
Abstract
Highly active antiretroviral therapy (HAART) successfully suppresses human immunodeficiency virus (HIV) replication and improves the quality of life of patients living with HIV. However, current HAART does not eradicate HIV infection because an HIV reservoir is established in latently infected cells and is not recognized by the immune system. The successful curative treatment of the Berlin and London patients following bone marrow transplantation inspired researchers to identify an approach for the functional cure of HIV. As a promising technology, gene editing-based strategies have attracted considerable attention and sparked much debate. Herein, we discuss the development of different gene editing strategies in the functional cure of HIV and highlight the potential for clinical applications prospects.
Graphical Abstract

Similar content being viewed by others
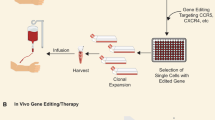
Introduction
Human immunodeficiency virus (HIV) infection remains a major public health problem worldwide, with an estimated 38.0 million people living with HIV (PLWH) as of 2019, including 1.7 million newly infected patients [1]. Although highly active antiretroviral therapy (HAART) can suppress viral replication to undetectable levels and extend the life of PLWH, this approach does not offer a permanent cure because the HIV reservoir cannot be eradicated once it is established [2, 3]. The HIV-1 reservoir comprises full-length, replication-competent, but transcriptionally inactive virus [4]; once HAART is interrupted, this latent HIV reservoir can rebound [5]. Moreover, patients taking long-term HAART may experience major side effects, and the drugs can be expensive, limiting wide application and compliance [6]. Accordingly, achieving long-term host-mediated control of viral replication and remission of the symptoms of HIV-1 infection without long-term administration of HAART, i.e., functional cure of HIV, is an urgent priority in global HIV research.
In 2009, Hütter et al. [7] reported the “Berlin patient,” who had HIV and acute leukemia; after chemotherapy, radiotherapy, and myeloablative allogeneic hematopoietic stem cell transplant (HSCT), this patient was cured of leukemia, and the HIV viral load was undetectable. In a similar case (the “London patient”), the patient was a PLWH with Hodgkin’s lymphoma, and the functional cure of HIV was achieved after administration of HSCT [8]. Notably, both of these patients received HSCT from CCR5 Δ32 homozygous donors [9]. However, owing to the rarity of CCR5 Δ32 gene mutations and tropism changes in HIV strains after cell transplantation, this strategy is difficult to replicate and apply in clinical practice. Despite these limitations, gene editing strategies may be promising for achieving functional cure of HIV.
Gene editing refers to the modification (e.g., deletion, insertion, frameshift mutation) of target genes by nuclease to alter gene function or phenotype. The gene editing techniques used in HIV therapy mainly include RNA interference [RNAi; small interfering RNA (siRNA) and short hairpin RNA (shRNA)]; programmable nuclease-based editing, such as zinc finger nucleases (ZFNs), transcription activator-like (TAL) effector nucleases (TALENs), and clustered regulatory interspaced short palindromic repeat (CRISPR; Fig. 1); and recombinant enzymes in vitro, which may be the most advanced systems currently available for inactivation or eradication of HIV genomes.

Schematic of different gene therapy strategies against HIV. A RNA interference (RNAi) processes the double stranded RNA (dsRNA) region of pathogenic RNA into small or short interfering RNAs (siRNAs), siRNAs bind to cell proteins to form RNA induced silencing complex (RISC), which can cleave foreign RNA sequences from siRNA. B Each ZFN consists of a cleavage domain of FokI, which is fused with a zinc finger protein (ZFP), which has been customized with a gender specific “left” or “right” half site. The combination of two ZFNs makes the dimerization and DNA cleavage possible. C The TALEN elements are targeted to specific DNA sites by DNA recognition module, and then cleaved under the action of FokI nuclease. D CRISPR/Cas system consists of CRISPR sequence elements and Cas gene family. The proteins encoded by these genes have the functional domain of nuclease activity, which can specifically cut the DNA sequence. E Application of different gene editing techniques in HIV eradication
In this review, we summarize the most recent progress in different gene editing technologies and their applications in the functional cure of HIV.
RNAi
RNAi is an endogenous cellular mechanism triggered by double-stranded RNA (dsRNA), which leads to the degradation of homologous RNAs [10]. When exogenous genes, such as viral genes, are randomly integrated into the host cell genome and transcribed, dsRNA is often generated [11, 12]. This dsRNA is recognized by an RNase type III enzyme, Dicer, and cleaved into siRNA, which can subsequently be unwound and assembled by certain enzymes (e.g., endonucleases, exonucleases, and helicases) to form effector complexes called RNA-induced silencing complexes (RISCs) [13, 14]. RISCs can cleave the sequence of the foreign RNA from which the siRNA was derived, thereby preventing translation [15]. Generally, under the control of Pol III promoters, siRNAs can also be expressed in cells using a DNA template to transcribe shRNAs [16, 17].
RNA-based therapeutic approaches are quickly emerging as adjunctive treatment methods for controlling HIV (Table 1). In 2001, Elbashir et al. [18] transfected cells with siRNA to selectively silence corresponding genes in mammalian cells. The findings of their study provided new tools for exploring gene functions of HIV in mammalian cells and for gene-specific therapeutics. Additionally, Park et al. [19] designed six long dsRNAs containing the HIV-1 gag and env genes to study RNAi-mediated gene editing in HIV-1-infected cells; they found that all of these dsRNAs could suppress HIV-1 replication. Chao et al. [20] also demonstrated that HIV-1-enhanced long noncoding RNA silencing using RNAi prevented HIV recrudescence in T cells and microglia upon cessation of azidothymidine treatment in vitro.
