
Abstract
Background
Bats were identified as a natural reservoir of emerging and re-emerging infectious pathogens threatening human health and life.
Methods
This study collected 21 fecal samples of Hipposideros armiger in Mengla County of Xishuangbanna Prefecture Yunnan Province to combine one pool for viral metagenomic sequencing.
Results
Two nearly complete genomes of parechoviruses, BPeV11 and BPeV20, were sequenced. Genome analysis revealed that BPeV11 and BPeV20 follow a 3-3-4 genome layout: 5′ UTR-VP0-VP3-VP1-2A-2B-2C-3A-3B-3C-3D-3′ UTR. The prevalence of BPev11 and BPev20 by Nested-PCR showed that 1 of 21 fecal samples was positive. Based on amino acid identity comparison and phylogenetic analysis of P1, 2C, and 3D, BPeV11 and BPeV20 were closely related to but distinct from FPeVs.
Conclusion
It was probably proposed to be a novel species in the genus Parechovirus of the family Picornaviridae. The isolation of BPev11 and BPev20 from H. armiger in China is the first complete genome of parechovirus isolations from bat feces of the genus Hipposideros.
Similar content being viewed by others

Background
The Picornaviridae is a family of viruses with single-stranded, highly diverse positive-sense, non-segmented RNA genomes with a poly(A) tail. The family contains > 30 genera and > 75 species, but many viruses are presently awaiting classification. Picornaviruses may cause subclinical infections of humans and animals or conditions ranging from inapparent or mild febrile illness to severe heart, liver, and central nervous system diseases [1, 2]. Parechovirus, a genus of the family Picornaviridae, was recently classified into six species: Parechovirus A, Parechovirus B, Parechovirus C, Parechovirus D, Parechovirus E, and Parechovirus F. Parechovirus A, including 19 genotypes (HPeV-1 to -19) of Human parechovirus (HPeV) [3], is only a species of Parechovirus genus which can cause various human diseases ranging from asymptomatic or mild gastrointestinal and respiratory illness to severe infections involving the central nervous system[4,5,6]. Parechovirus B includes 6 genotypes of Ljungan virus ((rodent host). Ljungan virus has been associated with the aetiological agent of myocarditis, diabetes, and possibly other human diseases [7, 8]. Parechovirus C [9] (formerly Sebokele virus 1) contains only one genotype. Parechovirus D [10] (ferret parechovirus), Parechovirus E [11] (falcon parechovirus), and Parechovirus F [12] (gecko parechovirus) are the same as Parechovirus C. The knowledge of picornaviruses host range, geographical distribution and genome organization has recently exploded due to the use of high-throughput sequencing and the identification of novel picornaviruses from various species [10].
Bats, the only flying mammal and account for more than 20% of the subsistent mammals, were recently identified as a natural reservoir of emerging and re-emerging infectious pathogens [13], many of which could spillover into animal and human populations, such as severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), coronaviral disease-19, Nipah virus, Hendra virus, and Ebola virus [14, 15]. Hence, investigating viruses in bats is critical for improved control and prevention of large epidemics.
In this study, two nearly complete genomes of parechoviruses were sequenced and analyzed from the fecal samples of Hipposideros armiger in Mengla County of Xishuangbanna Prefecture Yunan Province by metagenomic analysis.
Methods
Sample collection and pool preparation
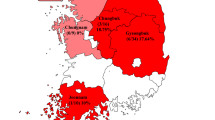
During 2017, a total of 21 fresh fecal samples were collected from wild H. armiger. The study was conducted in Mengla County of Xishuangbanna Prefecture Yunnan province, which is connected with Laos in the east and south and faces Myanmar across lancang River in the west, as shown in Fig. 1. All samples were collected with disposable materials, shipped on dry ice and stored at − 80 °C for further study. The collected fecal samples were mixed into a group, suspended in 600 µl of Dulbecco's phosphate-buffered saline (DPBS), and then vigorously vortex oscillation for 5 min. The 500 µl supernatants were then collected from each pool after centrifugation (5 min, 15,000 g, 4 °C).

