of the influenza virus infection in children are related to the cytokine storm [77,78,79]. The airway epithelial TLR3 drives IFN-β production in response to IAV infection, as determined by genetic mapping of TLR3-associated mutations in children who acquire severe IAV-induced ARDS [80]. Therefore, cytokine storm is likely related to age, affecting immune response.
Obesity is an independent risk factor for increased disease severity and death during IAV infection. Obesity primes the innate immune system to respond to IAV with a heightened proinflammatory response and a blunted antiviral response, leading to increased tissue damage and decreased virus elimination [81]. Immune response to infection is impaired in obese individuals [82]. B cells exacerbate inflammation and insulin sensitivity by producing auto-antibodies in fat mice [83]. Increased inflammation, particularly elevated IL-6 levels, activation of Renin–angiotensin–aldosterone system (RAAS), rise in Angiotensin II(Ang II) levels, higher leptin, and increased ectopic fat favor influenza virus progression and severity [84]. Therefore, cytokine storm is related to obesity.
Influenza viruses rely on numerous host factors to support their replication [85,86,87,88]. Sialic acid is a determinant of the host range. Viral neuraminidase displays species-specific adaptation. Different expression patterns of detection and antiviral effector molecules in other species will drive the adaption of influenza viruses when they infect a new host. This adaption can involve changes that alter the binding partners and the relative expression or cellular location of the viral antagonist of the cellular innate immune response [89]. Influenza induces DNA damage, and DNA damage responses are activated; the host response causes DNA damage in lung epithelium after influenza infection; DNA repair modulates the severity of influenza-induced cytotoxicity, thereby affecting tissue damage and regeneration [90]. Targeting host factors involved in virus replication and controlling virus-induced host immune responses [91]. The extent of cellular coinfection by influenza viruses may be a critical determinant of both viral production kinetics and cellular infection outcomes in a host cell type-dependent manner [92]. Therefore, host factors can affect the host's immune response after an influenza virus infection.
Endothelial cells are central regulators of cytokine storms during influenza virus infection [93]. Vulnerability to secondary bacterial infection peaks at approximately one-week post-influenza infection; influenza virus infection facilitates secondary bacterial infection through phagocyte function (macrophage and neutrophil) or phagocyte-independent mechanisms, regulation of antimicrobial peptide, expression of IFN, immune cells (Th17 cells, NK cells, Treg cells, iLCs), and genetic susceptibility [94]. Studies indicate that wound healing was delayed when mice with healing wounds were infected with IAV in the lung; an inflammatory cytokine milieu characterizes the earliest phase of cutaneous wound repair. The viral lung infection suppresses the innate immune response in a healing wound, including cellular infiltrate, chemokines, growth factors, and cytokines; the cytokine and chemokine expression indicates a lung infection can induce changes in the dermal wound environment of cutaneous and subcutaneous wounds [95,96,97,98]. Phospholipids present in the pulmonary surfactant complex, palmitoyl-oleoyl-phosphatidylglycerol (POPG) and phosphatidylinositol (PI) could disrupt the virus particle binding to host cell plasma membrane receptors [99]. The antagonism of activation of TLRs and virus binding to the alveolar epithelium by resident constituents of the pulmonary surfactant system suggests that POPG and PI function in homeostasis to prevent inflammatory processes that result in reductions in gas exchange within the alveolar compartment. Antagonism of TLR activation inhibits proinflammatory signaling pathway initiation steps; lipids block TLR recognition of activated ligands directly or through TLR4 co-receptors cluster of differentiation 14(CD14) and Myeloid differentiation protein 2 (MD2) [99]. Therefore, boosting lipid synthesis or increasing the expression of CD14 and MD2 can inhibit cytokine production in the lung. A study indicates that upregulated expression of cellular adhesins by TGF-β, especially fibronectin-binding protein activated in influenza viral infection, increases host susceptibility to secondary bacterial pneumonia or coinfection [100]. Therefore, reducing fibronectin-binding protein expression and TGF-β production, which cannot cause secondary bacterial pneumonia and coinfection.