Because RNAi has become more widely used in studies of HIV infection, increasing numbers of siRNAs and shRNAs are being tested against HIV-1. For example, Lee et al. [21] detected three shRNAs that targeted three viral sites (rev, gag, and vif) and found that all three of the shRNAs inhibited the replication of the homologous HIVIIIB strain. Novina et al. [22] also showed that siRNAs could inhibit virus production by targeting the mRNAs of the HIV-1 cellular receptor CD4, the viral structural Gag protein, or green fluorescent protein substituted for the Nef regulatory protein.
Notably, a small antisense RNA that targets a conserved region within the HIV-1 long terminal repeat (LTR) can remodel the surrounding chromatin by increasing both histone and DNA methylation and causing loss of nuclear factor-κB recruitment in primary human CD4+ T cells [23]. Furthermore, Turner et al. [24] used a clinically validated lentiviral vector termed HIV7-IGFP and found that LTR-362as could reduce virus replication in cell culture and primary human CD4+ T cells in a dose-dependent manner. Subsequently, the “sh1005/sh516” combination vector, which expressed two anti-HIV shRNAs, i.e., one directed at the HIV coreceptor CCR5 (sh1005) and the other directed at the LTR R region of HIV-1, showed antiviral efficacy against both R5- and X4-tropic HIV-1 in hematopoietic stem/progenitor cells (HSPCs) in a humanized bone marrow/liver/thymus mouse model [25,26,27]. Transplantation of sh1005/sh516-transduced HSPCs resulted in stable marking in hematopoietic lineages and potent inhibition of HIV-1-mediated depletion of modified CD4+ T cells in vivo [28].
Combinations of unmodified and genetically modified cells have been explored in clinical trials [29]. In a study of HSPC-based gene therapy, four patients with acquired immunodeficiency syndrome (AIDS)-related lymphoma were given gene-modified HSPCs transduced with a lentiviral vector encoding three RNA-based anti-HIV-1 moieties (tat/rev shRNA, TAR decoy, and CCR5 ribozyme) in addition to unmodified cells [30]. The treatment was well tolerated, and persistent expression of the introduced siRNA was observed; however, the anti-HIV-1 efficacy could not be determined.
Small RNAs that use RNAi pathways to target HIV-1 have been shown to be successful at inhibiting virus replication and new cell infection in vitro and have been included in all gene combinations that have entered clinical trials to date [31]. Nevertheless, the clinical application of siRNAs has been hindered by their limited cellular uptake and low biological stability, and the use of siRNA nanocarriers may be essential to overcome these barriers [32, 33]. Moreover, HIV-1 can also develop resistance to small RNAs [34]. If further developments in RNAi technology are able to overcome these limitations, curative treatment of HIV may be possible.
ZFNs/TALENs
ZFNs are engineered restriction endonucleases designed to target specific DNA sequences within the genome [35]. Normally, ZFNs are made up of two functional domain modules composed of a programmable zinc-finger array and the nuclease domain of FokI, which are linked together by a linker peptide [36]. The DNA-binding domain and the cleavage domain of the type IIS FokI restriction endonuclease function independently of each other [37]. The zinc finger domain is a common DNA-binding domain encoded in the human genome that binds to DNA in a modular fashion [38]. Additionally, artificial nucleases can generate site-specific double-strand breaks (DSBs) within the genome predominantly by the error-prone nonhomologous end-joining (NHEJ) or homology-directed repair (HDR) pathway to promote genome editing [39]. TALENs are similar to ZFNs owing to the presence of amino acid repeats that are capable of binding DNA and the use of Fok1 as a nuclease effector [40].
To date, numerous studies have evaluated the application of ZFNs and TALENs to edit CCR5 and other genes in an attempt to halt HIV-1 infection (Table 2). In these studies, researchers have used NHEJ to knockout genes in ex vivo autologous cell therapy, and somatic cells are then isolated, modified, and introduced back into the body [41]. Holt et al. used engineered ZFNs to disrupt the CCR5 gene in human HSCs at a mean frequency of 17% of total alleles in a population and demonstrated retention of the ability to engraft NOD/SCID/IL2rγ-null mice (an effective HIV/AIDS model) [42, 43]. Mice transplanted with ZFN-modified HSCs received a rapid selection of CCR5-negative cells and were shown to have significantly lower HIV-1 levels with preservation of human cells throughout their tissues [42]. In another study, Perez et al. [44] used engineered ZFNs to disrupt endogenous CCR5; transient expression of CCR5 ZFN permanently and specifically destroyed approximately 50% of the CCR5 alleles in primary human CD4+ T cells. HIV-1-infected mice transplanted with ZFN-modified CD4+ T cells were then found to have lower viral loads and higher CD4+ T cell counts than mice transplanted with wild-type CD4+ T cells [44]. The same approach has been successfully applied to edit C-X-C chemokine receptor 4 (CXCR4). ZFN-modification of CXCR4 in CD4+ T cells was found to be stable, and HIV-1-infected mice transplanted with CXCR4 ZFN-modified CD4+ T cells showed lower viral loads than mice transplanted with unmodified CD4+ T cells [45].
This approach has also been tested in clinical trials. Tebas et al. recruited 12 patients who had chronic aviremic HIV infection and infused these patients with autologous CD4-enriched T cells modified at the CCR5 gene locus using ZFNs. During the period of HAART interruption, the decrease in circulating CCR5-modified cells (− 1.81 cells/day) was significantly lower than that of unmodified cells (− 7.25 cells/day). In addition, the blood level of HIV DNA decreased in most patients. However, they observed serious adverse events related to the infusion of ZFN-modified autologous CD4+ T cells, which were attributed to a transfusion reaction [46]. The above-mentioned CCR5-specific ZFN developed by Sangamo BioSciences was tested in a phase I clinical study using a recombinant adenoviral vector for delivery (ClinicalTrials.gov number NCT00842634). However, the limited number of DNA targets available owing to restricted binding of zinc finger protein and the cytotoxicity caused by off-target cleavage hinder the development of ZFN-mediated therapies [47].
