
Abstract
Background
The human-pathogenic North American West Nile virus strain (WNVNY99), responsible for the outbreak in New York city in 1999, has caused 41000 infections and 1739 human deaths to date. A new strain of West Nile virus emerged in New South Wales, Australia in 2011 (WNVNSW2011), causing a major encephalitic outbreak in horses with close to 1000 cases and 10-15% mortality. Unexpectedly, no human cases have so far been documented.
Findings
We report here, using human monocyte-derived dendritic cells (MoDCs) as a model of initial WNV infection, that the pathogenic New York 99 WNV strain (WNVNY99) replicated better than WNVNSW2011, indicative of increased viral dissemination and pathogenesis in a natural infection. This was attributed to suppressed viral replication and type I interferon (IFN) response in the early phase of WNVNY99 infection, leading to enhanced viral replication at the later phase of infection. In addition, WNVNY99 induced significantly more pro-inflammatory cytokines in MoDCs compared to WNVNSW2011.
Conclusions
Our results suggest that the observed differences in replication and induction of IFN response between WNVNY99 and WNVNSW2011 in MoDCs may be indicative of their difference in virulence for humans.
Similar content being viewed by others

Findings
West Nile virus (WNV) is the leading cause of arboviral encephalitis in the Americas with over 41000 infections and 1739 human deaths (http://www.cdc.gov/westnile/index.html, accessed 25/11/2014) since the outbreak of a human virulent strain in New York in 1999 (WNVNY99 strain) [1,2]. There is currently no effective treatment or approved WNV vaccine for use in humans. A strain of WNV called Kunjin (WNVKUN) has been circulating in Australia since it was discovered in 1960, and have caused very few symptomatic infections in humans or horses [3]. This was until a new strain of WNV emerged in New South Wales in 2011 (WNVNSW2011) causing a major encephalitic outbreak in over 1000 horses [4,5]. WNVNSW2011 has likely emerged through mutations of previously circulating WNVKUN, and gained at least two known virulence determinants found in the human-pathogenic WNVNY99 strain [4]; i) the N-linked glycosylation on the E protein associated with increased virulence in mice [6], and ii) the phenylalanine residue at position 653 of NS5 which is a potent inhibitor of STAT1 phosphorylation [7].
The horses affected by the WNVNSW2011 outbreak presented with similar clinical symptoms to horses infected with WNVNY99, therefore it was rather unexpected that the WNVNSW2011 outbreak had no reported human cases. This prompted us to investigate the viral growth kinetics and immune induction profiles of WNVNSW2011 compared to WNVNY99 using cultures of primary human dendritic cells (DCs) as a model of initial infection in humans [8-11]. Human MoDCs were used as an ex vivo model of initial WNV infection in this study, because it has been shown that large numbers of bone marrow monocytes differentiate into DCs soon after WNV infection in the dermis [12]. Human MoDCs have been previously used to show that WNV replication was required for type I IFN induction, and this was a result of IRF3 translocation to the nucleus after dsRNA stimulation of RIG-1, MDA5 or TLR3 [13]. While comparison of WNV strains has been previously performed in DCs to show that WNV strains with glycosylated envelope (E) protein have increased infection and replication rates [14], WNVNY99 and WNVNSW2011 both have glycosylated E [4], and are therefore expected to be equally efficient in viral entry into MoDCs through attachment to DC-SIGN or DC-SIGNR receptors [11]. Therefore, additional differences must exist that result in either productive infection of DCs or effective viral clearance by the innate immune response.
To investigate this, we used MoDCs by isolating peripheral blood mononuclear cells (PBMCs) from human buffy coat from three donors, which were tested to be negative for arboviral infections, then performed CD14+ magnetic selection to isolate monocytes. The isolated monocytes were cultured with GM-CSF and IL-4 for 6 days to differentiate into immature MoDCs, which were then matured upon WNV infection represented by increased CD80 and CD86 expression. Briefly, at 48 hours post infection (hpi), 78.7% of WNVNY99-infected MoDCs and 84.5% of WNVNSW2011-infected MoDCs showed CD80 upregulation, and 93.9% of WNVNY99-infected MoDCs, 95.8% of WNVNSW2011-infected MoDCs showed CD86 upregulation.