Map of the Yunnan province of China showing sampling locations for the current study. Orange coordinates show Mengla County of Xishuangbanna Prefecture Yunnan Province. The county names are shown
Viral metagenomic analysis
The 500 µl supernatant was purified through a 0.45-μm filter (Millipore) to remove eukaryotic and bacterial cell-sized particles. The filtrate was treated for 60 min at 37 °C with a DNases mixture (Turbo DNase from Ambion, Baseline-ZERO from Epicentre), benzonase (Novagen), and RNase (Fermentas) to digest unprotected nucleic acid. Nucleic acids (total DNA and RNA) were then extracted using a QIAamp Viral RNA Mini Kit (QIAGEN) following the manufacturer’s instructions. Total nucleic acids were subjected to RT reactions with SuperScript III reverse transcriptase (Invitrogen), following second-strand cDNA synthesis with Large (Klenow) fragment (NEB). The library was then constructed using Nextera XT DNA Sample Preparation Kit (Illumina) and sequenced using the MiSeq Illumina platform with 250 base pair-ends with dual barcoding.
Bioinformatics analysis
Paired-end reads of 250 bp generated by MiSeq sequencing were debarcoded using vendor software from Illumina. An in-house analysis pipeline running on a 32-node Linux cluster was used to process the data. Reads were considered duplicates if bases 5 to 55 were identical and only one random copy of duplicates was kept. Clonal reads were removed, and low-sequencing-quality tails were trimmed using Phred. Adaptors were trimmed using VecScreen with the default parameters, which uses NCBI BLASTn with specific parameters designed for adapter removal. The cleaned reads were then compared to an in-house non-virus non-redundant (NVNR) protein database to remove false-positive viral hits using DIAMOND BLASTx search with default parameters [16]. The NVNR database was compiled using non-viral protein sequences extracted from an NCBI nr fasta file (based on annotation taxonomy, excluding the virus kingdom). Then, taxonomic classification for DIAMOND results was parsed using MEGAN to perform the LCA-assignment algorithm according to default parameters. Gene assembly, prediction, and annotation were completed with Geneious software [17].
Nested PCR
Nested PCR was performed using rTaq DNA Polymerase (Takara) to amplify complete DNA or RNA and determine whether exist the target viruses. The specific primer sequences are shown in Table 1.
Phylogenetic analysis
The predicted potential proteins were aligned with their corresponding homologs of reference viruses using the MUSCLE multiple sequence alignment program with default settings [18]. The RdRp is the only conserved-sequence domain across all RNA viruses and was used for phylogenetic inference [19]. All phylogenetic analysis was performed based on a Bayesian method implemented in MrBayes version 3.2.7 [20, 21]. In the MrBayes analyses, we used two simultaneous runs of Markov chain Monte Carlo sampling, and the runs were terminated upon convergence (standard deviation of the split frequencies < 0.01) [22]. The visualization and beautification of the phylogenetic trees were achieved by Figtree version 1.4.4 (available from http://tree.bio.ed.ac.uk/software/figtree/).
Prediction of protein domains and functions
All protein prediction was conducted by Geneious prime version 2019.2.3 [19]. The conserved domains were determined using the NCBI conserved domain search in combination with the Pfam conserved domain search [23, 24]. The cleavage sites of the BPev11 and BPev20 polyproteins were predicted by sequence alignment comparisons to the polyproteins of other viruses within the genus Parechovirus (See Additional file 1) using Geneious prime version 2019.2.3. The same approach was implemented in order to predict the polyprotein cleavage sites of three other closely related fish picornaviruses (Wenling bighead beaked sandfish picornavirus, Guangdong spotted longbarbel catfish picornavirus, West African lungfish picornavirus, and clownfish picornavirus) that had not previously been annotated [12, 25]. Pairwise genetic comparisons of the aa sequences of the P1, 2C, and 3D regions of BPev11 and BPev20 polyprotein were compared to those parechoviruses (Fig. 2B) using the Sequence Demarcation Tool v1.2 [26], with the MUSCLE alignment option implemented.

