
Abstract
Background
Respiratory syncytial virus (RSV), human Rhinovirus (HRV) and human Metapneumo Virus (HMPV) are important viral pathogens causing acute respiratory tract infections in the hospitalized patients. Sensitive and accurate detection of RSV, HRV and HMPV is necessary for clinical diagnosis and treatment.
Results
A locked nucleic acid (LNA)-based multiplex closed one-tube nested real-time RT-PCR (mOTNRT-PCR) assay was developed for simultaneous detection of RSV, HRV and HMPV. The sensitivity, specificity, reproducibility and clinical performance of mOTNRT-PCR were evaluated and compared with individual real time PCR (RT-qPCR) assay using clinical samples. The analytical sensitivity of mOTNRT-PCR assay was 5 copies/reaction for RSV, HRV and HMPV, respectively, and no cross-reaction with other common respiratory viruses was observed. The coefficients of variation (CV) of intra-assay and inter-assay were between 0.51 to 3.67%. Of 398 nasopharyngeal aspirates samples tested, 109 (27.39%), 150 (37.69%) and 44 (11.06%) were positive for RSV, HRV and HMPV, respectively, whereas 95 (23.87%), 137 (34.42%) and 38 (9.55%) were positive for RSV, HRV and HMPV, respectively, by individual RT-qPCR assay. Thirty three samples that were positive by mOTNRT-PCR but negative by RT-qPCR were confirmed as true positives by sequencing using reported traditional two-step nested PCR assay.
Conclusion
mOTNRT-PCR assay reveals extremely higher sensitivity than that of RT-qPCR assay for detecting RSV, HRV and HMPV in clinical settings.
Similar content being viewed by others

Background
Acute respiratory tract infections causes high morbidity and mortality in hospitalized patients and nearly 80% of infections are viral [1,2,3]. Respiratory syncytial virus (RSV), human rhinovirus (HRV) and human metapneumovirus (HMPV) are the main causative agents of acute respiratory tract infections in humans worldwide across all age group [4,5,6]. RSV and HMPV are negative-sense single-stranded RNA viruses of the family Pneumoviridae. HRV is a small and positive single-stranded RNA virus belonging to the Picornaviridae family. RSV, HMPV and HRV can cause disease ranging from mild upper respiratory infection to more severe lower respiratory infection in children and adults. In particularly, the young children, the elderly, severely immunecompromised adults are at high risk of serious disease following RSV, HMPV or HRV infection [4,5,6,7]. Thus, early and accurate detection of RSV, HRV and HMPV is extremely important for clinical diagnosis and treatment.
Multiplex PCR or traditional real time PCR (RT-qPCR) assays have been developed worldwide for the detection of RSV, HRV and HMPV [8,9,10,11,12,13,14]. However, the limit of detection of these assays is not yet adequate for assessing clinical specimens with a low viral load. In the present study, we adopted the design of locked nucleic acid (LNA)-modified primers and develop a multiplex one-tube nested real time RT-PCR (mOTNRT-PCR) assay for simultaneous detection of RSV, HRV and HMPV with the advantages of higher sensitivity, more convenience and cost-effectiveness than individual RT-qPCR.
Results
Sensitivity and specificity for mOTNRT-PCR assay
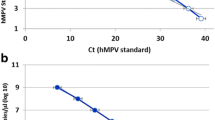
As shown in (Fig. 1a, b and c), the sensitivities of mOTNRT-PCR assay for RSV, HRV and HMPV were 5 copies /reaction. The standard curves acquired from the mOTNRT-PCR was shown in (Fig. 1d). The correlation coefficient (R2) of the standard curves of RSV, HRV and HMPV in multiplex reaction were 0.997, 0.994 and 0.995, respectively. The amplification efficiencies (E) for the different targets were 98.5, 95.7 and 92.5%, respectively, which was considered as acceptable for a multiplex screening assay.