TALENs represent second-generation designer nucleases that significantly reduce off-target effects and thereby decrease cytotoxicity compared with ZFNs [48]. In a recent study, Shi used sequence analysis of polymerase chain reaction amplicons expressing the target regions for TALENs and ZFNs 48 h post-transfection, showing significant mutations in the CCR2 region of ZFN-treated cells, which had high homology with CCR5, but no mutations in the TALEN-treated cell population [49]. In contrast to ZFNs, TALEN delivery is often achieved using selected recombinant viral vectors, such as adenoviral vectors, adeno-associated virus (AAV) vectors, and lentiviral vectors, for in vivo experiments [50]. AAV-mediated delivery of TALENs and mega TALs (fusion of the TALE binding domain and mega nuclease cleavage domain) was found to enable editing of the CCR5 gene in primary human T cells [51, 52]. However, the adenoviral and lentiviral plasmid vectors harboring TALEN sequences can be easily rearranged after transduction [53]. In addition, few viral vectors have been developed for HIV TALEN transgenes, and further research is still needed [50]. In later studies, Mock et al. introduced a new type of TALEN that can be effectively introduced into T cells through mRNA electroporation (a transient gene transfer technology). Their results showed that this approach resulted in highly efficient knockout of CCR5 (> 90% in PM1 T cells and > 50% in primary T cells) [54]. Nevertheless, further studies are needed to evaluate the potential applications of TALENs in the functional cure of HIV, and the production of TALENs seems to be more challenging than that of ZFNs [55].
CRISPR/Cas9
CRISPR tools are derived from an adaptive defense system found in most bacteria. The bacterial CRISPR/Cas9 system is composed of two elements: the nuclease protein Cas9, which cuts double-stranded DNA, and a single guide RNA (sgRNA) molecule that guides the Cas9 protein to a specific DNA sequence [56]. After cutting the double-strand DNA open, repair can occur through two basic mechanisms, i.e., NHEJ, a mechanism that allows the cell to randomly insert or delete nucleotides at the break site, and HDR, a mechanism that enables insertion of a template DNA to correct mutations at the DNA break site [57, 58]. CRISPR/cas9-induced DSBs are mainly repaired by NHEJ mechanisms [59].
Ebina et al. [60] successfully suppressed HIV-1 gene expression in Jurkat cells by targeting HIV-1 LTR with CRISPR/cas9 for the first time in 2013. Subsequently, the CRISPR/cas9 system has been used in the exploration of HIV treatments (Table 3). For example, Hu et al. [61] found that the CRISPR/Cas9 system can be used to identify the specific targets of complete excision and integration of the pre-HIV genome, leading to inactivation of viral gene expression and replication in HIV latently infected cells; which is a potential therapeutic advance in eliminating barriers of all pro-viruses in HIV-1 infected people. In addition to the HIV provirus, other researchers have focused on the HIV receptor. Indeed, Wang et al. [62] used a lentivirus expressing CCR5-sgRNA and Cas9 to knockout the coreceptor CCR5 in CD4+ T cells, making them resistant to HIV-1. Two different gRNA combinations targeting both CXCR4 and CCR5 were designed by Guo’s team. The CRISPR-sgRNA-Cas9 system successfully induced CXCR4 and CCR5 gene editing in various cell lines and primary CD4+ T cells, indicating that this CRISPR/Cas9 approach could have applications in the functional cure of HIV/AIDS [63].
Using two transgenic mouse models, Kaminski injected plasmid vectors expressing cas9 and various gRNAs into the tail vein or peritoneum. A large basic HIV DNA fragment was excised from the HIV-1 provirus and then detected in the spleen, liver, heart, lungs, and lymphocytes of mice, indicating that integrated HIV-1 provirus could be eliminated in vivo using CRISPR/cas9 in many different cells and tissues [64]. Furthermore, Yin et al. [65] demonstrated the feasibility and efficiency of this approach using AAV combined with multiple sgRNAs and Staphylococcus aureus Cas9 to destroy HIV-1 provirus in three different animal models. These findings established a foundation for the design of clinical trials in humans.
Based on these data showing that CRISPR/Cas9 can be used to edit the pro-HIV genome or CCR5 receptor in vivo and animal models, dsRNA are now attempting to achieve autologous HSCT through gene editing technology in clinical trials. For example, Deng et al. [66] successfully established a CRISPR/Cas9-mediated CCR5 ablating system in long-term HSCs and showed that this system conferred HIV-1 resistance in vivo. Subsequently, the team reported the first case of successful allogeneic transplantation and long-term engraftment of CRISPR/Cas9-edited HSPCs to a patient with HIV and acute leukemia. The patient’s symptoms of leukemia were then reported to be in complete remission [67], demonstrating that long-term persistence of CRISPR-edited allogeneic HSPCs is possible. However, NHEJ repair is error prone and introduces short insertions and deletions (indels), which remain after cas9/sgRNA cleavage and often interfere with the function of the target DNA [68]. Most of these indels are indeed lethal to HIV-1, although some indels have been shown to lead to the emergence of replication-active HIV-1 resistant to cas9/sgRNA [68, 69]. This resistance may accelerate the escape of HIV-1, which could limit the application of cas9/sgRNA in HIV-1 treatment [70].
In vitro-engineered recombinase
Although the CRISPR/cas9 system has some advantages over other technologies, it can cause unpredictable damage via the DNA repair mechanism and can result in virus escape. As an alternative, HIV genome editing may be achieved using engineered recombinase enzymes [71]. As a novel gene editing technology that can safely remove HIV provirus from cells, LTR-specific recombinase (TRE recombinase) was recently reported [72] and may represent a new strategy for HIV eradication. TRE, an engineered version of cyclization recombination enzyme (Cre) recombinase, was designed to target a 34-bp sequence within the HIV-1 LTR (loxLTR) sequence [73]. Expression of TRE in HIV-1-infected cells containing loxLTR sequences results in the removal of the integrated proviral DNA in infected cultured cells [73]. Moreover, TRE-mediated antiviral effects have been demonstrated in TRE-transduced primary CD4+ T cells or TRE-transduced CD34+ HSCs in HIV-infected humanized RAG2−/− γC−/− mice [74].
