
Abstract
Background
Retroviruses exist as exogenous infectious agents and as endogenous retroviruses (ERVs) integrated into host chromosomes. Such endogenous retroviruses (ERVs) are grouped into three classes roughly corresponding to the seven genera of infectious retroviruses: class I (gamma-, epsilonretroviruses), class II (alpha-, beta-, delta-, lentiretroviruses) and class III (spumaretroviruses). Some ERVs have counterparts among the known infectious retroviruses, while others represent paleovirological relics of extinct or undiscovered retroviruses.
Results
Here we identify an intact ERV in the Anuran amphibian, Xenopus tropicalis. XtERV-S has open reading frames (ORFs) for gag, pol (polymerase) and env (envelope) genes, with a small additional ORF in pol and a serine tRNA primer binding site. It has unusual features and domain relationships to known retroviruses. Analyses based on phylogeny and functional motifs establish that XtERV-S gag and pol genes are related to the ancient env-less class III ERV-L family but the surface subunit of env is unrelated to known retroviruses while its transmembrane subunit is class I-like. LTR constructs show transcriptional activity, and XtERV-S transcripts are detected in embryos after the maternal to zygotic mid-blastula transition and before the late tailbud stage. Tagged Gag protein shows typical subcellular localization. The presence of ORFs in all three protein-coding regions along with identical 5’ and 3’ LTRs (long terminal repeats) indicate this is a very recent germline acquisition. There are older, full-length, nonorthologous, defective copies in Xenopus laevis and the distantly related African bullfrog, Pyxicephalus adspersus. Additional older, internally deleted copies in X. tropicalis carry a 300 bp LTR substitution.
Conclusions
XtERV-S represents a genera-spanning member of the largely env-less class III ERV that has ancient and modern copies in Anurans. This provirus has an env ORF with a surface subunit unrelated to known retroviruses and a transmembrane subunit related to class I gammaretroviruses in sequence and organization, and is expressed in early embryogenesis. Additional XtERV-S-related but defective copies are present in X. tropicalis and other African frog taxa. XtERV-S is an unusual class III ERV variant, and it may represent an important transitional retroviral form that has been spreading in African frogs for tens of millions of years.
Similar content being viewed by others


Background
Retroviruses (RVs) are a diverse family of viruses with seven genera. The alpha-, beta-, gamma-, delta-, epsilon-, lenti-, and spumaRVs are distinguished by variations in sequence, genomic organization and life cycle. RVs replicate through a DNA intermediate generated by the virus-encoded reverse transcriptase (RT) [1]. These DNA copies integrate into the genomes of infected cells and can be passed to progeny cells. RVs thus can exist as infectious virions that can be horizontally transmitted through infection, and as endogenous retroviruses (ERVs) that have integrated into the host germline. ERVs represent the relics of past infections, and up to 10% of vertebrate genomes are RV-derived [2]. ERVs are grouped into three clusters largely based on RT sequence relationships to the infectious Retroviridae: class I (gamma- and epsilonRVs) and class II (alpha-, beta-, delta- and lentiRVs) are orthoRVs, and class III is most closely related to spumaRVs [2]. While many ERVs have counterparts among present day infectious RVs, others do not and serve as paleovirological records of extinct, or so far undiscovered, infectious viruses.
After their acquisition, ERVs are inactivated by mutations acquired at the neutral mutation rate of their host genomes. Over extended evolutionary timescales, ERVs accumulate mutations that render them defective, eventually becoming unrecognizable as RVs. Rarely, some ERV domains can be co-opted by their hosts to serve cellular functions, and these sequences are preserved by purifying selection preventing the mutational decay experienced by genetic sequences under neutral selection [3]. Examples of such domesticated genes include viral envelope (env) genes co-opted to serve in placenta formation (termed syncytins) [4, 5], env and gag genes that can serve anti-viral functions like Fv4 and Fv1 [6, 7], and regulatory sequences that affect host gene expression [8,9,10].
As documented through studies on the expanding number of sequenced genomes, ERVs are widely distributed in vertebrates, and genome analyses have catalogued the viral subtypes present in different species [11,12,13] and have also tracked cross-species transmissions [14,15,16,17]. The identification of ancient paleo-retroviruses encountered in this evolutionary record has led investigators to reconstruct the genomes of their progenitors [18, 19], viruses that may not have extant infectious counterparts.
Frogs are a diverse and mainly carnivorous subgroup of amphibians. They are classed in the vertebrate order Anura which dates to the Permian, 265 million years ago. Frogs show a wide geographic distribution and occupy diverse habitats ranging from the tropics to subarctic regions, although most species are found in tropical rainforests. There are at least 5424 recorded species, making them one of the five most diverse orders of vertebrates [20].
Xenopus, commonly known as the clawed frog, is a genus of aquatic frogs native to sub-Saharan Africa. Of the twenty-nine Xenopus species, the most well-studied are Xenopus laevis and Xenopus tropicalis (formerly Silurana tropicalis). X. laevis has been extensively used as a vertebrate model in developmental biology, cell biology, toxicology, neuroscience and gene expression, but its usefulness in genetic studies and for genetic manipulation has been complicated by its allotetrapoid genome (2n = 36). X. tropicalis offers advantages as an experimental model system as it is a smaller frog with a shorter generation time, and, because it is the only one of the 29 extant Xenopus species with a diploid genome (2n = 20), X. tropicalis was the first Xenopus species selected for genomic sequencing [21]. The subsequent sequencing of X. laevis has extended its utility by providing a model for the evolution of vertebrate polyploidy [22]. Analyses of these two genomes found a high diversity of transposable elements, including four superfamilies of LTR retroelements [21, 22].