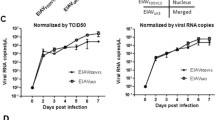
To compare the ability of WNVNY99 and WNVNSW2011 to productively infect these initial target cells, MoDCs were infected with WNVNY99 and WNVNSW2011 (passage 1 stocks grown in Vero76 cells) at a multiplicity of infection (MOI) of 1 and cell culture supernatant and total cellular RNA was harvested at 24, 48 and 72 hpi. Our results revealed that WNVNY99 produced similar titers to WNVNSW2011 at 24 hpi for donor 1 (Figure 1a) and donor 2 (Figure 1c), but less virus compared to WNVNSW2011 for donor 3 (Figure 1e). At later times of infection (48 and 72 hpi), WNVNY99 consistently produced higher titers than WNVNSW2011 (Figure 1a, c, and e). A similar trend was observed by qRT-PCR analysis of intracellular vRNA levels, showing reduced RNA replication for WNVNY99 compared to WNVNSW2011 at 24 hpi, but increased RNA replication for WNVNY99 compared to WNVNSW2011 at 48 and 72 hpi (Figure 1b, d, and f). It was also interesting to note that for all three donors, WNVNSW2011 viral titers and vRNA failed to increase after 24 hpi (Figure 1a-f), indicating that WNVNSW2011 could not productively replicate in MoDCs while WNVNY99 could. In addition, we observed significant variability between human donors, as cells isolated from donor 3 (Figure 1e and f) were more resistant to WNV infection than donors 1 and 2 (Figure 1a-d), which may reflect the high variability in disease symptoms observed in natural human WNV infections [9,15]. When the data from all three donors were combined, WNVNY99 produced significantly more virus particles at 48 and 72 hpi (p < 0.05, Figure 1g), and replicated to significantly higher vRNA levels at 72 hpi (p < 0.05, Figure 1h). This confirms the hypothesis that WNVNY99 was able to replicate more efficiently than WNVNSW2011 in MoDCs, which could possibly translate into increased dissemination in the body, and increased virulence. The enhanced replication of WNVNY99 compared to WNVNSW2011 was similarly shown in previous studies conducted in our laboratory using commonly used immortalised cell lines [4,16].

Replication of WNV NY99 and WNV NSW2011 in human MoDCs. MoDCs from donors 1 (a, b), 2 (c, d) and 3 (e, f) were infected with WNVNY99 and WNVNSW2011 at MOI = 1. Cell culture supernatant was collected at 24, 48 and 72 hours post-infection and virus titer was determined by plaque assay on BHK cells (a, c, e). Total cellular RNA was collected at 24, 48 and 72 hpi and vRNA levels as determined by qRT-PCR were normalised to a combination of three endogenous controls (TBP, GAPDH and PPIA), and was expressed as fold change from uninfected samples (b, d, f). The data from all donors (donors 1 to 3) were combined and error bars represent standard error of the mean (g, h). A Student’s t-test was performed to determine statistical significance at each time point (*: P ≤ 0.05).
It was hypothesized that differences in replication were associated with host innate immune response generated against the viruses. Therefore, qRT-PCR analysis of IFNβ, OAS1, MxA, TNFα, IL-6 and viral RNA (vRNA) expression was performed in the context of WNVNY99 and WNVNSW2011 infections. Interestingly, at the early phase of infection (24 hpi), MoDCs infected with WNVNY99 had reduced levels of vRNA (p < 0.05, Figure 2a), IFNβ mRNA (Figure 2b), OAS1 mRNA (Figure 2c) and MxA mRNA (Figure 2d) compared to MoDCs infected with WNVNSW2011. In contrast, at the later phase of infection (48 and 72 hpi), RNA levels for WNVNY99 were increased (Figure 1h), which likely explains the increased induction of IFNβ (Figure 3a), OAS1 (Figure 3b) and MxA mRNAs (Figure 3c), as well as a significant increase in induction of proinflammatory cytokines TNFα (48 hpi p < 0.05, Figure 3d) and IL-6 (72 hpi p < 0.05, Figure 3e).