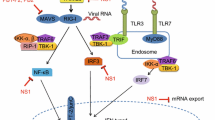
Influenza virus infections are associated with a cytokine storm and an exaggerated innate immune response [101]. A study indicates that p21 restricts IAV by perturbing the viral polymerase complex and activating the host's natural immune response. p21 directly interacts with HP-1 to inhibit K-48 ubiquitination-mediated degradation after IAV infection. p21 promotes IRF3 activation via the recruitment of HO-1 through the inhibition of K48-linked ubiquitination degradation, resulting in increased expression of type I IFNs. P21 acts as a positive regulator of type I IFN during IAV infection, a new role in innate immunity [102]. Therefore, enhancing p21 expression restricts the influenza A virus. A study found that disruption of SOCS3 expression provided significant protection against IAV infection in IAV early disease, attenuated acute lung injury, and silenced SOCS3 enhanced STAT3 activity and regulated NF-κB and IL-6 so that IAV circumvent IL-6/STAT3-mediated immune response through upregulating SOCS3 [103]. Therefore, disrupting influenza virus infection by restricting SOCS3 expression. Sphingosine 1-phosphate receptor 1 (S1PR1) is expressed by lymphocytes and endothelial cells and is known to control lymphocyte egress from lymph nodes [104]. Sphingosine 1-phosphate (S1P) agonist therapy suppresses innate immune cell recruitment and cytokine-chemokine production. S1P therapy could suppress detrimental innate immune responses without hindering virus control [93, 105]. Sphingosine analog AAL-R inhibits cellular and cytokine/chemokine responses and activates natural inflammatory infiltrate [105]. S1PR1 agonist is through inhibition downstream of myeloid differentiation primary response gene 88 and IFN-β promoter stimulator-1 signaling for blunting cytokine storm [106]. Therefore, activating S1PR1 signaling or using sphingosine analog to blunt cytokine storm for protecting the infected host from the consequences of influenza infection (Fig. 2).

The relationship of virus, immunity response, cytokine and lung. (1) Factor: from virus (NA activity, NA stalk length and transmembrane), from host (age, obesity and host factor), and these factors can affect immune response; (2) Phospholipid and p21 locate in the pulmonary epithelium, S1PR1 locate in the pulmonary endothelium, SOCS3 as inhibitory cytokine, pulmonary endothelium is central orchestrator of cytokine amplification. When virus invade body, p21 expression reduced, SOCS3 and S1PR1 signaling improved, immune cells are activated, frequent the number of cytokines is reduced, lung injury was decreased; (3) When body is infected with influenza virus, phospholipid increased, then the number of cytokines is increased, immune cells are inhibited, lead to cytokine storm and finally cause lung injury; (4) When virus invaded, TGF-β is activated through virus NA, then upregulated expression of cellular adhesins, immune cells are inhibited, and lead to cytokine storm, cause bacterial pneumonia
Targeting neuraminidase and NA-CD83 mediate the immune response
Recently, more and more research has shown that targeting NA helps suppress influenza virus infection. NA inhibitors are licensed as influenza therapeutics inhibiting NA activity [107,108,109,110]. KW is derived from the brown algae Kjellmaniella crassifolia, which blocks IAV invasion and release by targeting viral NA and cellular EGFR pathways [111]. Eliciting neutralizing antibodies that recognize variable epitopes on the HA head is the dominant way influenza vaccines protect individuals from influenza and prevent the spread of influenza through populations. Adjuvanted vaccines induce more robust CD4+ T cells and prolific germinal center reactions and activate naïve B cells with new specificities [112, 113]. By modulating NA stalk length in recombinant IAVs, anti-stem Abs inhibit virus release from infected cells by blocking NA, accounting for their in vitro neutralization activity. NA inhibitors enhance anti-stem-based Fc-dependent immune cell activation, raising the possibility of therapeutic synergy between NA inhibitors and anti-stem mAb treatment in humans, extending the NA stalk to enhance immunogenicity [114, 115]. A study found isolating three clonally related mAbs that bind to the influenza virus NA by inserting a long CDR H3 into the enzymatic active site, occupying the sialic acid substrate site and inhibiting all influenza A virus NA subtypes and influenza B virus NA [116]. NA is an important and protective antigen. NA is a promising target for future influenza vaccines, based on immunity optimally to enhance the breadth of influenza virus vaccines and increase vaccine efficacy [117,118,119]. A study found that it creates an NA inhibitor zanamivir-targeted cytotoxic drug and a viral NA-targeted CAR T cell, which can kill viral NA-expressing cells without damaging healthy cells [120]. The recombinant neuraminidase surface glycoprotein can enhance and broaden protection against the influenza virus [121]. Anti-NA antibodies are less dependent on the HA due to receptor binding for helping aid in the control of viruses [122,123,124]. Improving vaccine design for identifying NA antigenic drift and novel epitopes of anti-NA antibodies [125]. Moreover, the Chinese medicinal herb and compound could inhibit the influenza virus by targeting NA [109, 126, 127].