In the course of screening non-mammalian vertebrates for conserved and functionally important RV domains, we identified an unusual 8.0 kb ERV in X. tropicalis that we termed XtERV-S because it has a serine tRNA primer binding site (PBS). This ERV has gag, pol and env genes with open reading frames (ORFs), one additional ORF in pol and identical 5’ and 3’ LTRs, suggesting it is a recent germline acquisition. Older intact but defective and nonorthologous copies are also present in X. laevis and the African bullfrog, Pyxicephalus adspersus. XtERV-S is expressed during early development, its Gag protein shows expected cellular localization, and its LTR shows some activity in human 293T cells. Phylogenetic and functional motif comparisons indicate that the XtERV-S pol and gag genes are related to the ancient class III family of ERVs represented by ERV-L. However, XtERV-S, unlike mammalian ERV-Ls, has an env with an ORF. The surface subunit of this env, SUenv, is not related to known RVs although its transmembrane subunit, TMenv, is class I-like. The sequence homologies, presence of viral genus-specific functional motifs, and the distribution of older copies in other African frog species indicates that XtERV-S is a genera-spanning ancestral form that has been circulating in these species for at least 36 million years.
Results
Identification of the X. tropicalis endogenous retrovirus XtERV-S
An intact provirus, XtERV-S, was initially identified in an unplaced scaffold in the sequenced genome of X. tropicalis (NW_016684263.1:c1706-9791 X. tropicalis unplaced genomic scaffold_1181, X._tropicalis_v9.1). A molecular clone of the full-length provirus was assembled from three overlapping PCR products (Fig. 1; Table 1). The 5′ PCR fragment includes flanking sequence that corresponds to the scaffold sequence and maps to chromosome 7 (XTR7; NC_030683.2:127894395-127895901) in the most recent assembly (UCB_Xtro_10.0).

Cloning of XtERV-S. Positions are shown for PCR products PCR1-3 (grey boxes) and the locations of the primers (dashed lines and black arrowheads). Also shown are the PCR product sizes and the positions of restriction sites used for cloning and assembly of the provirus in the pBluescript SK(+) vector
XtERV-S is 8012 bp in length predicting a packaged genome of 7597 bp (Fig. 2). The coding regions have no fatal stop codons. XtERV-S has a genomic structure similar to that of simple RVs: LTR-gag-pro-pol-env-LTR (Figs. 1, 2), with a novel additional ORF in pol. The gag and pol regions are separated by an in-frame stop codon analogous to the organization found in mammalian gamma- and epsilonRVs, where expression of pol occurs through translation suppression of the gag termination codon. The Env protein is likely expressed from a spliced transcript from a start site that overlaps the pol stop with a -1 frameshift. The genome contains the functional motifs common to all RVs and has some motifs diagnostic of specific RV genera (Fig. 2; Table 2).

The complete nucleotide and deduced amino acid sequence of the XtERV-S proviral genome. The sequence is shown from the beginning of the 5′ U3 region to the end of the 3′ U5. The LTR sequence is in black italics and its inverse repeats are double underlined. The gag, pol and env ORFs are in red, blue and purple, respectively, and termination codons are marked by an asterisk. The Orf-x2 sequence is in light green. The positions of the functional motifs are bolded and highlighted and include the following in order: PBS (primer binding site); basic regions of Gag; MHR (major homology region); GQR motif; PSAP late domain; PR (protease); RT/RNH (reverse transcriptase/RNase H); CWIC (isomerase domain); furin site; ISD (immunosuppressive domain); MSD (membrane spanning domain); PPT (polypurine tract); polyA (polyadenylation signal). Arrows indicate the splice donor and acceptor sites. The dUTPase region of pol is underlined and bolded
LTR
XtERV-S has LTRs of 705 bp that are 100% identical and flanked by the trinucleotides 5′-TGT and 3′-ACA, integrase recognition motifs conserved in class III ERVs [23]. The 3’ cellular flank could not be amplified due to the highly repetitive downstream sequence in scaffold_1181, so we could not identify a target site duplication (TSD). Both LTRs contain recognizable promoter and polyadenylation signals (Fig. 2). The presumptive core promoter has a CAAT box (position 197–200), a GC dinucleotide (position 221–255) and a TATAA box (283–287). The CAAT and TATAA boxes are 78 nt apart. A polyadenylation signal (position 574–579) is followed by a GT-rich sequence stretch (position 601–610) typically required for binding of the CstF (cleavage stimulatory factor), responsible for cleavage of RNA and addition of poly-A tails. Upstream of the 3′ LTR is an AG-rich polypurine tract (PPT). XtERV-S has a short 158 bp leader region downstream of the 5′ LTR. The 3′ end of the LTR is followed by a tRNA-related primer binding site (PBS) complementary to the 3′-terminal 19 nucleotides of tRNASer(AGA/TGA).
gag
The XtERV-S gag ORF encodes a putative 530 amino acid (aa) protein of approximately 60 kDa (Fig. 2) that is related to ERV-L type gag genes (see below). While XtERV-S does not have distinguishable matrix (MA), nucleocapsid (NC) and capsid (CA) proteins, it contains key functional motifs found in ortho- and/or spumaRV Gag proteins (Fig. 2; Table 2). These motifs include the “late” or “L” domain motif, PSAP, required for virus budding and release, and the major homology region (MHR) found in orthoRV but not spumaRV CAs [24, 25].
The 5’ end of the XtERV-S Gag lacks a myristoylation signal that functions in some RVs to target Gag to the plasma membrane [26]; instead, it contains polybasic regions (aa positions 6–10 and 130–149) (Fig. 2), which are also found in various RVs where they mediate MA/plasma membrane interactions [27, 28]. A zinc finger Cys-His box motif present in 1–2 copies in the NC of all orthoRVs functions in RNA binding, but is absent from XtERV-S and spumaRVs [29]. Instead, XtERV-S Gag contains a single glycine-glutamine-arginine (GQR) domain (Fig. 2; Table 2); this motif is also present in fish foamy virus (FV)-like ERVs and is hypothesized to function in nucleic acid binding and nuclear localization analogous to the GR boxes found in infectious FVs [30,31,32].
pro, pol
The organization of the deduced XtERV-S Pol sequence is typical of gamma-, epsilon-, spuma- and lentiRVs with the order: PRO-RT-RNAseH-IN (Fig. 2). pro is in the same reading frame as pol, which is characteristic of gamma-, epsilon-, spumaRVs and class III ERV-L, but not lentiviruses.