The sensitivity analysis and standard curves of the mOTNRT-PCR assay. a, b, and c are the amplification curves of RSV, HRV and HMPV, respectively using serial 10-fold dilutions of the mixed recombinant plasmids from 108 to 100 copies/μL, and d is the standard curves of the mOTNRT-PCR assay. The R2 of RSV, HRV and HMPV were 0.997, 0.994, 0.995 and the E of RSV, HRV and HMPV were 98.5, 95.7 and 92.5%, respectively
A total of 159 RSV, HRV and HMPV-negative by mOTNRT-PCR out of 398 clinical specimens were used to determine the specificity of mOTNRT-PCR in this study. These samples were previously tested [15] and confirmed to be positive for either of a variety of other respiratory pathogens including, parainfluenza virus (PIV), mycoplasma pneumonia (MP), Chlamydia (including CP and CT), human Bocavirus (HBOV), adenovirus (ADV), coronavirus (COV), Chlamydia, influenza virus types A (Flu A), influenza virus FluA-H3, FluA-H1N1, and influenza virus types B (Flu B). No unspecific amplification or detection of mOTNRT-PCR was observed with these specimens (data not shown), suggesting the high specificity of mOTNRT-PCR assay.
Reproducibility of the mOTNRT-PCR assay
Three different concentration mixtures of 106, 104 and 102 copies/μL of each standard plasmid were respectively tested in triplicates within the same run. The inter-assay variability was evaluated by testing three dilutions of plasmids (106,104 and 102 copies/μL) on three different days within 1 week. The CV values of intra-assay and inter-assay were 0.51–3.66% (Table 1) and 0.83–3.67% (Table 2), respectively. Overall, these results show that the assay is reliable with different Ct values.
Clinical performance of the mOTNRT-PCR vs the RT-qPCR
Totally, 398 nasopharyngeal aspirates (NPAs) selected from children hospitalized with respiratory infection was examined by mOTNRT-PCR assay and previously published RT-qPCR assays [8,9,10]. mOTNRT-PCR assay detected 109 (27.39%) RSV infections with the Ct range from 12.37 to 35.00, 150 (37.69%) HRV with the Ct range from 12.70 to 33.49 and 44 (11.06%) HMPV with the Ct range from 10.79 to 35.00. Among them, 62 (15.58%) virus co-infections were found, including 36 (9.05%) RSV and HRV, 7 (1.76%) RSV and HMPV, 17 (4.27%) HRV and HMPV and 2 (0.50%) RSV, HRV and HMPV, respectively. Whereas RT-qPCR detected 95 (23.87%) RSV infections with the Ct range from 17.04 to 39.56, 137 (34.42%) HRV with the Ct range from 19.52 to 34.59 and 38 (9.55%) HMPV with the Ct range from 13.23 to 38.04. The comparisons between the results of two assays are summarized in Table 3, showing 96.48%, 96.73% and 98.49% agreements in detecting RSV, HRV and HMPV, respectively. Compared with the RT-qPCR assays, the specificities of the mOTNRT-PCR assay in detection of RSV, HRV and HMPV were more than 95% and the sensitivities of the mOTNRT-PCR assay were 100%. Thirty three samples that were positive by mOTNRT-PCR but negative by RT-qPCR were confirmed as true positives by sequencing using the traditional two-step nested PCR assay [16]. The detailed sequencing results were shown in Additional files 1, 2 and 3.
Discussion
In recent years, many ordinary multiple PCR and mono RT-qPCR assays have been routinely applied in the well-equipped laboratory for the diagnosis of RSV, HRV and HMPV. Mono RT-qPCR [11, 12, 14] cannot simultaneously detect RSV, HRV and HMPV infections. Multiple PCR assays [8, 13] enable detection of co-infection but are either more likelihood of cross-contamination or insensitive. The two-step nested PCR assay has the high sensitivity [16], however, is more likelihood of cross-contamination. LNA modification can be performed on any base of the oligonucleotide strand resulting in increased melting temperature and improved amplification sensitivity and specificity [17]. LNA has been used in many applications including the detection of single nucleotide polymorphism (SNP) analyses [18], real-time PCR probes [19], microarray probes [20] and PCR primers [17]. In this study, we used LNA-modified primers to develop a mOTNRT-PCR assay for simultaneous detection of RSV, HRV and HMPV. To our best knowledge, this is the first report on the simultaneous detection of RSV, HRV and HMPV in a closed one-tube using LNA.