CD83 is a type I transmembrane glycoprotein expressed in mature dendritic cells. It is an activation marker for DCs and has been suggested to be a sialic acid-binding Ig-like lectin adhesion receptor. It is involved in two forms: membrane-bond CD83(mCD83) and soluble CD83(sCD83), mCD83 regulates maturation, activation, and homeostasis, sCD83 have an immune suppressive function [128,129,130,131]. CD83 regulates lymphocyte maturation, activation, homeostasis, and antibody response to immunization and infection [132]. CD83 in lymphocyte homeostasis and antibody production during IAV infection [133]. DCs infected with the influenza virus upregulate proinflammatory cytokines, including CD83, and simultaneously downregulate anti-inflammatory cytokines [134]. We previously used the influenza H9N2 virus-infected mice model and found that NA treatment directly increased CD83 on the cell membrane surface of DCs and enhanced NF-κB signaling. We prove that CD83 is a sialylated protein embedded and masked in the cell membrane. Sialylation of CD83 delivers inhibiting signaling to DCs. NA deactivates the regulatory CD83 pathway by removing sialic acid and releasing excessive cytokines, causing lung injury. Antibody blocking CD83 prevents NA access, or soluble CD83 decoy NA, can mitigate cytokine storms, reduce lung injury induced by the influenza virus, and alleviate influenza symptoms [135] (Fig. 3). The NA-CD83 axis may serve as a new potential target for treating the influenza virus.

Neuraminidase-CD83 axis. When mice were infected with influenza virus, mainly component is neuraminidase, CD83 is a sialylated protein and sialylated CD83 delivers inhibiting signaling to DCs, CD83 expression level was upregulated on dendritic cells and macrophages in the lung. NA removed sialic acid and released superfluous cytokines, frequent causing lung injury; when using anti-CD83Ab for restrained NA across, reduced cytokines production, and reduction of lung injury
Conclusion
In recent years, interest has been aroused in understanding the immuno-modulating mechanisms of influenza virus neuraminidase. NA was a sialidase enzyme that cleaves sialic acids from cell surface proteins and carbohydrate chains on nascent virions. Cytokines are essential for intercellular communication and viral clearance in the immune system, but excessive cytokines can cause severe immune pathology. There are many medications available to prevent and protect against influenza viruses. Cytokine storm is responsible for mortality and morbidity. From virions structure, biology factors (age, obesity), host factor (DNA damage), lung (endothelial cells, lipid, pulmonary surfactant system), and cytokines/chemokines, these factors are involved in affecting immune response. The targeted NA through NA stalk length, NA activity, vaccines, anti-Ab antibodies, and so on. CD83 controls T cell and B cell maturation and regulates immune activity. NA upregulates CD83 expression in DCs and NA deactivates the regulatory CD83 pathway by removing sialic acid and releasing excessive cytokines, causing lung injury, so the path may inform novel and potential clinical strategies to target influenza virus pathogenesis strategically.
Availability of data and materials
Not applicable.
References
Javanian M, Barary M, Ghebrehewet S, Koppolu V, et al. A brief review of influenza virus infection. J Med Virol. 2021;93(8):4638–46.
Gaitonde DY, Moore FC, Morgan MK. Influenza: diagnosis and treatment. Am Fam Phys. 2019;100(12):751–8.
Bridges CB, Kuehnert MJ, Hall CB. Transmission of influenza: implications for control in health care settings. Clin Infect Dis. 2003;37(8):1094–101.
Tamerius J, Nelson MI, Zhou SZ, Viboud C, et al. Global influenza seasonality: reconciling patterns across temperate and tropical regions. Environ Health Perspect. 2011;119(4):439–45.
Kalil AC, Thomas PG. Influenza virus-related critical illness: pathophysiology and epidemiology. Crit Care. 2019;23(1):258.
To J, Torres J. Viroporins in the influenza virus. Cells. 2019;8(7):654.
Zhai SL, Zhang H, Chen SN, Zhou X, et al. Influenza D virus in animal species in Guangdong Province, Southern China. Emerg Infect Dis. 2017;23(8):1392–6.
Centers for Disease C, Prevention. Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1)—United States, May-August 2009. MMWR Morb Mortal Wkly Rep. 2009;58(38):1071–4.
Shaw ML, Stone KL, Colangelo CM, Gulcicek EE, et al. Cellular proteins in influenza virus particles. PLoS Pathog. 2008;4(6):e1000085.
Hutchinson EC, Charles PD, Hester SS, Thomas B, et al. Conserved and host-specific features of influenza virion architecture. Nat Commun. 2014;5:4816.