RV Pol proteins can be alternatively produced by readthrough suppression, ribosomal frameshifting, or, in the case of spumaRVs, use of a separate start codon for pol, which is in a different reading frame. The XtERV-S gag and pol genes are in the same frame and are separated by a stop codon, TGA, that can be subject to translational suppression (Fig. 2) [33, 34]. The pol ORF is thus predicted to start at or before the gag stop codon at position 2457.
Pro-pol spans 3489 bp, potentially encoding a 1162 aa polyprotein (Fig. 2). This region contains the conserved and properly spaced key residues for common functional motifs [35,36,37] (Table 2). Pro contains a catalytic region with the active aspartate site (DTG) and the active site flap (amino acid position 66–76) [38,39,40]. The pol gene encodes, in order: RT, a tether domain derived from a second degenerate RNaseH-related sequence [41], RNAseH and IN. The RT catalytic domain uses YIDD as the active YXDD site, which is typical of class III ERVs like ERV-L, but not FVs (Additional file 1: Fig. S1).
The XtERV-S pol includes an additional ORF of 276 bp in the -2 reading frame within IN. This ORF substantially overlaps the position of the orf-x sequence first identified in JSRV [42], with comparable ORFs in ERVs of other species like the bat DrERV and armadillo DnERV (Env1.1) [43, 44] (Additional file 2: Fig. S2). However, the XtERV-S ORF, Orf-x2, is shorter, with a 5′ end truncated by a stop codon, and has little sequence homology to the others.
A dUTPase gene is found in some RVs, but is located in different positions in four lineages: within pro in betaRVs, upstream of IN in nonprimate lentiRVs, after IN in some endogenous ERVs or at the 5′ end of gag in some Equid ERVs [45, 46]. Homology modeling using I-TASSER [47] of the XtERV-S Pol identified a segment positioned after IN as having structural similarities to other viral UTPase proteins (PBD 3ZEZ and PBD 5Y5O), and this position is common to class III ERVs, like ERV-L (Fig. 2).
env
The env ORF encodes a putative 441 aa precursor with obvious surface (SU) and transmembrane (TM) domains along with a 19 residue signal peptide (Fig. 3). Based on ESTs such as GenBank # CF222458.1 and Genbank ab inito gene prediction bioinformatics tools, there are potential splice donor/acceptor env sites at bases 576 and 5928 (Fig. 2). This positioning is unusual for RVs because the resulting transcripts would not contain the PBS; this configuration is typical of spumaRVs but not orthoRVs [48]. The env start overlaps the pol stop and is in the -1 reading frame. The SU and TM domains of RVs are typically cleaved by the furin protease at the consensus site K/R-X-K/R-R; XtERV-S contains a similar but nonstandard sequence, RNWKR, at the putative N-terminus of TM (Fig. 2).

Hydrophobicity plot of the XtERV-S Env. The SU and TM subunits of envelope are separated by a furin site (RNWKR) at position 251–255. The SU CWIC domain (position 40–43) and its interacting TM CX6CC (position 344–352) domains are indicated in red. The TM subunit contains the following: FP (fusion peptide), two heptad repeats (HR1, HR2), ISD (immunosuppressive domain), MSD (membrane spanning domain), CT (cytoplasmic tail). N-linked glycosylation sites are marked with a Y
The XtERV-S SU shows no discernible sequence homology to known RVs or ERVs, but its TM resembles gammaRVs in having an immunosuppressive domain (ISD) and a CX6CC motif that functions to establish a covalent disulfide link with a CXXC motif in the Env SU [49, 50]. The XtERV-S SU has a CWIC element positioned near the SU N-terminus (Fig. 2). TM has the domain structure typical of gamma- and alphaRVs (Table 2). A hydrophobic stretch is the likely fusion peptide but is 22 residues downstream of the putative furin site, an organization that is characteristic of alphaRVs, although the the alphaRV peptide is flanked by C residues not present in XtERV-S. The fusion peptide is followed by an N-heptad repeat, an ISD, a chain reversal region containing CX6CC, and a C-heptad repeat [50]. The ISD contains the sequence QNRAA/SLD which is typical of nonmammalian gammaRVs [51]. The TM ectodomain is followed by a membrane spanning motif [52] (Fig. 3) and an unusually short cytoplasmic tail of 27 residues [53].
RV env genes have 4–30+ potential N-linked glycosylation sites; XtERV-S has eight in SU and one in TM. In some gammaRVs, the first heptad repeat pattern in TM is disrupted by a “stutter” that is associated with the presence of a glycosylation site [51]. This “stutter”-associated glycosylation site is not found in infectious mammalian RVs but is present in XtERV-S and is also present in some fish FVs, some alphaRVs [51], some mammalian syncytins, and some other non-RV virus envelopes [54,55,56,57], as also shown below.
XtERV-S related sequences in X. tropicalis and other species
The most recent X. tropicalis genome assembly (UCB_Xtro_10.0) contains two different full length XtERV-S-related copies on chromosome XTR4: XtERV-S2(Xt-S2) (8009 bp; NC_030680.2:c12439548-12431540) and XtERV-S3 (Xt-S3) (7961 bp; NC_030680.2:c11872885-11864925). Xt-S2 and Xt-S3 differ from XtERV-S at 28 and 95 nt positions, respectively (Additional file 3: Fig. S3). Xt-S2 carries intact open reading frames for Gag, Pol and Env proteins but has an in-frame deletion in Pol relative to XtERV-S, while Xt-S3 has fatal mutations in gag, pol and env. These XTR4 ERVs have nearly identical flanking sequences including the same target site duplication, CCCTA, consistent with a local genomic duplication. A 5 bp TSD is also characteristic of ERV-L. The LTRs of Xt-S2 and Xt-S3 differ by 1 and 7 nts, respectively indicating their recent acquisition (Additional file 8: Table S1).