Because the loxLTR sequence is not highly conserved among different HIV-1 subtypes, it is not suitable as a target for eradication of provirus from most HIV-1-infected individuals. Therefore, Karpinski et al. reported the development and application of broad-spectrum recombinase 1 (brec1), which shows activity against most primary HIV-1 isolates. Brec1 can specifically recognize and recombine a highly conserved target site (loxBTR) located in the LTR sequence of most HIV-1 isolates to remove provirus from HIV-1-infected cells. Indeed, more than 72% of HIV-infected individuals worldwide have HIV-1 subtypes M, A, B, or C, and 90% of these patients are expected to harbor the exact loxbtr sequence targeted by Brec1 [75]. Brec1 is derived from the mature Cre/loxP system, which facilitates directed evolution by substrate linkage. The engineered recombinase has significant advantages over traditional knockouts (e.g., ZFNs, TALENs, and CRISPR/Cas9), for which gene deletion and off-target effects can be lethal. By contrast, engineered recombinases are independent of cellular pathways and do not activate DNA repair pathways during genome editing. Nevertheless, the safety of engineered recombinases and their side effects in edited cells still need to be evaluated.
Discussion and perspective
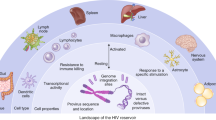
When HIV infects a new host, it spreads to the lymph nodes and blood within 1–2 weeks. During this process, the HIV reservoir is established throughout the whole body, including the central nervous system, lymphoid tissue (i.e., the spleen, thymus, lymph nodes, and intestinal-related lymphoid tissue), bone marrow, lungs, kidneys, liver, adipose tissue, gastrointestinal tract, and urogenital system [76,77,78,79]. The lymph nodes are the main reservoir, with a large number of target cells, high level of activation, and high level of replication, resulting in infection of new cells [80]. Gut-associated lymphoid tissue (GALT) contains 60% of human lymphocytes and plays important roles in the pathogenesis of HIV infection through Th17 cell depletion, bacterial translocation, and local host cell activation [81,82,83,84]. The main reservoirs are resting CD4+ T cells [85]. In addition, other cell types are known to be infected with HIV, establishing the HIV reservoir. For example, macrophages, microglia, and astrocytes in the central nervous system may be infected with HIV, and microglia are considered the main reservoir in the brain [86, 87]. The effectiveness of antiretroviral drugs is anatomically and pharmacologically limited in the brain region, contributing to the persistence of the virus in the brain [88, 89].
There is no standard method to test the HIV reservoir. Some studies have characterized the reservoir based on the level of total HIV DNA [80, 90, 91]. Interestingly, during acute and early HIV infection, gastrointestinal CD4+ T cells have been shown to contain 13-fold higher levels of HIV DNA than blood CD4+ T cells [92]. Additionally, in the intestinal tract, non-CD4 T cells contain less HIV DNA than CD4+ T cells; however, the infection level of non-T leukocytes in the GALT is higher than that in the blood [93]. Although HAART can quickly maintain the viral load at a very low level when activated during acute and chronic infection, the continuous transcription of viral RNA can still be detected in lymphoid tissue at the main site of viral transcription [94]. Therefore, strategies aiming to clear the HIV reservoir are essential for achieving functional cure of HIV. As described in this review, targeted knockout of HIV integration fragments based on gene editing technology prevents the transcription and translation of HIV, thereby suppressing the formation of new virus particles. However, the biggest problem with gene editing technology is efficiency. Whether higher editing efficiency means greater risk of side effects remains unclear, and further studies are needed to verify the effectiveness and safety of these methods.
The editing efficacy and duration of editing effects, particularly in vivo, still warrant improvement, and the safety and off-target effects of these approaches are concerning. Therefore, gene editing technologies are the subject of many ethical discussions. After these disadvantages are overcome, gene editing technology is expected to be a promising approach, particularly when used in combination with other therapies that affect HIV replication. For example, chimeric antigen receptor (CAR) T-cell (CAR-T) therapy, as a new tool to target the HIV reservoir, may be combined with gene editing technology to promote immune system function, and this safe approach may be an effective strategy for achieving functional cure of HIV. Moreover, the combination of autologous cell gene editing and stem cell transplantation could present rejection related to allogeneic stem cell transplantation, and CRISPR/Cas9 combined with HAART can promote the so-called “shock and kill” strategy [95]. Taken together, these studies indicate that gene editing technology combined with other treatment strategies may be an effective approach for achieving functional cure of HIV.
Conclusion
In summary, gene editing technology has improved our understanding of the interactions between the host and virus, enabling the creation of new animal models of HIV infection and providing new strategies for the functional cure of HIV.
Availability of data and materials
Not applicable.
References
WHO. HIV/AIDS data and statistics, 2019.
Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sekaly RP. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15:893–900.
Mellberg T, Gonzalez VD, Lindkvist A, Eden A, Sonnerborg A, Sandberg JK, Svennerholm B, Gisslen M. Rebound of residual plasma viremia after initial decrease following addition of intravenous immunoglobulin to effective antiretroviral treatment of HIV. AIDS Res Ther. 2011;8:21.
Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37:377–88.
Castro-Gonzalez S, Colomer-Lluch M, Serra-Moreno R. Barriers for HIV cure: the latent reservoir. AIDS Res Hum Retrovir. 2018;34:739–59.