Tate MD, Job ER, Deng YM, Gunalan V, et al. Playing hide and seek: how glycosylation of the influenza virus hemagglutinin can modulate the immune response to infection. Viruses. 2014;6(3):1294–316.
Reading PC, Tate MD, Pickett DL, Brooks AG. Glycosylation as a target for recognition of influenza viruses by the innate immune system. Adv Exp Med Biol. 2007;598:279–92.
Tumpey TM, Maines TR, Van Hoeven N, Glaser L, et al. A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science. 2007;315(5812):655–9.
McCrone JT, Woods RJ, Martin ET, Malosh RE, et al. Stochastic processes constrain the within and between host evolution of influenza virus. Elife. 2018;7:e35962.
Dadonaite B, Gilbertson B, Knight ML, Trifkovic S, et al. The structure of the influenza A virus genome. Nat Microbiol. 2019;4(11):1781–9.
Sanders CJ, Vogel P, McClaren JL, Bajracharya R, et al. Compromised respiratory function in lethal influenza infection is characterized by the depletion of type I alveolar epithelial cells beyond threshold levels. Am J Physiol Lung Cell Mol Physiol. 2013;304(7):L481–8.
Short KR, Kroeze E, Fouchier RAM, Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis. 2014;14(1):57–69.
Rust MJ, Lakadamyali M, Zhang F, Zhuang X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol. 2004;11(6):567–73.
Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology. 2011;411(2):229–36.
Ohuchi M, Asaoka N, Sakai T, Ohuchi R. Roles of neuraminidase in the initial stage of influenza virus infection. Microbes Infect. 2006;8(5):1287–93.
McAuley JL, Gilbertson BP, Trifkovic S, Brown LE, et al. Influenza virus neuraminidase structure and functions. Front Microbiol. 2019;10:39.
Brown EG. Influenza virus genetics. Biomed Pharmacother. 2000;54(4):196–209.
Li L, Dai S, Gao GF, Wang J. Lattice-translocation defects in specific crystals of the catalytic head domain of influenza neuraminidase. Acta Crystallogr D Struct Biol. 2020;76(Pt 11):1057–64.
Ellis D, Lederhofer J, Acton OJ, Tsybovsky Y, et al. Structure-based design of stabilized recombinant influenza neuraminidase tetramers. Nat Commun. 2022;13(1):1825.
Einav T, Gentles LE, Bloom JD. SnapShot: influenza by the numbers. Cell. 2020;182(2):532-e1.
Jin H, Leser GP, Zhang J, Lamb RA. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997;16(6):1236–47.
Barman S, Adhikary L, Chakrabarti AK, Bernas C, et al. Role of transmembrane domain and cytoplasmic tail amino acid sequences of influenza a virus neuraminidase in raft association and virus budding. J Virol. 2004;78(10):5258–69.
Kundu A, Avalos RT, Sanderson CM, Nayak DP. Transmembrane domain of influenza virus neuraminidase, a type II protein, possesses an apical sorting signal in polarized MDCK cells. J Virol. 1996;70(9):6508–15.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80.
Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17(7):646–53.
Air GM. Influenza neuraminidase. Influenza Other Respir Viruses. 2012;6(4):245–56.
Kumari K, Gulati S, Smith DF, Gulati U, et al. Receptor binding specificity of recent human H3N2 influenza viruses. Virol J. 2007;4:42.
Chandrasekaran A, Srinivasan A, Raman R, Viswanathan K, et al. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol. 2008;26(1):107–13.
Sun Y, Tan Y, Wei K, Sun H, et al. Amino acid 316 of hemagglutinin and the neuraminidase stalk length influence virulence of H9N2 influenza virus in chickens and mice. J Virol. 2013;87(5):2963–8.
Wang X, Zeng Z, Zhang Z, Zheng Y, et al. The appropriate combination of hemagglutinin and neuraminidase prompts the predominant H5N6 highly pathogenic avian influenza virus in birds. Front Microbiol. 2018;9:1088.
Burmeister WP, Ruigrok RW, Cusack S. The 2.2 A resolution crystal structure of influenza B neuraminidase and its complex with sialic acid. EMBO J. 1992;11(1):49–56.
Xu X, Zhu X, Dwek RA, Stevens J, et al. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J Virol. 2008;82(21):10493–501.
Russell RJ, Haire LF, Stevens DJ, Collins PJ, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443(7107):45–9.
Zhu X, Turner HL, Lang S, McBride R, et al. Structural basis of protection against H7N9 influenza virus by human anti-N9 neuraminidase antibodies. Cell Host Microbe. 2019;26(6):729–38.