Genomic characterization and sequences identity matrices of BPeV11 and BPeV20. A Predicted genome organization of BPeV11 and BPeV20. P1 represents viral structural proteins, and P2 and P3 represent non-structural proteins. Positions of predicted cleavage sites are indicated along the polyprotein (green). Conserved picornaviral amino acid domains are presented with the blue bar. B Sequence identity matrices are shown below for P1, 2C, and 3D regions of BPeV11 and BPeV20 compared with viruses within genus Parechovirus
Result
Identification and prevalence of novel parechovirus sequences
The 21 fecal samples of H. armiger in the Yunnan Province were combined into one pool for viral metagenomic sequencing. The Illumina MiSeq outputted a total of 581,026 of 250-base pair-end reads and then removed duplicate, low-quality, and cellular reads. After de novo assembly, we acquired two contigs that be similar to parechovirus. Then, the two novel parechovirus sequences, BatPeVyn11 (BPeV11) and BatPeVyn20 (BPeV20) were identified by reads mapping. Nested-PCR was employed to investigate the prevalence of BPev11 and BPev20 using a primer pair. Amplicons of approximately 316 and 330 base pairs were obtained from 2 of 21 fecal samples (sample 11 and 20), respectively. The primer sequences are shown in Table 1.
Genomic characterization of BPeV11 and BPeV20
The nearly complete genomic sequence of BPeV11 is 7143 nucleotides (nt) in length, which is within the range found for other picornaviruses (6938–9035 nt). A single ORF (nt 522-7142) was predicted to encode a polyprotein precursor of 2207 aa, which can be artificially divided into three parts: P1, P2, and P3. Similarly, the nearly complete 7096-nt-long RNA genome of BPeV20 has a single ORF (nt 320 -7048), which encodes a polyprotein precursor of 2243 aa (Fig. 2A). BPev11 and BPev20 both follow a 3-3-4 genome layout: 5′ UTR-VP0-VP3-VP1-2A-2B-2C-3A-3B-3C-3D-3′ UTR (Fig. 2A). The Predicted cleavage sites for genes of BPev11 and BPev20 are shown in Fig. 2A and Table 1, based on a Muscle-alignment with fourteen previously annotated parechoviruses showing cleavage sites. In the polyprotein of BPeV11 and BPeV20, five conserved domains could be identified by an NCBI combined with a Pfam conserved domain search (Fig. 2A). The specific sites and lengths are listed in Table 2.
Phylogenetic analysis of BPeV11 and BPeV20
According to BLASTx search, the ORF sequence of BPev11 and BPev20 shared the 50.44% and 50.47% identity at aa level with that of Ferret parechovirus (FPeV) isolate MpPeV1 (GenBank no. NC_034453) collected from Mustela putorius furo, respectively [3]. The 2C region of the BPev11 exhibited the greatest (60%) aa identity to FPeV, while its P1 and 3D displayed 46 and 56% identity to FPeV, respectively (Fig. 2B). For BPev20, the 3D region showed the highest (59%) aa identity to FPeV, while its P1 and 2C displayed 46 and 56% identity to FPeV, respectively (Fig. 2B).
BPev11 and BPev20 were most closely to the genus Parechovirus of picornaviruses. Therefore, the representative members in the genus Parechovirus and other representative genera and species in Picornaviridae were selected as reference strains for the phylogenetic analysis. The phylogenetic relationships between BPev11 and BPev20 and the representative picornaviruses based on aa sequences of the different picornavirus coding regions (P1and 3CD) are shown in Fig. 3. Phylogenetic analysis of the P1 confirmed that BPev11 and BPev20 formed a monophyletic branch with the members within Parechoviruses E (two Ferret parechoviruses and an unclassified parechovirus), while GPeV in Parechovirus F were also involved in this monophyletic clade based on the 2C and 3CD phylogenetic trees. However, BPev11 and BPev20 have distant relatedness to the viruses in same branch. According to the species demarcation criteria proposed by the International Committee on Taxonomy of Viruses (ICTV), the divergence (number of differences per site between sequences) between members of different Parechovirus species ranges from 0.44–0.63 for P1 and 0.34–0.59 for 3CD. This criterion suggests BPev11 and BPev20 are probably classified as representatives of a new species in the Parechovirus genus. Nevertheless, the members of species Parechovirus D and Parechovirus F relatively lack, the identification of new species needs further research.