Zhou G, Li X, Qiao S, Shen Z, Zhou Y. Influence of side effects on ART adherence among PLWH in China: the moderator role of ART-related knowledge. AIDS Behav. 2018;22:961–70.
Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, Schneider T, Hofmann J, Kucherer C, Blau O, Blau IW, Hofmann WK, Thiel E. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–8.
Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, Martinez-Picado J, Nijhuis M, Wensing AMJ, Lee H, Grant P, Nastouli E, Lambert J, Pace M, Salasc F, Monit C, Innes AJ, Muir L, Waters L, Frater J, Lever AML, Edwards SG, Gabriel IH, Olavarria E. HIV-1 remission following CCR5Delta32/Delta32 haematopoietic stem-cell transplantation. Nature. 2019;568:244–8.
Peterson CW, Kiem HP. Lessons from London and Berlin: designing a scalable gene therapy approach for HIV cure. Cell Stem Cell. 2019;24:685–7.
Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14:475–88.
Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–8.
Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annu Rev Biophys. 2013;42:217–39.
Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–6.
Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32.
Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell. 2002;110:563–74.
Lee NS, Dohjima T, Bauer G, Li H, Li MJ, Ehsani A, Salvaterra P, Rossi J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol. 2002;20:500–5.
Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3.
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8.
Park WS, Miyano-Kurosaki N, Hayafune M, Nakajima E, Matsuzaki T, Shimada F, Takaku H. Prevention of HIV-1 infection in human peripheral blood mononuclear cells by specific RNA interference. Nucleic Acids Res. 2002;30:4830–5.
Chao TC, Zhang Q, Li Z, Tiwari SK, Qin Y, Yau E, Sanchez A, Singh G, Chang K, Kaul M, Karris MAY, Rana TM. The long noncoding RNA HEAL regulates HIV-1 replication through epigenetic regulation of the HIV-1 promoter. MBio. 2019;10:e02016-19.
Lee SK, Dykxhoorn DM, Kumar P, Ranjbar S, Song E, Maliszewski LE, Francois-Bongarcon V, Goldfeld A, Swamy NM, Lieberman J, Shankar P. Lentiviral delivery of short hairpin RNAs protects CD4 T cells from multiple clades and primary isolates of HIV. Blood. 2005;106:818–26.
Novina CD, Murray MF, Dykxhoorn DM, Beresford PJ, Riess J, Lee SK, Collman RG, Lieberman J, Shankar P, Sharp PA. siRNA-directed inhibition of HIV-1 infection. Nat Med. 2002;8:681–6.
Zhou J, Lazar D, Li H, Xia X, Satheesan S, Charlins P, O’Mealy D, Akkina R, Saayman S, Weinberg MS, Rossi JJ, Morris KV. Receptor-targeted aptamer-siRNA conjugate-directed transcriptional regulation of HIV-1. Theranostics. 2018;8:1575–90.
Turner AM, Ackley AM, Matrone MA, Morris KV. Characterization of an HIV-targeted transcriptional gene-silencing RNA in primary cells. Hum Gene Ther. 2012;23:473–83.
Shimizu S, Ringpis GE, Marsden MD, Cortado RV, Wilhalme HM, Elashoff D, Zack JA, Chen IS, An DS. RNAi-mediated CCR5 knockdown provides HIV-1 resistance to memory T cells in humanized BLT mice. Mol Ther Nucleic Acids. 2015;4:e227.
McIntyre GJ, Groneman JL, Yu YH, Jaramillo A, Shen S, Applegate TL. 96 shRNAs designed for maximal coverage of HIV-1 variants. Retrovirology. 2009;6:55.
Suryawanshi GW, Khamaikawin W, Wen J, Shimizu S, Arokium H, Xie Y, Wang E, Kim S, Choi H, Zhang C, Yu H, Presson AP, Kim N, An DS, Chen ISY, Kim S. The clonal repopulation of HSPC gene modified with anti-HIV-1 RNAi is not affected by preexisting HIV-1 infection. Sci Adv. 2020;6:eaay9206.
Ringpis GE, Shimizu S, Arokium H, Camba-Colon J, Carroll MV, Cortado R, Xie Y, Kim PY, Sahakyan A, Lowe EL, Narukawa M, Kandarian FN, Burke BP, Symonds GP, An DS, Chen IS, Kamata M. Engineering HIV-1-resistant T-cells from short-hairpin RNA-expressing hematopoietic stem/progenitor cells in humanized BLT mice. PLoS ONE. 2012;7:e53492.
Zhou J, Rossi JJ. Current progress in the development of RNAi-based therapeutics for HIV-1. Gene Ther. 2011;18:1134–8.
DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A, Mi S, Yam P, Stinson S, Kalos M, Alvarnas J, Lacey SF, Yee JK, Li M, Couture L, Hsu D, Forman SJ, Rossi JJ, Zaia JA. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med. 2010;2:36ra43.
Scarborough RJ, Gatignol A. RNA interference therapies for an HIV-1 functional cure. Viruses. 2017;10:8.
Adesina SK, Akala EO. Nanotechnology approaches for the delivery of exogenous siRNA for HIV therapy. Mol Pharm. 2015;12:4175–87.
Subramanya S, Kim SS, Manjunath N, Shankar P. RNA interference-based therapeutics for human immunodeficiency virus HIV-1 treatment: synthetic siRNA or vector-based shRNA? Expert Opin Biol Ther. 2010;10:201–13.
Rossi JJ, June CH, Kohn DB. Genetic therapies against HIV. Nat Biotechnol. 2007;25:1444–54.
Jabalameli HR, Zahednasab H, Karimi-Moghaddam A, Jabalameli MR. Zinc finger nuclease technology: advances and obstacles in modelling and treating genetic disorders. Gene. 2015;558:1–5.
Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA. 1996;93:1156–60.
Li L, Wu LP, Chandrasegaran S. Functional domains in Fok I restriction endonuclease. Proc Natl Acad Sci USA. 1992;89:4275–9.