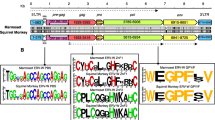
The X. tropicalis genome also contains 19 additional related, but deleted copies (Xt-S4–Xt-S22) having at least two LTR sequences and some internal sequence that usually includes the PBS, gag leader, the 5’ end of gag and the 3’ PPT (Fig. 4A; Additional file 8: Table S1). These insertions are all flanked by a 5 bp TSD (Additional file 8: Table S1). The LTRs in these copies are nearly identical to the XtERV-S LTR in the 3′ half, but have a 5′ 296 bp replacement (Fig. 4B). Based on LTR differences, the oldest of these copies was acquired ~ 25 mya while others are more recent acquisitions (Additional file 8: Table S1). There are also more than 140 solo LTRs with this altered LTR sequence (Additional file 8: Table S1).

Additional XtERV-S-related copies in X. tropicalis. A Schematic representation of XtERV-S-related deleted ERVs. Most of the 19 deleted copies are present in a single copy while Xt-S4—Xt-S14 are structurally similar. Identical line and bar colors represent sequence similarities. Dotted lines represent deletions. B Dot plot comparison of XtERV-S and the 5′ end of Xt-S5 (~ 1400 nt) shows similarities in the 3′ half of the LTR and the N-terminal region of gag
BLAST searches identified XtERV-S-related sequences in the X. laevis genome [22]. The two Xenopus species diverged 57 mya [58], and have minimally overlapping geographic ranges (Fig. 5). The single full-length X. laevis copy, XlERV-S (NC_030735:53003809-53021418), is 82% identical to XtERV-S but all three coding regions contain frameshifting deletions and insertions (Additional file 4: Fig. S4). Most notably, there are insertions of 3975 bp in pol, and 315 and 1924 bp in env. The two LTRs resemble the XtERV-S LTRs in the 3’ half and the 5’ half has no equivalent in X. tropicalis. Differences in these LTRs provide an age estimate for XlERV-S of 36 my (Additional file 8: Table S1). X. laevis is allotetraploid with two sets of chromosomes, L and S, that are homeologous and co-orthologous to X. tropicalis chromosomes and originated from the interbreeding of frogs with distinguishable genomes 34 mya [22]. XlERV-S maps to XLA6S and is therefore not orthologous to XtERV-S or to any of the deleted XtERV-S copies on its chromosome 6, XTR6. The X. laevis genome also carries more than 100 related solo LTRs.

Geographic distribution of X. tropicalis, X. laevis and P. adspersus. The areas represented by red and blue highlighting and black stripes represent the natural habitats of X. tropicalis, P. adspersus and X. laevis, respectively [98]. The phylogenetic tree from Timetree [58] places the divergence of P. adspersus and Xenopus sp. at ~ 204 mya and the divergence of two X. tropicalis and X. laevis at 57 mya
BLAST searches of other frog genomes identified an intact related provirus in another African Anuran, the African bull frog (P. adspersus isolate 1538 chromosome 4, CM016419:110619499-110628269) (Additional file 5: Fig. S5). This genus diverged from Xenopus approximately 200 mya and is sympatric with X. laevis (Fig. 5). This provirus has full-length but defective gag, pol and env genes, and the predicted proteins show high similarity to XtERV-S in Gag (51% identical and 71% similar: Blossum90) and Pol (67.4% identical and 84.2 similar: Blossum90). This ERV has a leucine PBS and a dUTPase positioned as in XtERV-S and ERV-L. The Env protein shows no homology to XtERV-S in SU, but contains a CWIC motif in a comparable location near the SU N-terminus. The gammaRV-like TM has a putative ISD, a CX6CC motif and a “stutter” in its heptad repeat but is only distantly related to the Xenopus ERVs (Fig. 6). The LTRs are 504 bp and show no significant similarity to the LTRs of XtERV-S or ERV-L, and the LTR differences produce an age estimate of 15 mya (Additional file 8: Table S1).

Unrooted phylogenetic trees of representative retroviruses (Additional file 9: Table S2) based on a MUSCLE multiple alignment and neighbor-joining method. Asterisks indicate bootstrap values greater than 70. Horizontal branch lengths are proportional to the degree of amino acid substitutions per site. The three trees represent RTpol (A), the MHR region of CAgag (B) and a segment of TMenv (C). The RT tree identifies the clusters representative of the seven RV genera. The arrowheads in the TMenv tree identify sequences with the N-linked glycosylation site associated with a heptad “stutter” (Additional file 7: Fig. S7)
Phylogenetic analysis of XtERV-S
Segments of RV genomes can have different phylogenetic histories because RV recombination is common and can involve distantly related RVs [16, 59, 60] or can occur between endogenous and exogenous viruses [61]. XtERV-S has a class III gag-pol and a class I env. We generated phylogenetic trees based on alignments of three regions of the genome: the RT core of pol, the MHR-containing region of gag, and the TM domain of env, including representative members of the seven retroviral genera where possible, and previously described and newly extracted ERVs from the genomes of nonmammalian vertebrates (Fig. 6; Additional file 9: Table S2).
The RT core is the most highly conserved region across all seven RV genera and this tree shows three groupings that correspond to the class I-III ERVs (Fig. 6A). XtERV-S is not closely related to the two previously identified Xenopus ERVs, XTERV1 and Xen-1 [62, 63], which are epsilonRVs. The XtERV-S RT clusters with the other African frog ERVs and with class III which includes ERVs from fish, amphibians and birds, FVs, and mammalian ERV-L and ERV-S. RT alignments identify obvious ERV-L lineage specific sequence stretches in XtERV-S (Table 2; Additional file 1: Fig. S1).
The gag gene is poorly conserved among RVs, but XtERV-S contains an MHR, shared by most orthoRVs and ERVs, and there are lineage specific sequence patterns in and around the MHR (Additional file 6: Fig. S6). This tree also groups the XtERV-S segment with ERV-L (Fig. 6B).