Phylogenetic tree of BPev11 and BPev20. Phylogenetic relationships between BPev11 and BPev20 and representative members of the genus Parechovirus and representative members of other genera in the family Picornaviridae, based on the predicted amino acid sequences of the different picornavirus coding regions: P1 (1211 aa) and 3D (566 aa). Scale bars indicate amino acid substitutions per site. BPev11 and BPev20 are labelled in red. Nodes with bootstrap values > 70 are noted
Discussion
With the rise of viral metagenomics analysis, new bat-borne viruses have been continuously discovered around the world, including influenza A virus [27], Phlebovirus [28], and Banyangvirus [29]. From the wide geographical locations of the various bat viruses detected so far, it is almost certain that we will see more and more disease outbreaks caused by bat viruses [14]. Bats harbour a more significant proportion of zoonotic viruses per host species than other mammalian orders [30, 31]. In some cases, outbreaks have been linked to bat roosting or foraging in close proximity to human settlements [32,33,34]. The driving factors for the increasing spillover events of bat viruses, particularly bat CoVs, are complex and most likely a combination of bat habitat disruption through climate change, increased urbanization pressure from humans, wildlife trade and animal markets [35, 36]. The SARS CoV outbreak in China, which caused more than 8000 cases of severe respiratory disease in humans resulting in 10% Mortality [37], was linked to Rhinolophus sp. bats and the wildlife trade [38]. Up to today, the virus members from families of Rhabdoviridae, Orthomyxoviridae, Paramyxoviridae, Coronaviridae, Togaviridae, Flaviviridae, Bunyaviridae, Reoviridae, Arenaviridae, Herpesviridae, Picornaviridae, Hepesviridae and Adenoviridae, have been isolated from different bat species [39], but no complete genome of Parechovirus has been found, only a 1080 nt parechovirus contig was recovered in a previous study [40]. As a natural reservoir, the bats deserve more studies to prevent the outbreaks of diseases caused by viruses.
This study collected 21 fecal samples of H. armiger from Mengla County of Xishuangbanna Prefecture, Yunnan province, to process metagenomic analysis, then acquired two nearly complete sequences BPev11 and BPev20. The isolation of BPev11 and BPev20 from H. armiger in China is the first complete genome of parechovirus isolations from bat feces of the genus Hipposideros. Nested-PCR was employed to investigate the prevalence of BPev11 and BPev20 using a primer pair. The positive rates of BPev11 and BPev20 both were 1/21. BPev11 showed an electrophoresis band in sample 11, while BPev20 showed an electrophoresis band in sample 20 (See Additional file 2). Parechovirus is a genus of RNA viruses with a poly(A) tail. Regrettably, after a series of trials, such as a 5′ Rapid Amplification for cDNA End (RACE) PCR and 3’ RACE, the 5’UTR and 3’UTR of BPev11 and BPev20 are not complete. Sequence identity matrices for P1, 2C, and 3D regions (Fig. 2B) and phylogenetic tree for P1 and 3D regions of BPev11 and BPev20 suggest BPev11 and BPev20 may be a novel species of genus Parechovirus. Although BPev11 and BPev20 are more distantly related to the viruses of Parechovirus A and Parechovirus B, further studying the host range restriction and pathogenicity is necessary.
Conclusions
This study found two nearly complete genomes of parechoviruses, BPeV11 and BPeV20, in 21 fecal samples of H. armiger collected in Mengla County of Xishuangbanna Prefecture Yunnan Province, China. The prevalence of BPev11 and BPev20 by Nested-PCR showed that 1 of 21 fecal samples was positive. Based on amino acid identity comparison and phylogenetic analysis of P1, 2C, and 3D, BPeV11 and BPeV20 were closely related to but distinct from FPeVs. It was probably proposed to be a novel species in the genus Parechovirus of the family Picornaviridae. Until now, this is the first time that complete genome of parechovirus has been found in bat feces. However, further research is needed to uncover BPeV11 and BPeV20 in the pathogenic mechanism in animals and humans.