Zhu C, Gupta A, Hall VL, Rayla AL, Christensen RG, Dake B, Lakshmanan A, Kuperwasser C, Stormo GD, Wolfe SA. Using defined finger-finger interfaces as units of assembly for constructing zinc-finger nucleases. Nucleic Acids Res. 2013;41:2455–65.
Desjarlais JR, Berg JM. Use of a zinc-finger consensus sequence framework and specificity rules to design specific DNA binding proteins. Proc Natl Acad Sci USA. 1993;90:2256–60.
Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55.
Maeder ML, Gersbach CA. Genome-editing technologies for gene and cell therapy. Mol Ther. 2016;24:430–46.
Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–47.
Watanabe S, Terashima K, Ohta S, Horibata S, Yajima M, Shiozawa Y, Dewan MZ, Yu Z, Ito M, Morio T, Shimizu N, Honda M, Yamamoto N. Hematopoietic stem cell-engrafted NOD/SCID/IL2Rgamma null mice develop human lymphoid systems and induce long-lasting HIV-1 infection with specific humoral immune responses. Blood. 2007;109:212–8.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–16.
Yuan J, Wang J, Crain K, Fearns C, Kim KA, Hua KL, Gregory PD, Holmes MC, Torbett BE. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and enrichment. Mol Ther. 2012;20:849–59.
Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, Holmes MC, Gregory PD, Ando DG, Kalos M, Collman RG, Binder-Scholl G, Plesa G, Hwang WT, Levine BL, June CH. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–10.
Allen AG, Chung CH, Atkins A, Dampier W, Khalili K, Nonnemacher MR, Wigdahl B. Gene editing of HIV-1 co-receptors to prevent and/or cure virus infection. Front Microbiol. 2018;9:2940.
Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng. 2013;110:1811–21.
Shi B, Li J, Shi X, Jia W, Wen Y, Hu X, Zhuang F, Xi J, Zhang L. TALEN-mediated knockout of CCR5 confers protection against infection of human immunodeficiency virus. J Acquir Immune Defic Syndr. 2017;74:229–41.
Benjamin R, Berges BK, Solis-Leal A, Igbinedion O, Strong CL, Schiller MR. TALEN gene editing takes aim on HIV. Hum Genet. 2016;135:1059–70.
Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405.
Sather BD, Romano Ibarra GS, Sommer K, Curinga G, Hale M, Khan IF, Singh S, Song Y, Gwiazda K, Sahni J, Jarjour J, Astrakhan A, Wagner TA, Scharenberg AM, Rawlings DJ. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med. 2015;7:307ra156.
Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C, Recchia A, Cathomen T, Goncalves MA. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63.
Mock U, Machowicz R, Hauber I, Horn S, Abramowski P, Berdien B, Hauber J, Fehse B. mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res. 2015;43:5560–71.
Drake MJ, Bates P. Application of gene-editing technologies to HIV-1. Curr Opin HIV AIDS. 2015;10:123–7.
Sternberg SH, Doudna JA. Expanding the Biologist’s Toolkit with CRISPR-Cas9. Mol Cell. 2015;58:568–74.
DeWitt MA, Corn JE, Carroll D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. 2017;121–122:9–15.
Song M. The CRISPR/Cas9 system: their delivery, in vivo and ex vivo applications and clinical development by startups. Biotechnol Prog. 2017;33:1035–45.
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–55.
Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:2510.
Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, Luo B, Alvarez-Carbonell D, Garcia-Mesa Y, Karn J, Mo X, Khalili K. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci USA. 2014;111:11461–6.
Wang W, Ye C, Liu J, Zhang D, Kimata JT, Zhou P. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS ONE. 2014;9:e115987.
Liu Z, Chen S, Jin X, Wang Q, Yang K, Li C, Xiao Q, Hou P, Liu S, Wu S, Hou W, Xiong Y, Kong C, Zhao X, Wu L, Li C, Sun G, Guo D. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4(+) T cells from HIV-1 infection. Cell Biosci. 2017;7:47.
Kaminski R, Bella R, Yin C, Otte J, Ferrante P, Gendelman HE, Li H, Booze R, Gordon J, Hu W, Khalili K. Excision of HIV-1 DNA by gene editing: a proof-of-concept in vivo study. Gene Ther. 2016;23:696.
Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, Li F, Xiao W, Zhao H, Dai S, Qin X, Mo X, Young WB, Khalili K, Hu W. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol Ther. 2017;25:1168–86.
Xu L, Yang H, Gao Y, Chen Z, Xie L, Liu Y, Liu Y, Wang X, Li H, Lai W, He Y, Yao A, Ma L, Shao Y, Zhang B, Wang C, Chen H, Deng H. CRISPR/Cas9-mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Mol Ther. 2017;25:1782–9.
Xu L, Wang J, Liu Y, Xie L, Su B, Mou D, Wang L, Liu T, Wang X, Zhang B, Zhao L, Hu L, Ning H, Zhang Y, Deng K, Liu L, Lu X, Zhang T, Xu J, Li C, Wu H, Deng H, Chen H. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N Engl J Med. 2019;381:1240–7.
Wang Z, Pan Q, Gendron P, Zhu W, Guo F, Cen S, Wainberg MA, Liang C. CRISPR/Cas9-derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep. 2016;15:481–9.
Yoder KE, Bundschuh R. Host double strand break repair generates HIV-1 strains resistant to CRISPR/Cas9. Sci Rep. 2016;6:29530.
Jern P, Russell RA, Pathak VK, Coffin JM. Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 2009;5:e1000367.
Buchholz F, Hauber J. In vitro evolution and analysis of HIV-1 LTR-specific recombinases. Methods. 2011;53:102–9.