XtERV-S encodes a TMenv typical of class I gammaRVs and ERVs [50]. This tree defines two subgroups (Fig. 6C) with XtERV-S grouping with some nonmammalian gammaRVs and several syncytins. The subgroup containing XtERV-S includes all of the TMs with the heptad-stutter associated glycosylation site (Fig. 2; Additional file 7: Fig. S7) [51].
Expression of XtERV-S in vivo and in cultured cells
The presence of intact and correctly positioned CAAT and TATA boxes along with a polyadenylation signal within the LTRs strongly suggests that XtERV-S can be transcribed. We cloned the XtERV-S LTR into a luciferase reporter vector. In the absence of established cell culture systems to test for Xenopus gene expression, we used human 293T cells and found that the XtERV-S LTR increased luciferase expression by four–fivefold compared to promoter-less reporter (Fig. 7A), but was 20–60 fold lower than the MoMLV and CMV promoters (Fig. 7A). This reduced expression directed by the XtERV-S LTR may be due to its partial mutational inactivation or to some incompatibility of this LTR in 293T cells.

XtERV-S is transcriptionally active. A Functional analysis of the XtERV-S LTR cloned into a luciferase reporter vector and transfected into 293T cells. Luciferase expression is compared to the promoter-less vector pGL3 basic and to vectors using the Moloney mouse leukemia virus (MoMLV) LTR and CMV promoter. B Confocal examination of GFP-tagged XtERV-S Gag protein in 293T cells shows accumulation in the cytoplasm at the plasma membrane (red arrows) and localization in nucleus (yellow arrows). C ESTs mapping to XtERV-S are expressed embryonically in X. tropicalis. ESTs map to the gag, pol and env regions. D RNAseq reads map mostly to LTR and the gag-pol ORFs. The chart shows the total number of reads per kilobase per million (RPKM) mapped to the provirus and the reads mapping to LTR, gag, pol, and env. The developmental stages and events are indicated, including MBT (mid-blastula transition); the red arrow represents the transcriptional transition from maternal mRNA to zygote genome transcribed mRNAs; the green arrow represents the beginning of the tadpole stage
The XtERV-S gag with a GFP tag can express stable protein under the constitutive CMV promoter in transfected 293T cells (Fig. 7B). This protein accumulated in the cytoplasm at the plasma membrane, but also distributed to the nucleus. The Gag of multiple RVs is primarily found in the cytoplasm of infected cells, but for some RVs can be distributed to the nucleus, and in late stages of the viral life cycle accumulates at the plasma membrane for assembly [64]. ERV-L Gag has been found in the cytoplasm [65].
Expressed sequence tags (ESTs) related to XtERV-S and RNAseq data document the production of XtERV-S gag, pol and env transcripts (Fig. 7C, D), indicating that the LTR promoter is transcriptionally active in vivo. ESTs mapping to XtERV-S are detected from gastrulation to the tailbud embryo. These ESTs correspond to segments of the gag, pol, env and LTR (Fig. 7C). We mapped the RNAseq reads from the publicly available RNAseq datasets of adult tissues and distinct developmental stages to the XtERV-S genome (301–7916 bp) [66] (Fig. 7D). Most reads mapped to gag, pol or LTR (Fig. 7D).
The RNAseq data shows little or no expression of XtERV-S transcripts in adult X. tropicalis tissues including brain, liver, kidney heart, skeletal muscles and in 2 cell to stage 8 embryos (Fig. 7D). Initiation of XtERV-S transcription thus coincides with the maternal-to-zygotic mid-blastula transition (MBT) characterized by large scale activation of the zygotic genome (onset of transcription from the embryonic/zygotic genome) and destabilization of maternal mRNAs (stage 9; Fig. 7D) [67]. There is increasing expression during development stages 9 and 10 (late blastula—early gastrula) and robust expression through stages 11–28 (mid-gastrula, neurula and early tailbud). Expression decreases subsequent to stage 28 with little or no expression by stage 44–45 (late tailbud–tadpole) suggesting a developmental role and/or regulation of these transcripts. Embryonic expression of XtERV-S in mid-stage embryos differs from that of mouse ERV-L which peaks at the 2 cell stage and decreases at the 8 cell stage [65, 68]. A previous genome wide analysis that examined expression of X. tropicalis LTR retroelements similarly found that expression of this set of retroelements is activated at mid-blastula [69]. These data taken together show that XtERV-S is likely transcribed, that transcription is particularly active during specific stages of development, and that tagged transfected gag can produce protein that shows the expected cellular distribution. We do not however have evidence that infectious XtERV-S is produced.
Discussion
XtERV-S is a novel, intact RV ERV with unusual domain relationships to known RVs. It shows closest sequence homology in gag-pol with the ancient class III ERVs, while its env gene has an SUenv subunit that is unrelated to any known RV and a TMenv characteristic of class I gammaRVs in organization and functional motifs. Recombinant structures are common among RVs, and multiple instances of env-swapping have described the acquisition of class I envs by class II RVs isolated from multiple species [16, 59, 70] as well as env-swapping between different subgroups of class I RVs [71, 72], a phenomenon which occurs regularly during MLV-induced lymphomagenesis [61]. XtERV-S is thus an unusual example of an intact and apparently nondefective ERV genome with a class I env in a class III backbone. The TMenv subunit of this virus has the motifs necessary to establish a covalent disulfide SU/TM bond, an obviously successful and ancient env structure that is common in other virus families including filoviruses, influenza and coronaviruses [73,74,75]. XtERV-S may thus represent an ancient evolutionary RV form with a combination of viral genes not found in extant mammalian RVs but that may still be circulating in African frogs. This RV structure may be prove to be common in ancient ERVs and representative of infectious RVs yet to be discovered.