Availability of data and materials
All genome sequences have been deposited into GenBank under accessions OK149219- OK149220. Quality-filtered sequence reads have been deposited in the sequence read archive (SRA) under the accession number SRR15885421.
Abbreviations
- NCBI:
-
National Center for Biotechnology Information
- ICTV:
-
International Committee on Taxonomy of Viruses
- NVNR:
-
Non-virus non-redundant protein database
- BLAST:
-
Basic local alignment search tool
References
Picornaviridae. Virus Taxonomy.
Zell R, Delwart E, Gorbalenya AE, Hovi T, King AMQ, Knowles NJ, Lindberg AM, Pallansch MA, Palmenberg AC, Reuter G, et al. ICTV virus taxonomy profile: picornaviridae. J Gen Virol. 2017;98:2421–2.
Zhirakovskaia E, Tikunov A, Babkin I, Tikunova N. Complete genome sequences of the first parechoviruses a associated with sporadic pediatric acute gastroenteritis in Russia. Infect Genet Evol J Mol Epidemiol Evolut Genet Infect Dis. 2020;80: 104214.
Oberste MS, Gerber SI. Enteroviruses and parechoviruses: echoviruses, coxsackieviruses, and others. In: Kaslow RA, Stanberry LR, Le Duc JW, editors. Viral infections of humans: epidemiology and control. Boston: Springer; 2014. p. 225–52.
Esposito S, Rahamat-Langendoen J, Ascolese B, Senatore L, Castellazzi L, Niesters HG. Pediatric parechovirus infections. J Clin Virol. 2014;60:84–9.
de Crom SC, Rossen JW, van Furth AM, Obihara CC. Enterovirus and parechovirus infection in children: a brief overview. Eur J Pediatr. 2016;175:1023–9.
Niklasson B, Kinnunen L, Hörnfeldt B, Hörling J, Benemar C, Olof Hedlund K, Matskova L, Hyypiä T, Winberg G. A new picornavirus isolated from bank voles (Clethrionomys glareolus). Virology. 1999;255:86–93.
Niklasson B, Samsioe A, Papadogiannakis N, Gustafsson S, Klitz W. Zoonotic Ljungan virus associated with central nervous system malformations in terminated pregnancy. Birth Defects Res A Clin Mol Teratol. 2009;85:542–5.
Sridhar A, Karelehto E, Brouwer L, Pajkrt D, Wolthers KC. Parechovirus a pathogenesis and the enigma of genotype A-3. Viruses. 2019;11:1062.
Olijve L, Jennings L, Walls T. Human parechovirus: an increasingly recognized cause of sepsis-like illness in young infants. Clin Microbiol Rev. 2018;31:e00047.
Pankovics P, Boros Á, Mátics R, Kapusinszky B, Delwart E, Reuter G. Ljungan/Sebokele-like picornavirus in birds of prey, common kestrel (Falco tinnunculus) and red-footed falcon (F. vespertinus). Infect Genet Evol J Mol Epidemiol Evolut Genet Infect Dis. 2017;55:14–9.
Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, Wang W, Eden JS, Shen JJ, Liu L, et al. The evolutionary history of vertebrate RNA viruses. Nature. 2018;556:197–202.
Shi Z. Bat and virus. Protein Cell. 2010;1:109–14.
Wang L-F, Anderson DE. Viruses in bats and potential spillover to animals and humans. Curr Opin Virol. 2019;34:79–89.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Tanca A, Abbondio M, Palomba A, Fraumene C, Manghina V, Cucca F, Fiorillo E, Uzzau S. Potential and active functions in the gut microbiota of a healthy human cohort. Microbiome. 2017;5:79.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, Qin XC, Xu J, Holmes EC, Zhang YZ. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife. 2015;4: e05378.
El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–32.
Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48:D265–8.