Meinke G, Karpinski J, Buchholz F, Bohm A. Crystal structure of an engineered, HIV-specific recombinase for removal of integrated proviral DNA. Nucleic Acids Res. 2017;45:9726–40.
Sarkar I, Hauber I, Hauber J, Buchholz F. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007;316:1912–5.
Hauber I, Hofmann-Sieber H, Chemnitz J, Dubrau D, Chusainow J, Stucka R, Hartjen P, Schambach A, Ziegler P, Hackmann K, Schrock E, Schumacher U, Lindner C, Grundhoff A, Baum C, Manz MG, Buchholz F, Hauber J. Highly significant antiviral activity of HIV-1 LTR-specific tre-recombinase in humanized mice. PLoS Pathog. 2013;9:e1003587.
Taylor BS, Hammer SM. The challenge of HIV-1 subtype diversity. N Engl J Med. 2008;359:1965–6.
Chaillon A, Gianella S, Dellicour S, Rawlings SA, Schlub TE, De Oliveira MF, Ignacio C, Porrachia M, Vrancken B, Smith DM. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J Clin Invest. 2020;130:1699–712.
Whitney JB, Hill AL, Sanisetty S, Penaloza-MacMaster P, Liu J, Shetty M, Parenteau L, Cabral C, Shields J, Blackmore S, Smith JY, Brinkman AL, Peter LE, Mathew SI, Smith KM, Borducchi EN, Rosenbloom DI, Lewis MG, Hattersley J, Li B, Hesselgesser J, Geleziunas R, Robb ML, Kim JH, Michael NL, Barouch DH. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature. 2014;512:74–7.
Cohen MS, Shaw GM, McMichael AJ, Haynes BF. Acute HIV-1 Infection. N Engl J Med. 2011;364:1943–54.
Mzingwane ML, Tiemessen CT. Mechanisms of HIV persistence in HIV reservoirs. Rev Med Virol. 2017;27:e1924.
Avettand-Fenoel V, Hocqueloux L, Ghosn J, Cheret A, Frange P, Melard A, Viard JP, Rouzioux C. Total HIV-1 DNA, a marker of viral reservoir dynamics with clinical implications. Clin Microbiol Rev. 2016;29:859–80.
Clayton F, Snow G, Reka S, Kotler DP. Selective depletion of rectal lamina propria rather than lymphoid aggregate CD4 lymphocytes in HIV infection. Clin Exp Immunol. 1997;107:288–92.
Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med. 2009;60:471–84.
Guadalupe M, Reay E, Sankaran S, Prindiville T, Flamm J, McNeil A, Dandekar S. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J Virol. 2003;77:11708–17.
Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–31.
Marcello A. Latency: the hidden HIV-1 challenge. Retrovirology. 2006;3:7.
Garcia M, Buzon MJ, Benito JM, Rallon N. Peering into the HIV reservoir. Rev Med Virol. 2018;28:e1981.
Sung JM, Margolis DM. HIV persistence on antiretroviral therapy and barriers to a cure. Adv Exp Med Biol. 2018;1075:165–85.
Canestri A, Lescure FX, Jaureguiberry S, Moulignier A, Amiel C, Marcelin AG, Peytavin G, Tubiana R, Pialoux G, Katlama C. Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin Infect Dis. 2010;50:773–8.
Dahl V, Gisslen M, Hagberg L, Peterson J, Shao W, Spudich S, Price RW, Palmer S. An example of genetically distinct HIV type 1 variants in cerebrospinal fluid and plasma during suppressive therapy. J Infect Dis. 2014;209:1618–22.
Rouzioux C, Tremeaux P, Avettand-Fenoel V. HIV DNA: a clinical marker of HIV reservoirs. Curr Opin HIV AIDS. 2018;13:389–94.
Rouzioux C, Avettand-Fenoel V. Total HIV DNA: a global marker of HIV persistence. Retrovirology. 2018;15:30.
Mehandru S, Poles MA, Tenner-Racz K, Manuelli V, Jean-Pierre P, Lopez P, Shet A, Low A, Mohri H, Boden D, Racz P, Markowitz M. Mechanisms of gastrointestinal CD4+ T-cell depletion during acute and early human immunodeficiency virus type 1 infection. J Virol. 2007;81:599–612.
Yukl SA, Shergill AK, Ho T, Killian M, Girling V, Epling L, Li P, Wong LK, Crouch P, Deeks SG, Havlir DV, McQuaid K, Sinclair E, Wong JK. The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: implications for viral persistence. J Infect Dis. 2013;208:1212–20.
Cadena AM, Ventura JD, Abbink P, Borducchi EN, Tuyishime H, Mercado NB, Walker-Sperling V, Siamatu M, Liu PT, Chandrashekar A, Nkolola JP, McMahan K, Kordana N, Hamza V, Bondzie EA, Fray E, Kumar M, Fischinger S, Shin SA, Lewis MG, Siliciano RF, Alter G, Barouch DH. Persistence of viral RNA in lymph nodes in ART-suppressed SIV/SHIV-infected Rhesus Macaques. Nat Commun. 2021;12:1474.
Saayman SM, Lazar DC, Scott TA, Hart JR, Takahashi M, Burnett JC, Planelles V, Morris KV, Weinberg MS. Potent and targeted activation of latent HIV-1 using the CRISPR/dCas9 activator complex. Mol Ther. 2016;24:488–98.
Choi JG, Bharaj P, Abraham S, Ma H, Yi G, Ye C, Dang Y, Manjunath N, Wu H, Shankar P. Multiplexing seven miRNA-based shRNAs to suppress HIV replication. Mol Ther. 2015;23:310–20.
Ronsard L, Yousif AS, Ramesh J, Sumi N, Gorman M, Ramachandran VG, Banerjea AC. In-vitro subtype-specific modulation of HIV-1 trans-activator of transcription (Tat) on RNAi silencing suppressor activity and cell death. Viruses. 2019;11:976.