Early IFN/ISG response to infection with WNV NY99 and WNV NSW2011 in human MoDCs. MoDCs from three separate donors were infected with WNVNY99 or WNVNSW2011 at MOI = 1 and total cellular RNA was isolated at 24 hours post-infection. vRNA (a), IFNβ (b), OAS1 (c) and MxA (d) mRNA levels as determined by qRT-PCR were normalised to a combination of three endogenous controls (TBP, GAPDH and PPIA), and expressed as fold change from uninfected samples. The results show mean values from three different donors (donors 1 to 3) and error bars represent standard error of the mean. A Student’s t-test was performed to determine statistical significance (*: P ≤ 0.05).

Late IFN/ISG and pro-inflammatory cytokine response to infection with WNV NY99 and WNV NSW2011 in human MoDCs. MoDCs from three separate donors were infected with WNVNY99 or WNVNSW2011 at MOI = 1 and total cellular RNA was isolated at 48 and 72 hours post-infection. IFNβ (a), OAS1 (b), MxA (c), TNFα (d) and IL-6 (e) mRNA levels as determined by qRT-PCR were normalised to a combination of three endogenous controls (TBP, GAPDH and PPIA), and expressed as fold change from uninfected samples. The results show mean values from three different donors (donors 1 to 3) and error bars represent standard error of the mean. A Student’s t-test was performed to determine statistical significance at each time point (*: P ≤ 0.05).
The upregulation of TNFα and IL-6 is likely to occur via viral dsRNA activation of TLR3 [9,17,18]. Studies in vivo have shown that TLR3 knockout mice were more resistant against WNV encephalitis, and this was linked to decreased TNFα and IL-6 expression [18]. The proposed mechanism of this is that TNFα and IL-6 secreted into the bloodstream from infected leukocytes may induce endocytosis and degradation of tight junction proteins (Claudin-1, JAM-1 and occludin) of the blood brain barrier (BBB), causing breakdown of the BBB and allowing WNV entry into the brain [18-22].
Our finding that WNVNY99, but not WNVNSW2011, suppresses early viral RNA replication in MoDCs is supported by previous reports. Scherbik et al. [23] showed that the pathogenic lineage 1 strain of WNV (Eg101), but not the non-pathogenic lineage 2 strain (W956IC), suppressed early viral RNA replication (but increased viral protein expression), resulting in reduced host innate immune responses at the early phase of infection. This effect of early IFN suppression was also shown for pathogenic vs. non-pathogenic Tick Borne Encephalitis virus (TBEV) strains [24]. These two studies suggest that pathogenic viral strains more effectively suppress early viral RNA replication but increase translation, which has three outcomes that favour virulence; i) decreasing the amount of viral dsRNA to decrease IFN induction, ii) increased viral non-structural protein production to block the JAK/STAT signalling cascade, and iii) more effective viral protein-dependent remodelling of cellular membranes which house replication complexes [23,25].
Taken together, our results have identified that a possible factor leading to higher human virulence of WNVNY99 is likely to be decreased viral RNA replication and lower induction of IFNβ, OAS1 and MxA early in MoDCs infection. This likely facilitates the enhanced replication of WNVNY99 vRNA and dissemination of particles later in infection.
References
Roehrig JT. West nile virus in the United States - a historical perspective. Viruses. 2013;5:3088–108.
Brinton MA. The molecular biology of West Nile Virus: a new invader of the western hemisphere. Ann Rev Microbiol. 2002;56:371–402.
Hall RA, Broom AK, Smith DW, Mackenzie JS. The ecology and epidemiology of Kunjin virus. Curr Top Microbiol Immunol. 2002;267:253–69.
Frost MJ, Hick P, Read A, Hobson-Peters J, May FJ, Doggett SL, et al. Characterization of virulent West Nile virus Kunjin strain, Australia, 2011. Emerg Infect Dis. 2012;18:792–800.
Roche SE, Wicks R, Garner MG, East IJ, Paskin R, Moloney BJ, et al. Descriptive overview of the 2011 epidemic of arboviral disease in horses in Australia. Aust Vet J. 2013;91:5–13.
Beasley DW, Whiteman MC, Zhang S, Huang CY, Schneider BS, Smith DR, et al. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J Virol. 2005;79:8339–47.
Laurent-Rolle M, Holbrook MR, Barrett AD, Mason PW, Bloom ME, García-Sastre A, et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J Virol. 2010;84:3503–15.