In the mOTNRT-PCR assay, the LNA-modified outer primers of RSV, HRV and HMPV had higher annealing temperatures than the inner primers thus possibly allowing an independent reaction during the amplification. The purpose of this design was to maximize the difference in the annealing temperatures (64 °C vs 54 °C) of the outer primer modified by LNA and inner primer sets, allowing one-step nested amplification to be carried out. The 10 °C difference in the melting temperature between outer and inner primers was applicable for developing a one-tube nested PCR successfully in our previous reports [21, 22]. According to the rule of TSP [21, 22], the LNA-outer primers were carried out at a high annealing temperature (64 °C) in the initial cycles of the mOTNRT-PCR, enabling hybridization of only the outer primers, and later cycles were carried out at a lower annealing temperature (54 °C), which enables hybridization of both the inner primers to the amplicons and the antisense oligonucleotides to the outer primers.To minimize the primer competition and ensure that the final amplification products were inner primer products of RSV, HRV and HMPV, the working concentration of each primer were optimized.The reaction conditions of the mOTNRT-PCR assay were also optimized, and this enabled the detection of RSV, HRV and HMPV with extremely high sensitivity (5copies /reaction) while maintaining good specificity as shown in this study (Fig. 1).
This mOTNRT-PCR assay was further evaluated and compared with RT-qPCR assay using 398 clinical samples. As shown in Table 3, the positive sample numbers of RSV, HRV and HMPV detected by RT-qPCR were 95 (23.87%), 137 (34.42%) and 38 (9.55%), respectively. In comparison with RT-qPCR, additional 14 of RSV positive sample,13 of HRV positive sample and 6 of HMPV positive sample were detected only by mOTNRT-PCR assay, respectively. Sequencing of traditional two-step nested PCR [16] product confirmed these were true positives, suggesting the mOTNRT-PCR assay is more sensitive than RT-qPCR. The originally reported sensitivity of RT-qPCR for detecting RSV [8], HRV [9] in a multiplex PCR format and HMPV in mono RT-qPCR [10] was 5 TCID 50 /ml, not shown and 5–10 copies/reaction, respectively. Even we assume the sensitivities in the original research are comparable to the analytical sensitivities in our study, our data of clinical sensitivities are superior to the those obtained from original reports, as evidenced by the difference in the minimal Ct values for RSV (12.37 vs 17.04), HRV (12.70 vs 19.52) and HMPV (10.79 vs 13.23) between mOTNRT-PCR and corresponding RT-qPCR assays.
mOTNRT-PCR assay can be carried out in one closed tube on real-time PCR machines (CFX96 Touch (Bio-Rad), Roche Light Cycler 480 or ABI 7900HT) with similar results (data not shown), indicating the wide adaptability of mOTNRT-PCR assay. The total detection time is 2.7 h for 96 samples and the cost for one sample is approximately $3.9 (excluding nucleic acid extraction), which is about one thirds of that of monoRT-qPCR assays for detection of RSV, HRV and HMPV in separate individual reaction.
Viral co-infection has been described previously in 5–45% of infected patients in different studies [23,24,25]. Co-infection of RSV and HRV were the most frequently found which was consistent with the previous report [25]. However, the clinical severity was vague in patients with single or multiple respiratory virus infection in this study due to the limited access to the clinical information. Future work includes further evaluation of mOTNRT-PCR assay using a large number of samples from different population and integrates the clinical data for more comprehensive analysis,which may be beneficial to the patient’s clinical treatment and prognosis.
Conclusions
We established a mOTNRT-PCR assay allowing simultaneous detection of RSV, HRV, and HMPV. The mOTNRT-PCR assay offers the advantages of higher sensitivity, and more cost-effectiveness than individual RT-qPCR assay, which might be of great potential for clinical use.