Scherbatskoy EC, Subramaniam K, Al-Hussinee L, Imnoi K, Thompson PM, Popov VL, Ng TFF, Kelley KL, Alvarado R, Wolf JC, et al. Characterization of a novel picornavirus isolated from moribund aquacultured clownfish. J Gen Virol. 2020;101:735–45.
Muhire BM, Varsani A, Martin DP. SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE. 2014;9: e108277.
Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, et al. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci U S A. 2012;109:4269–74.
Mourya DT, Yadav PD, Basu A, Shete A, Patil DY, Zawar D, Majumdar TD, Kokate P, Sarkale P, Raut CG, Jadhav SM. Malsoor virus, a novel bat phlebovirus, is closely related to severe fever with thrombocytopenia syndrome virus and heartland virus. J Virol. 2014;88:3605–9.
Kohl C, Brinkmann A, Radonic A, Dabrowski PW, Nitsche A, Muhldorfer K, Wibbelt G, Kurth A. Zwiesel bat banyangvirus, a potentially zoonotic Huaiyangshan banyangvirus (Formerly known as SFTS)-like banyangvirus in Northern bats from Germany. Sci Rep. 2020;10:1370.
Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. Host and viral traits predict zoonotic spillover from mammals. Nature. 2017;546:646–50.
Luis AD, Hayman DTS, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JRC, Mills JN, Timonin ME, Willis CKR, Cunningham AA, et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc Biol Sci. 2013;280:20122753.
Geldenhuys M, Mortlock M, Weyer J, Bezuidt O, Seamark ECJ, Kearney T, Gleasner C, Erkkila TH, Cui H, Markotter W. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS ONE. 2018;13: e0194527.
Memish Z, Mishra N, Olival K, Fagbo S, Kapoor V, Epstein J, AlHakeem R, Durosinloun A, Al Asmari M, Islam A, et al. Middle east respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg Infect Dis J. 1819;2013:19.
Poon LLM, Chu DKW, Chan KH, Wong OK, Ellis TM, Leung YHC, Lau SKP, Woo PCY, Suen KY, Yuen KY, et al. Identification of a novel coronavirus in bats. J Virol. 2005;79:2001–9.
Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P. Global trends in emerging infectious diseases. Nature. 2008;451:990–3.
Keesing F, Belden LK, Daszak P, Dobson A, Harvell CD, Holt RD, Hudson P, Jolles A, Jones KE, Mitchell CE, et al. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature. 2010;468:647–52.
Berger A, Drosten C, Doerr HW, Stürmer M, Preiser W. Severe acute respiratory syndrome (SARS)–paradigm of an emerging viral infection. J Clin Virol Off Publ Pan Am Soc Clin Virol. 2004;29:13–22.
Ge X-Y, Li J-L, Yang X-L, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–8.
Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–45.
Mishra N, Fagbo SF, Alagaili AN, Nitido A, Williams SH, Ng J, Lee B, Durosinlorun A, Garcia JA, Jain K, et al. A viral metagenomic survey identifies known and novel mammalian viruses in bats from Saudi Arabia. PLoS ONE. 2019;14: e0214227.
Acknowledgements
We are grateful for the valuable support from National Key Research and Development Programs of China for Virome in Important Wildlife, Jiangsu Provincial Key Research and Development Projects.
Funding
This research was funded by the National Key Research and Development Programs of China for Virome in Important Wildlife [No. 2017YFC1200201], Jiangsu Provincial Key Research and Development Projects [No. BE2017693].
Author information
Authors and Affiliations
Contributions
WZ conceived the study. JZ and ZY performed most of the experiments. All authors participated in part of the experiments, and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
BPev11 and BPev20 MAFFT-alignment with other parechovirus polyproteins.
Additional file 2.
The result of Nested PCR about BPev11 and BPev20.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zeng, J., Yang, Z., Guo, W. et al. Identification and genome characterization of novel parechovirus sequences from Hipposideros armiger in China. Virol J 19, 80 (2022). https://doi.org/10.1186/s12985-022-01806-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-022-01806-1