Ayala-Suarez R, Diez-Fuertes F, Calonge E, De La Torre Tarazona HE, Gracia-Ruiz de Alda M, Capa L, Alcami J. Insight in miRNome of long-term non-progressors and elite controllers exposes potential RNAi role in restraining HIV-1 infection. J Clin Med. 2020;9:2452.
Kong W, Biswas A, Zhou D, Fiches G, Fujinaga K, Santoso N, Zhu J. Nucleolar protein NOP2/NSUN1 suppresses HIV-1 transcription and promotes viral latency by competing with Tat for TAR binding and methylation. PLoS Pathog. 2020;16:e1008430.
Ru R, Yao Y, Yu S, Yin B, Xu W, Zhao S, Qin L, Chen X. Targeted genome engineering in human induced pluripotent stem cells by penetrating TALENs. Cell Regen. 2013;2:5.
Yi G, Choi JG, Bharaj P, Abraham S, Dang Y, Kafri T, Alozie O, Manjunath MN, Shankar P. CCR5 gene editing of resting CD4(+) T cells by transient ZFN expression from HIV envelope pseudotyped nonintegrating lentivirus confers HIV-1 resistance in humanized mice. Mol Ther Nucleic Acids. 2014;3:e198.
Fadel HJ, Morrison JH, Saenz DT, Fuchs JR, Kvaratskhelia M, Ekker SC, Poeschla EM. TALEN knockout of the PSIP1 gene in human cells: analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. J Virol. 2014;88:9704–17.
Mock U, Riecken K, Berdien B, Qasim W, Chan E, Cathomen T, Fehse B. Novel lentiviral vectors with mutated reverse transcriptase for mRNA delivery of TALE nucleases. Sci Rep. 2014;4:6409.
Strong CL, Guerra HP, Mathew KR, Roy N, Simpson LR, Schiller MR. Damaging the integrated HIV proviral DNA with TALENs. PLoS ONE. 2015;10:e0125652.
Manotham K, Chattong S, Setpakdee A. Generation of CCR5-defective CD34 cells from ZFN-driven stop codon-integrated mesenchymal stem cell clones. J Biomed Sci. 2015;22:25.
DiGiusto DL, Cannon PM, Holmes MC, Li L, Rao A, Wang J, Lee G, Gregory PD, Kim KA, Hayward SB, Meyer K, Exline C, Lopez E, Henley J, Gonzalez N, Bedell V, Stan R, Zaia JA. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol Ther Methods Clin Dev. 2016;3:16067.
Chattong S, Chaikomon K, Chaiya T, Tangkosakul T, Palavutitotai N, Anusornvongchai T, Manotham K. Efficient ZFN-mediated stop codon integration into the ccr5 locus in hematopoietic stem cells: a possible source for intrabone marrow cell transplantation. AIDS Res Hum Retrovir. 2018;34:575–9.
Liu X, Wang M, Qin Y, Shi X, Cong P, Chen Y, He Z. Targeted integration in human cells through single crossover mediated by ZFN or CRISPR/Cas9. BMC Biotechnol. 2018;18:66.
Ji H, Lu P, Liu B, Qu X, Wang Y, Jiang Z, Yang X, Zhong Y, Yang H, Pan H, Zhao L, Xu J, Lu H, Zhu H. Zinc-finger nucleases induced by HIV-1 Tat excise HIV-1 from the host genome in infected and latently infected cells. Mol Ther Nucleic Acids. 2018;12:67–74.
Nerys-Junior A, Braga-Dias LP, Pezzuto P, Cotta-de-Almeida V, Tanuri A. Comparison of the editing patterns and editing efficiencies of TALEN and CRISPR-Cas9 when targeting the human CCR5 gene. Genet Mol Biol. 2018;41:167–79.
Qi C, Jia X, Lu L, Ma P, Wei M. HEK293T cells are heterozygous for CCR5 delta 32 mutation. PLoS ONE. 2016;11:e0152975.
Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med. 2018;215:985–97.
Ye C, Wang W, Cheng L, Li G, Wen M, Wang Q, Zhang Q, Li D, Zhou P, Su L. Glycosylphosphatidylinositol-anchored anti-HIV scFv efficiently protects CD4 T cells from HIV-1 infection and deletion in hu-PBL mice. J Virol. 2017;91:e0138916.
Gaj T, Staahl BT, Rodrigues GMC, Limsirichai P, Ekman FK, Doudna JA, Schaffer DV. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic Acids Res. 2017;45:e98.
Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737.
Yin H, Song CQ, Suresh S, Wu Q, Walsh S, Rhym LH, Mintzer E, Bolukbasi MF, Zhu LJ, Kauffman K, Mou H, Oberholzer A, Ding J, Kwan SY, Bogorad RL, Zatsepin T, Koteliansky V, Wolfe SA, Xue W, Langer R, Anderson DG. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol. 2017;35:1179–87.
Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, Shen AH, Ren C, Esrick EB, Manis JP, Dorfman DM, Williams DA, Biffi A, Brugnara C, Biasco L, Brendel C, Pinello L, Tsai SQ, Wolfe SA, Bauer DE. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. 2019;25:776–83.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Science and Technology Major Project of China (2017ZX09304027), the project of Shanghai Municipal Key Clinical Specialty (no. GWV-10.1-XK02), the Shanghai “Rising stars of Medical Talent” Youth Development Program, Specialist Program (no. 2019-72).
Author information
Authors and Affiliations
Contributions
JX designed the framework and wrote the manuscript. XZ wrote the RNAi part, SG searched documents and made tables. HL and JC revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xun, J., Zhang, X., Guo, S. et al. Editing out HIV: application of gene editing technology to achieve functional cure. Retrovirology 18, 39 (2021). https://doi.org/10.1186/s12977-021-00581-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-021-00581-1