Methods
Clinical samples
Totally, 398 nasopharyngeal aspirates (NPAs) were collected from inpatients with respiratory tract infections at Children’s Hospital of Hebei, China, between July, 2017 and February, 2018. Of those 167 (41.96%) were female and 231 (58.04%) were male. Ages were ranged from 36 days to 11 years old and 386 (96.98%) were under 5 years old. 1.8 ml of nasopharyngeal aspiratewas collected in 2 ml of transport mediumand stored at − 80 °C. And the study was conducted with the approval of the Ethics Committee of Children’s hospital of Hebei Province, and written informed consents were obtained from the children’s parents.
Nucleic acid extraction
Total RNA was extracted from 200 μL of clinical sample using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany). The extracts were eluted into 50 μL RNase-free water and stored at − 80 °C.
Primers and probes design for mOTNRT-PCR
Inner primer and probe sequences were derived from previously published RT-qPCR assays targeting nucleoprotein gene for RSV [8], the 5′untranslated region for HRV [9] and the nucleoprotein gene for HMPV [10]. The outer primer sequences of RSV, HRV and HMPV were derived from previously published [8,9,10, 14, 26, 27] with base modifications by LNA [17]. LNA modification can increase maximum annealing temperature of primers [28].
Primers and probes were tested to determine whether they could be used together in a multiplex assay. The specificity of these primer and probe sequences were tested by the BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). And we use oligo7 to evaluate the hairpin of internal primers, primer-dimer potential, G-C content, and melting temperatures of the primers and probes. The primers and probes were obtained from Sangon Biotech (Shanghai, China). The fluorescent reporter dyes for RSV, HRV and HMPV were FAM, Cy5 and VIC, respectively. The sequences of primer and probe are showed in Table 4.
Preparation of plasmid standards
A 374 bp (nt1277–1650, GenBank accession no. MF737204.1), 397 bp fragment (nt84–480, GenBank accession no. EU096006.1) and 475 bp fragment (nt35–509, GenBank accession no.AY145278.1) corresponding to the nucleoprotein gene of RSV, 5′untranslated region of HRV and nucleoprotein gene of HMPV were respectively cloned by TsingKe Biotech Corp (Beijing, China). The recombinant plasmid was quantified using a Qubit ® dsDNA HS Assay Kits (Life technologies Invitrogen). Plasmid copy number was calculated using the following formula: Plasmid copy number = (copy number / μL) = {[6.02 × 1023 × plasmid concentration (ng/μL) × 10− 9]} / [Plasmid length× 660]. 10-fold dilutions of mixed recombinant plasmid ranging from 108 to 100 copies /μl were used as standards for the sensitivity analysis of mOTNRT-PCR.
mOTNRT-PCR amplification
For mOTNRT-PCR assay, the reaction mixture was prepared containing: 5 μL of 5 × PCR buffer, 2μLof One Step RT-PCR Enzyme Mix (Qiagen, Hilden, Germany), 1 mM dNTP mix, 0.1μLof RRI (Takara, Dalian, China), which is the inhibitor of RNase activity, 0.1 μL of each of 0.5 μM RSV, HMPV LNA-outer primer mix, 0.25 μL of 0.5 μM HRV LNA-outer primer mix, 1 μL of each of 5 μM RSV, HMPV inner primer mix, 2.5 μL of 5 μM HRV inner primer mix, 0.5 μL of each of 10 μM RSV, HMPV probe, 1.25μLof 10 μM HRV probe, 5 μL of template nucleic acid and RNAse-free water to reach 25 μL. PCR amplification was performed on the CFX96 Touch (Bio-Rad) and the conditions were: 50 °C for 30 min, a 15 min denaturation step at 95 °C, and 10 cycles of 94 °C for 30s, 64 °C for 40s and 72 °C for 40s, followed by 40 cycles of 94 °C for 30 s, 54 °C for 30s and 72 °C for 30s, with fluorescent readings taken at the the annealing phase of the last 40 cycles. Cycle threshold (Ct) values were calculated using the software at the automatic threshold setting. Positive and negative controls were included in each run. The results were defined as positive if the Ct value was not higher than 35.