Krammer F, Smith GJD, Fouchier RAM, Peiris M, et al. Influenza. Nat Rev Dis Primers. 2018;4(1):3.
Wu R, Zhang H, Yang K, Liang W, et al. Multiple amino acid substitutions are involved in the adaptation of H9N2 avian influenza virus to mice. Vet Microbiol. 2009;138(1–2):85–91.
Zhang Z, Hu S, Li Z, Wang X, et al. Multiple amino acid substitutions involved in enhanced pathogenicity of LPAI H9N2 in mice. Infect Genet Evol. 2011;11(7):1790–7.
Kim H, Webster RG, Webby RJ. Influenza virus: dealing with a drifting and shifting pathogen. Viral Immunol. 2018;31(2):174–83.
Morens DM, Taubenberger JK. Making universal influenza vaccines: lessons from the 1918 pandemic. J Infect Dis. 2019;219(Suppl_1):S5–13.
Heaton NS, Sachs D, Chen CJ, Hai R, et al. Genome-wide mutagenesis of influenza virus reveals unique plasticity of the hemagglutinin and NS1 proteins. Proc Natl Acad Sci U S A. 2013;110(50):20248–53.
Das SR, Hensley SE, Ince WL, Brooke CB, et al. Defining influenza A virus hemagglutinin antigenic drift by sequential monoclonal antibody selection. Cell Host Microbe. 2013;13(3):314–23.
Sandbulte MR, Westgeest KB, Gao J, Xu X, et al. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc Natl Acad Sci U S A. 2011;108(51):20748–53.
Sandbulte MR, Gao J, Straight TM, Eichelberger MC. A miniaturized assay for influenza neuraminidase-inhibiting antibodies utilizing reverse genetics-derived antigens. Influenza Other Respir Viruses. 2009;3(5):233–40.
Zeller MA, Chang J, Vincent AL, Gauger PC, et al. Spatial and temporal coevolution of N2 neuraminidase and H1 and H3 hemagglutinin genes of influenza A virus in US swine. Virus Evol. 2021;7(2):veab090.
Kirkpatrick Roubidoux E, McMahon M, Carreno JM, Capuano C, et al. Identification and characterization of novel antibody epitopes on the N2 neuraminidase. mSphere. 2021;6(1):e00958-20.
Benton DJ, Martin SR, Wharton SA, McCauley JW. Biophysical measurement of the balance of influenza a hemagglutinin and neuraminidase activities. J Biol Chem. 2015;290(10):6516–21.
Arai Y, Elgendy EM, Daidoji T, Ibrahim MS, et al. H9N2 influenza virus infections in human cells require a balance between neuraminidase sialidase activity and hemagglutinin receptor affinity. J Virol. 2020;94(18):10–1128.
Benton DJ, Gamblin SJ, Rosenthal PB, Skehel JJ. Structural transitions in influenza haemagglutinin at membrane fusion pH. Nature. 2020;583(7814):150–3.
de Vries E, Du W, Guo H, de Haan CAM. Influenza A virus hemagglutinin-neuraminidase-receptor balance: preserving virus motility. Trends Microbiol. 2020;28(1):57–67.
Cohen M, Zhang XQ, Senaati HP, Chen HW, et al. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol J. 2013;10:321.
Byrd-Leotis L, Cummings RD, Steinhauer DA. The interplay between the host receptor and influenza virus hemagglutinin and neuraminidase. Int J Mol Sci. 2017;18(7):1541.
Creytens S, Pascha MN, Ballegeer M, Saelens X, et al. Influenza neuraminidase characteristics and potential as a vaccine target. Front Immunol. 2021;12:786617.
Guo XJ, Thomas PG. New fronts emerge in the influenza cytokine storm. Semin Immunopathol. 2017;39(5):541–50.
Julkunen I, Melen K, Nyqvist M, Pirhonen J, et al. Inflammatory responses in influenza A virus infection. Vaccine. 2000;19(Suppl 1):S32-7.
Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008;19(1):3–19.
Saenz RA, Quinlivan M, Elton D, Macrae S, et al. Dynamics of influenza virus infection and pathology. J Virol. 2010;84(8):3974–83.
Gu Y, Hsu AC, Pang Z, Pan H, et al. Role of the innate cytokine storm induced by the influenza A virus. Viral Immunol. 2019;32(6):244–51.
Liu Q, Zhou YH, Yang ZQ. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol. 2016;13(1):3–10.