The ERVs related to this sequence in the genomes of various African frogs include recent and ancient copies. The intactness of the XtERV-S ORFs and its identical 5′ and 3′ LTRs show it to be a recent insert in the early stages of retroviral endogenization. On the other hand, the divergent LTRs of the mutationally damaged nonorthologous copy found in XlERV-S date it to 36 MYA, and similarly ancient ERVs that are related but not identical are found in the African bullfrog (P. adspersus). These data suggest that related infectious RVs have long been spreading in Anuran populations and were fairly recently active in X. tropicalis, although we have no evidence that Xenopus carries such infectious viruses or that XtERV-S can produce virus. These frogs are all African, but the two Xenopus species have a limited shared geographical distribution (Fig. 5). While the distribution of the African bullfrog largely overlaps the territory of sub-Saharan Africa occupied by X. laevis, their ecological niches differ as Xenopus is fully aquatic whereas the bullfrog resides largely in dry savannas and shrub land; both species, however, reproduce in aquatic settings suggesting the possibility of trans-species transmission.
RV family relationships are determined by sequence identities and by the presence of conserved functional motifs that can be genus specific in their presence/absence, sequence variations and position. These features identify XtERV-S gag and pol as class III. The class III ERVs most closely related to XtERV-S are largely degenerate human ERVs, mouse ERVs with intact gag and pol genes, and ERVs found in nonmammalian vertebrates. Class III ERVs most prominently include ERV-L, an env-less ancient proviral lineage that entered mammalian genomes more than 100 mya [76] as well as divergent and generally degenerate subtypes like ERV-S [77], which has associated env sequences, but notably differs from ERV-L and XtERV-L in gag-pol, particularly in the absence of an identifiable dUTPase.
Class III ERVs are most closely related to the infectious spumaRVs, but XtERV-S is not particularly FV-like [30, 78]. While the location of the predicted env splice sites excludes the PBS, as is also the case for FVs, the novel XtERV-S SUenv lacks FV features like a putative gag-interacting WXXW, a second furin site and an internal promoter. Also, while XtERV-S, like FVs, contains an FV-like consensus Gag p3 cleavage site (VXXV) [79], the location of this site downstream of the Gag stop suggests that any possible ancestral link no longer has any functional significance.
While the XtERV-S SUenv sequence is not closely related to any other RV, it has a gammaRV-like CWIC motif that can potentially establish a covalent bond with its gammaRW-like TMenv. Gamma-like Envs can be subgrouped on the basis of a “stutter” found in the N-terminal heptad repeat [51]. This motif, present in XtERV-S, is shared by other class III ERVs, some alphaRVs, env ERVs in some spiny-rayed fish and some mammalian envs domesticated to serve as syncytins. Syncytins are Env-encoding ERVs independently co-opted from different orthoretroviruses for a convergent physiological role in the formation of the syncytial layers at the placental fetal-maternal interface. More than 11 syncytins are found in different mammalian lineages [4], and the heptad stutter cluster in the TMenv tree includes some but not all of these syncytins, a feature that is not related to taxa or to placenta type. This stutter has a presumed functional role in entry mechanisms involving endocytosis [80]. That this motif has important functionality and ancient origins is supported by its presence in the envelope genes of filoviruses, arenaviruses, influenza and coronaviruses [54,55,56,57].
The expression of retroviral LTRs in vertebrates depends on genetic and epigenetic factors including tissue type, ontogenic stages, age and sex [62, 81,82,83,84]. The LTR is transcriptionally active and Gag protein in transfected cells duplicates patterns reported for orthoRVs, but we have no evidence that XtERV-S can produce viral proteins or virus in vivo. XtERV-S expression is obviously under regulation as it is largely restricted to development stages 9 to 34. This expression coincides with transcriptional activation of the zygotic genome through the early tailbud stage suggesting these transcripts may have possible role in development. Many other ERVs and ERV-derived genes are expressed during embryogenesis (or in epididymis) including ERV-L [85,86,87] and while some of this expression has been co-opted to serve host regulatory functions, as for HERV-H [88], the timing of this expression may also represent a strategy to maximize or regulate proliferation in undifferentiated cells in early development to ensure preservation of ERV lineages within the host genome [89]. Further studies focused on these early developmental stages should clarify the extent of XtERV-S expression and uncover possible roles in development.
The different classes and families of ERVs are derived from independent genome invasion events followed by their differential amplification. ERV characterization has long focused on the many invasions that occurred after the divergence of mammalian orders. Most ERV families that have retained function are lineage-specific although important functional motifs have ancient roots and are found in dead ERVs. Here we described a set of ERVs that have ancient members along with recent acquisitions that retain some functionality. Elucidating the ancient origins of Retroviridae benefits from the increasing attention directed to nonmammalian vertebrates.
Conclusions
We have identified a recently acquired intact ERV in X. tropicalis. Characterization of XtERV-S based on phylogenetics and the presence or absence of functional motifs that can be retrovirus or virus subtype specific shows that the gag and pol genes of XtERV-S are representative of the largely env-less class III ERVs. This provirus, however, carries a class I env gene with a novel surface subunit and a transmembrane subunit. XtERV-S expression is developmentally regulated with transcripts that are expressed between the mid-blastula maternal—zygotic transition and the tailbud stage. Additional much older defective copies are found in X. tropicalis as well as other African frog taxa indicating that this virus subtype has been circulating in these species for at least 36 million years, and may be representative of a yet to be discovered infectious retrovirus. Exploring XtERV-S expression and replication in X. tropicalis and also in vitro cell culture provides us an opportunity to understand the biology of ancient ERV-L and related family of endogenous retroviruses.
Methods
Cloning of XtERV-S
The XtERV-S genomic sequence was amplified from the genomic DNA of 20 pooled stage 12 X. tropicalis embryos provided by Dr. Frank L. Conlon (University of North Carolina, Raleigh, NC). Primers listed in Table 1 were designed from the XtERV-S proviral sequence identified in X. tropicalis v9.1 scaffold 1181, GenBank NW_016684263.1. PCR was performed using TaKaRa LA as per the manufacturer’s instructions (Clontech/TaKaRa, Mountain View, CA) using the strategy indicated in Fig. 1. The viral genome was amplified in three fragments that were cloned separately into the Xho1/Not1 site of the pBluescript SK(+) vector (Agilent Biosciences, Santa Clara, CA, USA). These fragments were sequenced and then ligated to each other to generate the full length XTERV-S proviral clone. The GenBank Accession number for XtERV-S is MW779451.