Sensitivity and specificity of the mOTNRT-PCR assay
Sensitivity analysis of mOTNRT-PCR assay was carried out using 10-fold dilutions of mixed plasmid ranging from 108 to 100 copies /μl. Multiplex PCR kit (Qiagen, Hilden, Germany) was used in the sensitivity analysis. In a 25 μL reaction system, 12.5 μL of 2 × QIAGEN Multiplex PCR Master Mix, 0.1 μL of each of 0.5 μM RSV, HMPV LNA-outer primer mix, 0.25 μL of 0.5 μM HRV LNA-outer primer mix, 1 μL of each of 5 μM RSV, HMPV inner primer mix, 2.5 μL of 5 μM inner primer mix, 0.5 μL of each of 10 μM RSV, HMPV probe, 1.25 μL of 10 μM HRV probe, 5 μL of mixed plasmid and 0.3 μL RNAse-free water were added. PCR amplification was performed on the CFX96 Touch (Bio-Rad) and the conditions were: a 15 min denaturation step at 95 °C, and 10 cycles of 94 °C for 30s, 64 °C for 40s and 72 °C for 40s, followed by 40 cycles of 94 °C for 30 s, 54 °C for 30s and 72 °C for 30s, with fluorescent readings taken at the the annealing phase of the last 40 cycles.The specificity was evaluated by using 159 RSV, HRV and HMPV-negative by mOTNRT-PCR out of 398 clinical specimens in this study. These specimens were retrospectively tested by Respiratory Pathogen 13 Detection Kit (13× kit) [15].
Reproducibility of inter-assay and intra-assay
The intra-assay coefficients of variation (CV) of this assay was tested using three plasmids (106, 104 and 102 copies /μl) of each standard plasmid in three replicates and inter-assay reproducibility was tested in three different days within a week. The variability of the Ct values was assessed.
Detection of clinical samples
The mOTNRT-PCR assay for the detection of RSV, HRV and HMPV was evaluated using a total of 398 nasopharyngeal aspirates (NPAs) selected from children hospitalized with respiratory infection. For comparison, the previously published RT-qPCR assays [8,9,10] was also performed in parallel.
Sequencing
Sequencing of traditional two-step nested PCR [16] product was used to resolve discrepant results between the two assays. Briefly, the product was sequenced in TsingKe Biotech Corp (Beijing, China) using both forward and reverse primers, respectively [16] and the sequencing results were compared with the sequences in GenBank for pathogen identification by using the BLAST algorithm.
Abbreviations
- ADV:
-
Adenovirus
- COV:
-
Coronavirus
- Ct:
-
Cycle threshold
- CV:
-
Coefficients of variation
- Flu A:
-
Influenza virus types A
- Flu B:
-
Influenza virus types B
- HBOV:
-
Human Bocavirus
- HMPV:
-
Human metapneumo virus
- HRV:
-
Human rhinovirus
- LNA:
-
Locked nucleic acid
- mOTNRT-PCR:
-
Multiplex one-tube nested real time RT-PCR
- MP:
-
Mycoplasma pneumonia
- NPAs:
-
Nasopharyngeal aspirates
- PCR:
-
Polymerase chain reaction
- PIV:
-
Parainfluenza virus
- RSV:
-
Respiratory syncytial virus
- RT-qPCR:
-
Traditional real time PCR
- SNP:
-
Single nucleotide polymorphism
- TSP:
-
Temperature switch PCR
References
Williams BG, Gouws E, Boschi-Pinto C, Bryce J, Dye C. Estimates of world-wide distribution of child deaths from acute respiratory infections. Lancet Infect Dis. 2002;2:25–32.
Kofteridis DP, Papadakis JA, Bouros D, Nikolaides P, Kioumis G, Levidiotou S, Maltezos E, Kastanakis S, Kartali S, Gikas A. Nosocomial lower respiratory tract infections: prevalence and risk factors in 14 Greek hospitals. Eur J Clin Microbiol Infect Dis. 2004;23:888–91.