Johnston LJ, Halliday GM, King NJ. Langerhans Cells Migrate to Local Lymph Nodes Following Cutaneous Infection with an Arbovirus. J Investigative Dermatology. 2000;114:560–8.
Suthar MS, Diamond MS, Gale JM. West Nile virus infection and immunity. Nat Rev Microbiol. 2013;11:115.
Elizabeth JS, Drew W, Nicholas C, Benhur L, Lesley SM, George L, et al. Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J Leukoc Biol. 2002;71:445–57.
Davis CW, Nguyen HY, Hanna SL, Sanchez MD, Doms RW, Pierson TC. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J Virol. 2006;80:1290–301.
Davison AM, King NJ. Accelerated Dendritic Cell Differentiation from Migrating Ly6Clo Bone Marrow Monocytes in Early Dermal West Nile Virus Infection. J Immunol. 2011;186:2382–96.
Silva MC, Guerrero-Plata A, Gilfoy FD, Garofalo RP, Mason PW. Differential activation of human monocyte-derived and plasmacytoid dendritic cells by West Nile virus generated in different host cells. J Virol. 2007;81:13640–8.
Martina BEE, Koraka P, van den Doel P, Rimmelzwaan GF, Haagmans BL, Osterhaus ADME. DC-SIGN enhances infection of cells with glycosylated West Nile virus in vitro and virus replication in human dendritic cells induces production of IFN-alpha and TNF-alpha. Virus Res. 2008;135:64.
Samuel MA, Diamond MS. Pathogenesis of West Nile Virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol. 2006;80:9349–60.
Setoh YX, Prow NA, Rawle DJ, Tan CSE, Edmonds JH, Hall RA, Khromykh AA: Systematic analysis of viral genes responsible for differential virulence between American and Australian West Nile virus strains Journal of general virology 2015, Epub Jan 2015. doi:10.1099/vir.0.000069. [Epub ahead of print]). http://www.ncbi.nlm.nih.gov/pubmed/25626681.
Boehme KW, Compton T. Innate sensing of viruses by toll-like receptors. J Virol. 2004;78:7867–73.
Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–73.
Arjona A, Foellmer HG, Town T, Leng L, McDonald C, Wang T, et al. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest. 2007;117:3059–66.
Diamond MS. West Nile encephalitis virus infection: viral pathogenesis and the host immune response. New York: Springer; 2009.
Xu Z, Waeckerlin R, Urbanowski MD, van Marle G, Hobman TC. West Nile virus infection causes endocytosis of a specific subset of tight junction membrane proteins. PLoS One. 2012;7:e37886.
Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Exp Med. 2010;207:111–26.
Scherbik SV, Pulit-Penaloza JA, Basu M, Courtney SC, Brinton MA. Increased early RNA replication by chimeric West Nile virus W956IC leads to IPS-1-mediated activation of NF-κB and insufficient virus-mediated counteraction of the resulting canonical type I interferon signaling. J Virol. 2013;87:7952–65.
Overby AK, Popov VL, Niedrig M, Weber F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J Virol. 2010;84:8470–83.
Courtney S, Scherbik S, Stockman B, Brinton M. West Nile virus infections suppress early viral RNA synthesis and avoid inducing the cell stress granule response. J Virol. 2012;86:3647–57.
Acknowledgements
We acknowledge the help of the Australian Red Cross Blood Service for providing buffy coat, and Jesse Fyrk (Australian Red Cross Blood Service Research Assistant) for organising and distributing the buffy coat on request. This work was supported by the National Health and Medical Research Council of Australia (APP1045188).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
DR: generation of data, data analysis and interpretation, preparation of MS; YXS: conception and design, data analysis and interpretation, preparation of MS; JE: conception and design, interpretation of data; AK: conception and design, interpretation of data, preparation and revision of MS. All authors read and approved the final manuscript.
Daniel J Rawle and Yin Xiang Setoh contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Rawle, D.J., Setoh, Y.X., Edmonds, J.H. et al. Comparison of attenuated and virulent West Nile virus strains in human monocyte-derived dendritic cells as a model of initial human infection. Virol J 12, 46 (2015). https://doi.org/10.1186/s12985-015-0279-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-015-0279-3