The XTERV-S and Moloney mouse leukemia virus (MoMLV) proviral LTR sequence was PCR amplified from pNCA [90] using primers listed in Table 1, and the fragment was cloned between the KpnI-BglII and BglII-HindIII sites of the pGL3 basic luciferase reporter plasmid (Promega, Madison, WI). The XtERV-S gag gene was amplified using primers listed in Table 1 and cloned into the eGFP-C1 vector (Clontech/TaKaRa) to produce GFP-Gag.
Homology modeling
The XtERV-S Pol sequence was submitted to the I-TASSER [47] program which identifies homologs based on a multiple threading approach—identifying templates from PDB, iterative structure assembly simulation, model selection and refinement, and structure-based function annotation.
Cell culture and luciferase assay
293T cells were grown and maintained in DMEM (Lonza, Walkersville, MD) containing 10% fetal bovine serum and supplemented with penicillin–streptomycin and L-Glutamine. 293T cells were transfected separately with the luciferase reporter vector carrying XtERV-S LTR, the promoter less—pGL3 basic control vector, CMV luciferase (Promega) and MoMLV LTR luciferase. Transfections were performed using Lipofectamine 3000 (Thermo Fisher Scientific, Atlanta, GA) and repeated three or more times and normalized to β-galactosidase activity expressed from a cotransfected pCMV-β (Clontech/TaKaRa). Cells transfected with reporter vectors were lysed in luciferase reporter cell lysis buffer and assayed for luciferase and β-galoctosidase activity as described previously [91].
Confocal imaging
293T cells were cultured on 25-mm coverslips and transfected with 200 ng of either pEGFP-C1 or pEGFP-XtERV-S Gag plasmid in 12 well cell culture plates. One day later, cells were fixed with 3.7% formaldehyde and permeabilized with PBS containing 0.1% Triton X-100. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific). Coverslips were mounted onto glass slides with ProLong antifade kit (Thermo Fisher Scientific) and examined with a Leica laser-scanning microscope.
Sequence analysis and phylogenetic trees
NCBI Blastn was used to search for additional copies of XtERV-S in the genomes of X. tropicalis, X. laevis and the African bullfrog, P. adspersus. Sequence analysis was performed using Geneious Prime 2021.0.3 (https://www.geneious.com). XtERV-S Env hydrophobicity plots were drawn using DNASTAR Lasergene 17 (DNASTAR Inc., Madison, WI).
Three phylogenetic trees were constructed in MEGA-X [92] using the Neighbor-Joining method [93]. The three trees were based on the RT domain of pol, the MHR region of gag and a segment of TMenv; these segments correspond to the following positions in XtERV-S: RT: 3311–3871, gag:1841–2260, TM: 7126–7590. RV sequences used for the trees are listed in Additional file 9: Table S2. The evolutionary distances were computed using the JTT matrix-based method [94]. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). All positions with less than 95% site coverage were eliminated so fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position.
Mapping and quantitation of the RNAseq reads to XTERV-S proviral region
Publicly available RNA-seq datasets for adult tissues (Accession No. SRX191164-68, 5 runs (brain, liver, kidney, heart and skeletal muscle, 39 Gbases) and distinct developmental stages from (Accession No. SRA051954—40 runs compromising 92 Gbases [66] were downloaded using the fastq-dump utility of the NCBI SRA Toolkit. Reads were then aligned to the XtERV-S genome using Bowtie2 [95], and the output was converted into indexed BAM files with Samtools [96]. Finally, Bedtools [97] was used to count the reads aligned to each particular region of the XtERV-S genome. The reads were mapped to the proviral sequence between the regions 301–7916 nt positions.
Availability of data and materials
The XtERV-S sequence is deposited in GenBank under accession number: MW779451. Other sequences used and analyzed in the current study are publicly available in GenBank, accession numbers are provided in this article and its Additional files.
Abbreviations
- ERV:
-
Endogenous retrovirus
- ORF:
-
Open reading frame
- RT:
-
Reverse transcriptase
- env :
-
Envelope
- gag :
-
Group specific antigen
- XTR:
-
Xenopus tropicalis Chromosome
- LTR:
-
Long terminal repeat
- TSD:
-
Target site duplication
- PPT:
-
Polypurine tract
- PBS:
-
Primer binding site
- MA:
-
Matrix
- CA:
-
Capsid
- NC:
-
Nucleocapsid
- MHR:
-
Major homology region
- pro :
-
Protease
- RNaseH:
-
Ribonuclease H
- IN:
-
Integrase
- FV:
-
Foamy virus
- TM:
-
Transmembrane
- SU:
-
Surface subunit
- ISD:
-
Immunosuppressive domain
- GFP:
-
Green fluorescense protein
- MBT:
-
Mid-blastula transition
- RV:
-
Retrovirus
- HERV:
-
Human endogenous retrovirus
- DAPI:
-
4,6-Diamidino-2-phenylindole
- MSD:
-
Membrane spanning domain
- CT:
-
Cytoplasmic tail
- RPKM:
-
Reads per kilo base per million
- JSRV:
-
Jaagsiekte sheep retrovirus
References
Taxonomy V. Family-retroviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier; 2012. p. 477–95.
Johnson WE. Endogenous retroviruses in the genomics era. Annu Rev Virol. 2015;2:135–59.
Feschotte C, Gilbert C. Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet. 2012;13:283–96.
Dupressoir A, Lavialle C, Heidmann T. From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta. 2012;33:663–71.