Mahony JB. Detection of respiratory viruses by molecular methods. Clin Microbiol Rev. 2008;21:716–47.
Simoes EAF. Respiratory syncytial virus infection. Lancet. 1999;354:847–52.
Lu X, Holloway B, Dare RK, Kuypers J, Yagi S, Williams JV, Hall CB, Erdman DD. Real-time reverse transcription-PCR assay for comprehensive detection of human rhinoviruses. J Clin Microbiol. 2009;46:533–9.
Arnott A, Vong S, Sek M, Naughtin M, Beaute J, Rith S, Guillard B, Deubel V, Buchy P. Genetic variability of human metapneumovirus amongst an all ages population in Cambodia between 2007 and 2009. Infect Genet Evol. 2013;15:43–52.
You HL, Chang SJ, Yu HR, Li CC, Chen CH, Liao W. Simultaneous detection of respiratory syncytial virus and human metapneumovirus by one-step multiplex real-time RT-PCR in patients with respiratory symptoms. BMC Pediatr. 2017;17:89.
Sanghavi SK, Bullotta A, Husain S, Rinaldo CR. Clinical evaluation of multiplex real-time PCR panels for rapid detection of respiratory viral infections. J Med Virol. 2012;84:162–9.
Hammitt LL, Kazungu S, Welch S, Bett A, Onyango CO, Gunson RN, Scott JA, Nokes DJ. Added value of an oropharyngeal swab in detection of viruses in children hospitalized with lower respiratory tract infection. J Clin Microbiol. 2011;49:2318–20.
Maertzdorf J, Wang CK, Brown JB, Quinto JD, Chu M, de Graaf M, van den Hoogen BG, Spaete R, Osterhaus AD, Fouchier RA. Real-time reverse transcriptase PCR assay for detection of human metapneumoviruses from all known genetic lineages. J Clin Microbiol. 2004;42:981–6.
Hu A, Colella M, Tam JS, Rappaport R, Cheng SM. Simultaneous detection, subgrouping, and quantitation of respiratory syncytial virus a and B by real-time PCR. J Clin Microbiol. 2003;41:149–54.
Do DH, Laus S, Leber A, Marcon MJ, Jordan JA, Martin JM, Wadowsky RM. A one-step, real-time PCR assay for rapid detection of rhinovirus. J Mol Diagn. 2010;12:102–8.
J L, Mao NY, Zhang C, Yang MJ, Wang M, Xu WB, Ma XJ. The development of a GeXP-based multiplexreverse transcription-PCR assay for simultaneousdetection of sixteen human respiratory virus types/subtypes. BMC Infect Dis. 2012;12:189.
Dare R, Sanghavi S, Bullotta A, Keightley MC, George KS, Wadowsky RM, Paterson DL, McCurry KR, Reinhart TA, Husain S, Rinaldo CR. Diagnosis of human metapneumovirus infection in immunosuppressed lung transplant recipients and children evaluated for pertussis. J Clin Microbiol. 2007;45:548–52.
Zhao MC, Li GX, Zhang D, Zhou HY, Wang H, Yang S, Wang L, Feng ZS, Ma XJ. Clinical evaluation of a new single-tube multiplex reverse transcription PCR assay for simultaneous detection of 11 respiratory viruses, mycoplasma pneumoniae and chlamydia in hospitalized children with acute respiratory infections. Diagn Micr Infec Dis. 2017;88:115–9.
Bellau-Pujol S, Vabret A, Legrand L, Dina J, Gouarin S, Petitjean-Lecherbonnier J, Pozzetto B, Ginevra C, Freymuth F. Development of three multiplex RT-PCR assays for the detection of 12 respiratory RNA viruses. J Virol Methods. 2005;126:53–63.
Ballantyne KN, van Oorschot RAH, Mitchell RJ. Locked nucleic acids in PCR primers increase sensitivity and performance. Genomics. 2008;91:301–5.