Teijaro JR. The role of cytokine responses during influenza virus pathogenesis and potential therapeutic options. Curr Top Microbiol Immunol. 2015;386:3–22.
Gu Y, Zuo X, Zhang S, Ouyang Z, et al. The mechanism behind influenza virus cytokine storm. Viruses. 2021;13(7):1362.
Li X, Shao M, Zeng X, Qian P, et al. Signaling pathways in the regulation of cytokine release syndrome in human diseases and intervention therapy. Signal Transduct Target Ther. 2021;6(1):367.
Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383(23):2255–73.
Karki R, Kanneganti TD. The “cytokine storm”: molecular mechanisms and therapeutic prospects. Trends Immunol. 2021;42(8):681–705.
Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3:499–522.
Le VB, Schneider JG, Boergeling Y, Berri F, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med. 2015;191(7):804–19.
Joseph C, Togawa Y, Shindo N. Bacterial and viral infections associated with influenza. Influenza Other Respir Viruses. 2013;7(Suppl 2):105–13.
Zhou Y, Fu X, Liu X, Huang C, et al. Use of corticosteroids in influenza-associated acute respiratory distress syndrome and severe pneumonia: a systemic review and meta-analysis. Sci Rep. 2020;10(1):3044.
Klomp M, Ghosh S, Mohammed S, Nadeem KM. From virus to inflammation, how influenza promotes lung damage. J Leukoc Biol. 2021;110(1):115–22.
Keef E, Zhang LA, Swigon D, Urbano A, et al. Discrete dynamical modeling of influenza virus infection suggests age-dependent differences in immunity. J Virol. 2017;91(23):10–1128.
Tanner AR, Dorey RB, Brendish NJ, Clark TW. Influenza vaccination: protecting the most vulnerable. Eur Respir Rev. 2021;30(159):200258.
Thompson WW, Shay DK, Weintraub E, Brammer L, et al. Influenza-associated hospitalizations in the United States. JAMA. 2004;292(11):1333–40.
Jefferson T, Jones MA, Doshi P, Del Mar CB, et al. Neuraminidase inhibitors for preventing and treating influenza in adults and children. Cochrane Database Syst Rev. 2014;4:CD008965.
Coates BM, Staricha KL, Koch CM, Cheng Y, et al. Inflammatory monocytes drive influenza A virus-mediated lung injury in juvenile mice. J Immunol. 2018;200(7):2391–404.
Jefferson T, Rivetti A, Di Pietrantonj C, Demicheli V. Vaccines for preventing influenza in healthy children. Cochrane Database Syst Rev. 2018;2:CD004879.
Lim HK, Huang SXL, Chen J, Kerner G, et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med. 2019;216(9):2038–56.
Hulme KD, Noye EC, Short KR, Labzin LI. Dysregulated inflammation during obesity: driving disease severity in influenza virus and SARS-CoV-2 infections. Front Immunol. 2021;12:770066.
Milner JJ, Beck MA. The impact of obesity on the immune response to infection. Proc Nutr Soc. 2012;71(2):298–306.
Shaikh S, Haas K, Beck M, Teague H. The effects of diet-induced obesity on B cell function. Clin Exp Immunol. 2015;179(1):90–9.
Bhattacharya I, Ghayor C, Perez Dominguez A, Weber FE. From influenza virus to novel corona virus (SARS-CoV-2)—the contribution of obesity. Front Endocrinol (Lausanne). 2020;11:556962.
Brooke CB, Ince WL, Wrammert J, Ahmed R, et al. Most influenza a virions fail to express at least one essential viral protein. J Virol. 2013;87(6):3155–62.
Kosik I, Yewdell JW. Influenza hemagglutinin and neuraminidase: Yin(–)Yang Proteins coevolving to thwart immunity. Viruses. 2019;11(4):346.
McCraw DM, Gallagher JR, Torian U, Myers ML, et al. Structural analysis of influenza vaccine virus-like particles reveals a multicomponent organization. Sci Rep. 2018;8(1):10342.
Smith BJ, Huyton T, Joosten RP, McKimm-Breschkin JL, et al. Structure of a calcium-deficient form of influenza virus neuraminidase: implications for substrate binding. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 9):947–52.
Long JS, Mistry B, Haslam SM, Barclay WS. Host and viral determinants of influenza A virus species specificity. Nat Rev Microbiol. 2019;17(2):67–81.
Li N, Parrish M, Chan TK, Yin L, et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell Mol Life Sci. 2015;72(15):2973–88.
Yip TF, Selim ASM, Lian I, Lee SM. Advancements in host-based interventions for influenza treatment. Front Immunol. 2018;9:1547.