Karmakar S, Hrdlicka PJ. DNA strands with alternating incorporations of LNA and 2'-O-(pyren-1-yl)methyluridine: SNP-discriminating RNA detection probes. Chem Sci. 2013;4:3447–54.
Osterback R, Tevaluoto T, Ylinen T, Peltola V, Susi P, Hyypiä T, Waris M. Simultaneous detection and differentiation of human rhino and enteroviruses in clinical specmens by real-time PCR with locked nucleic acid probes. J Clin Microbiol. 2013;51:3960–7.
Castoldi M, Schmidt S, Benes V, Hentze MW, Muckenthaler M. miChip: an array-based method for microRNA expression profiling using locked nucleic acid capture probes. Nat Protoc. 2008;3:321–9.
Niu P, Qi S, Yu B, Zhang C, Wang J, Li Q, Ma X. Development of a highly sensitive real-time nested RT-PCR assay in a single closed tube for detection of enterovirus 71 in hand, foot and mouth disease. Arch Virol. 2016;161:3003–10.
Shen XX, Qiu FZ, Zhao HL, Yang MJ, Hong L, Xu ST, Zhou SF, Li GX, Feng ZS, Ma XJ. A novel and highly sensitive real-time nested RT-PCR assay in a single closed tube for detection of enterovirus. Diagn Microbiol Infect Dis. 2018;90:181–5.
Ebihara T, End o R, Kikuta H, Ishiguro N, Ishiko H, Hara M. Human metapneumovirus infection in Japanese children. J Clin Microbiol. 2004;42:126–32.
van Woensel JB, Bos AP, Lutter R, Rossen JW, Schuurman R. Absence of human metapneumovirus co-infection in cases of severe respiratory syncytial virus infection. Pediatr Pulmonol. 2006;41:872–4.
Ghazaly M, Nadel S. Characteristics of children admitted to intensive care with acute bronchiolitis. Eur J Pediatr. 2018;177:913–20.
Kim H, Kang N, An K, Kim D, Koo J, Kim MS. MRPrimerV: a database of PCR primers for RNA virus detection. Nucleic Acids Res. 2017;(45):D475–81.
Wisdom A, Leitch EC, Gaunt E, Harvala H, Simmonds P. Screening respiratory samples for detection of human rhinoviruses (HRVs) and enteroviruses: comprehensive VP4-VP2 typing reveals high incidence and genetic diversity of HRV species C. J Clin Microbiol. 2009;47:3958–67.
Latorra D, Arar K, Michael Hurley J. Design considerations and effects of LNA in PCR primers. Mol Cell Probes. 2003;17:253–9.
Acknowledgments
We acknowledge the children’s hospital of Hebei Province, China for providing clinical specimen.
Funding
This work was supported by grants from the National key research and development plan of China (2016YFC1202700, 2016YFC1200903 and 2017YFC1200503); China Mega-Project for Infectious Disease (2017ZX10302301–004 and 2017ZX10104001); and Hebei Key Project Plan for Medical Science Research (20180616).
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
XJM, ZSF and GXL designed the study. ZL and JW performed the experiments. MCZ, LW and SXD collected samples. FZQ, RQZ, CC, JJQ and TF analysed the results. LZ wrote this article. XJM revised it. The first three authors in the author list contributed equally in this study. All the authors have read and approved the final version of this manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All aspects of the study were performed in accordance with national ethics regulations and approved by the Institutional Review Boards of National Institute for Viral Disease Control and Prevention, Center for Disease Control and Prevention of China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
The sequence results of RSV. (DOC 142 kb)
Additional file 2:
The sequence results of HRV. (DOC 212 kb)
Additional file 3:
The sequence results of HMPV. (DOC 212 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Feng, Zs., Zhao, L., Wang, J. et al. A multiplex one-tube nested real time RT-PCR assay for simultaneous detection of respiratory syncytial virus, human rhinovirus and human metapneumovirus. Virol J 15, 167 (2018). https://doi.org/10.1186/s12985-018-1061-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-018-1061-0