Martin BE, Harris JD, Sun J, Koelle K, et al. Cellular co-infection can modulate the efficiency of influenza A virus production and shape the interferon response. PLoS Pathog. 2020;16(10):e1008974.
Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146(6):980–91.
Robinson KM, Kolls JK, Alcorn JF. The immunology of influenza virus-associated bacterial pneumonia. Curr Opin Immunol. 2015;34:59–67.
Gallucci RM, Sloan DK, Heck JM, Murray AR, et al. Interleukin 6 indirectly induces keratinocyte migration. J Invest Dermatol. 2004;122(3):764–72.
Novak ML, Koh TJ. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol. 2013;183(5):1352–63.
Crane MJ, Xu Y, Henry WL Jr, Gillis SP, et al. Pulmonary influenza A virus infection leads to suppression of the innate immune response to dermal injury. PLoS Pathog. 2018;14(8):e1007212.
Boniakowski AE, Kimball AS, Joshi A, Schaller M, et al. Murine macrophage chemokine receptor CCR2 plays a crucial role in macrophage recruitment and regulated inflammation in wound healing. Eur J Immunol. 2018;48(9):1445–55.
Voelker DR, Numata M. Phospholipid regulation of innate immunity and respiratory viral infection. J Biol Chem. 2019;294(12):4282–9.
Li N, Ren A, Wang X, Fan X, et al. Influenza viral neuraminidase primes bacterial coinfection through TGF-beta-mediated expression of host cell receptors. Proc Natl Acad Sci U S A. 2015;112(1):238–43.
Koutsakos M, Kedzierska K, Subbarao K. Immune responses to avian influenza viruses. J Immunol. 2019;202(2):382–91.
Ma C, Li Y, Zong Y, Velkov T, et al. p21 restricts influenza A virus by perturbing the viral polymerase complex and upregulating type I interferon signaling. PLoS Pathog. 2022;18(2):e1010295.
Liu S, Yan R, Chen B, Pan Q, et al. Influenza virus-induced robust expression of SOCS3 contributes to excessive production of IL-6. Front Immunol. 2019;10:1843.
Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–59.
Walsh KB, Teijaro JR, Wilker PR, Jatzek A, et al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci U S A. 2011;108(29):12018–23.
Teijaro JR, Walsh KB, Rice S, Rosen H, et al. Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc Natl Acad Sci U S A. 2014;111(10):3799–804.
Smee DF, Huffman JH, Morrison AC, Barnard DL, et al. Cyclopentane neuraminidase inhibitors with potent in vitro anti-influenza virus activities. Antimicrob Agents Chemother. 2001;45(3):743–8.
Jia R, Zhang J, Bertagnin C, Cherukupalli S, et al. Discovery of highly potent and selective influenza virus neuraminidase inhibitors targeting 150-cavity. Eur J Med Chem. 2021;212:113097.
Wang HX, Zeng MS, Ye Y, Liu JY, et al. Antiviral activity of puerarin as potent inhibitor of influenza virus neuraminidase. Phytother Res. 2021;35(1):324–36.
Davidson S. Treating influenza infection, from now and into the future. Front Immunol. 2018;9:1946.
Wang W, Wu J, Zhang X, Hao C, et al. Inhibition of influenza A virus infection by fucoidan targeting viral neuraminidase and cellular EGFR pathway. Sci Rep. 2017;7:40760.
Dormitzer PR, Galli G, Castellino F, Golding H, et al. Influenza vaccine immunology. Immunol Rev. 2011;239(1):167–77.
Pliasas VC, Menne Z, Aida V, Yin JH, et al. A novel neuraminidase virus-like particle vaccine offers protection against heterologous H3N2 influenza virus infection in the porcine model. Front Immunol. 2022;13:915364.
Kosik I, Angeletti D, Gibbs JS, Angel M, et al. Neuraminidase inhibition contributes to influenza A virus neutralization by anti-hemagglutinin stem antibodies. J Exp Med. 2019;216(2):304–16.
Broecker F, Zheng A, Suntronwong N, Sun W, et al. Extending the stalk enhances immunogenicity of the influenza virus neuraminidase. J Virol. 2019;93(18):10–1128.
Stadlbauer D, Zhu X, McMahon M, Turner JS, et al. Broadly protective human antibodies that target the active site of influenza virus neuraminidase. Science. 2019;366(6464):499–504.
Krammer F, Fouchier RAM, Eichelberger MC, Webby RJ, et al. NAction! How can neuraminidase-based immunity contribute to better influenza virus vaccines? MBio. 2018;9(2):10–128.
Eichelberger MC, Wan H. Influenza neuraminidase as a vaccine antigen. Curr Top Microbiol Immunol. 2015;386:275–99.
Wang Y, Lei R, Nourmohammad A, Wu NC. Antigenic evolution of human influenza H3N2 neuraminidase is constrained by charge balancing. Elife. 2021;10:e72516.
Liu X, Luo W, Zhang B, Lee YG, et al. Design of neuraminidase-targeted imaging and therapeutic agents for the diagnosis and treatment of influenza virus infections. Bioconjug Chem. 2021;32(8):1548–53.
Strohmeier S, Amanat F, Zhu X, McMahon M, et al. A novel recombinant influenza virus neuraminidase vaccine candidate stabilized by a measles virus phosphoprotein tetramerization domain provides robust protection from virus challenge in the mouse model. MBio. 2021;12(6):e0224121.
Tan J, O’Dell G, Hernandez MM, Sordillo EM, et al. Human anti-neuraminidase antibodies reduce airborne transmission of clinical influenza virus isolates in the guinea pig model. J Virol. 2022;96(2):e0142121.
Strohmeier S, Amanat F, Carreno JM, Krammer F. Monoclonal antibodies targeting the influenza virus N6 neuraminidase. Front Immunol. 2022;13:944907.
Wong SS, DeBeauchamp J, Zanin M, Sun Y, et al. H5N1 influenza vaccine induces a less robust neutralizing antibody response than seasonal trivalent and H7N9 influenza vaccines. NPJ Vaccines. 2017;2:16.
Kirkpatrick Roubidoux E, Sano K, McMahon M, Carreno JM, et al. Novel epitopes of the influenza virus N1 neuraminidase targeted by human monoclonal antibodies. J Virol. 2022;96(9):e0033222.
Kutkat O, Kandeil A, Moatasim Y, Elshaier Y, et al. In vitro and in vivo antiviral studies of new heteroannulated 1,2,3-triazole glycosides targeting the neuraminidase of influenza A viruses. Pharmaceuticals (Basel). 2022;15(3):351.
Xiao M, Xu L, Lin D, Lian W, et al. Design, synthesis, and bioassay of 4-thiazolinone derivatives as influenza neuraminidase inhibitors. Eur J Med Chem. 2021;213:113161.
Li Z, Ju X, Silveira PA, Abadir E, et al. CD83: activation marker for antigen presenting cells and its therapeutic potential. Front Immunol. 2019;10:1312.
Lechmann M, Berchtold S, Hauber J, Steinkasserer A. CD83 on dendritic cells: more than just a marker for maturation. Trends Immunol. 2002;23(6):273–5.
Scholler N, Hayden-Ledbetter M, Hellstrom KE, Hellstrom I, et al. CD83 is an I-type lectin adhesion receptor that binds monocytes and a subset of activated CD8+ T cells [corrected]. J Immunol. 2001;166(6):3865–72.
Zhou LJ, Tedder TF. Human blood dendritic cells selectively express CD83, a member of the immunoglobulin superfamily. J Immunol. 1995;154(8):3821–35.
Breloer M, Fleischer B. CD83 regulates lymphocyte maturation, activation and homeostasis. Trends Immunol. 2008;29(4):186–94.
Akauliya M, Gautam A, Maharjan S, Park BK, et al. CD83 expression regulates antibody production in response to influenza A virus infection. Virol J. 2020;17(1):194.
Wu Y, Mao H, Ling MT, Chow KH, et al. Successive influenza virus infection and Streptococcus pneumoniae stimulation alter human dendritic cell function. BMC Infect Dis. 2011;11:201.
Ma N, Li X, Jiang H, Dai Y, et al. Influenza virus neuraminidase engages CD83 and promotes pulmonary injury. J Virol. 2021;95(3):10–128.
Funding
This study was supported by the Foundation of People's Hospital of Dayi Country, Luzhou Science and Technology Program and Southwest Medical University (No.2019LZXNYDJ33). Funders had no role in study design, literature collection, review, analyses, interpretation, writing of the report, and so on.
Author information
Authors and Affiliations
Contributions
HJ and ZZ performed the literature search and drafted the manuscript. ZZ designed the manuscript. HJ generated figures. ZZ revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, H., Zhang, Z. Immune response in influenza virus infection and modulation of immune injury by viral neuraminidase. Virol J 20, 193 (2023). https://doi.org/10.1186/s12985-023-02164-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-023-02164-2