Lavialle C, Cornelis G, Dupressoir A, Esnault C, Heidmann O, Vernochet C, Heidmann T. Paleovirology of ‘syncytins’, retroviral env genes exapted for a role in placentation. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120507.
Ikeda H, Laigret F, Martin MA, Repaske R. Characterization of a molecularly cloned retroviral sequence associated with Fv-4 resistance. J Virol. 1985;55:768–77.
Best S, Le Tissier P, Towers G, Stoye JP. Positional cloning of the mouse retrovirus restriction gene Fv1. Nature. 1996;382:826–9.
Meyer TJ, Rosenkrantz JL, Carbone L, Chavez SL. Endogenous retroviruses: with Us and against Us. Front Chem. 2017;5:23.
Buzdin AA, Prassolov V, Garazha AV. Friends–enemies: endogenous retroviruses are major transcriptional regulators of human DNA. Front Chem. 2017;5:35.
Wang J, Xie G, Singh M, Ghanbarian AT, Raskó T, Szvetnik A, Cai H, Besser D, Prigione A, Fuchs NV, et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature. 2014;516:405–9.
Herniou E, Martin J, Miller K, Cook J, Wilkinson M, Tristem M. Retroviral diversity and distribution in vertebrates. J Virol. 1998;72:5955–66.
Gifford R, Tristem M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes. 2003;26:291–315.
Hayward A, Cornwallis CK, Jern P. Pan-vertebrate comparative genomics unmasks retrovirus macroevolution. Proc Natl Acad Sci USA. 2015;112:464–9.
Zhuo X, Feschotte C. Cross-species transmission and differential fate of an endogenous retrovirus in three mammal lineages. PLOS Pathog. 2015;11:e1005279.
Greenwood AD, Ishida Y, O’Brien SP, Roca AL, Eiden MV. Transmission, evolution, and endogenization: lessons learned from recent retroviral invasions. Microbiol Mol Biol Rev 2018;82.
van der Kuyl AC, Dekker JT, Goudsmit J. Discovery of a new endogenous type C retrovirus (FcEV) in cats: evidence for RD-114 being an FcEV(Gag-Pol)/baboon endogenous virus BaEV(Env) recombinant. J Virol. 1999;73:7994–8002.
Locatelli S, Peeters M. Cross-species transmission of simian retroviruses: how and why they could lead to the emergence of new diseases in the human population. AIDS. 2012;26:659–73.
Blanco-Melo D, Gifford RJ, Bieniasz PD. Reconstruction of a replication-competent ancestral murine endogenous retrovirus-L. Retrovirology. 2018;15:34.
Lee YN, Bieniasz PD. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007;3:e10.
Salientia. Frogs and toads. Version 11 January 2008. http://tolweb.org/Salientia/14938/2008.01.11. Accessed 23 Mar 2021.
Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V, Ovcharenko I, Putnam NH, Shu S, Taher L, et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328:633–6.
Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, Fukui A, Hikosaka A, Suzuki A, Kondo M, et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538:336–43.
Benachenhou F, Sperber GO, Bongcam-Rudloff E, Andersson G, Boeke JD, Blomberg J. Conserved structure and inferred evolutionary history of long terminal repeats (LTRs). Mob DNA. 2013;4:5.
Hutter S, Zurnic I, Lindemann D. Foamy virus budding and release. Viruses. 2013;5:1075–98.
Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106:87–102.
Gottlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1989;86:5781–5.
Hamard-Peron E, Juillard F, Saad JS, Roy C, Roingeard P, Summers MF, Darlix JL, Picart C, Muriaux D. Targeting of murine leukemia virus gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J Virol. 2010;84:503–15.
Maldonado JO, Martin JL, Mueller JD, Zhang W, Mansky LM. New insights into retroviral Gag-Gag and Gag-membrane interactions. Front Microbiol. 2014;5:302.
Llorens C, Munoz-Pomer A, Bernad L, Botella H, Moya A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol Direct. 2009;4:41.
Ruboyianes R, Worobey M. Foamy-like endogenous retroviruses are extensive and abundant in teleosts. Virus Evol. 2016;2:vew032.
Mullers E. The foamy virus Gag proteins: what makes them different? Viruses. 2013;5:1023–41.
Schliephake AW, Rethwilm A. Nuclear localization of foamy virus Gag precursor protein. J Virol. 1994;68:4946–54.
Feng YX, Levin JG, Hatfield DL, Schaefer TS, Gorelick RJ, Rein A. Suppression of UAA and UGA termination codons in mutant murine leukemia viruses. J Virol. 1989;63:2870–3.
Jones DS, Nemoto F, Kuchino Y, Masuda M, Yoshikura H, Nishimura S. The effect of specific mutations at and around the gag-pol gene junction of Moloney murine leukaemia virus. Nucleic Acids Res. 1989;17:5933–45.
Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–90.
Najmudin S, Coté ML, Sun D, Yohannan S, Montano SP, Gu J, Georgiadis MM. Crystal structures of an N-terminal fragment from moloney murine leukemia virus reverse transcriptase complexed with nucleic acid: functional implications for template-primer binding to the fingers domain11Edited by D. C Rees J Mol Biol. 2000;296:613–32.
Menéndez-Arias L, Sebastián-Martín A, Álvarez M. Viral reverse transcriptases. Virus Res. 2017;234:153–76.
Toh H, Kikuno R, Hayashida H, Miyata T, Kugimiya W, Inouye S, Yuki S, Saigo K. Close structural resemblance between putative polymerase of a Drosophila transposable genetic element 17.6 and pol gene product of Moloney murine leukaemia virus. EMBO J. 1985;4:1267–72.
Pearl LH, Taylor WR. A structural model for the retroviral proteases. Nature. 1987;329:351–4.
Rao JK, Erickson JW, Wlodawer A. Structural and evolutionary relationships between retroviral and eucaryotic aspartic proteinases. Biochemistry. 1991;30:4663–71.
Malik HS, Eickbush TH. Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 2001;11:1187–97.
Rosati S, Pittau M, Alberti A, Pozzi S, York DF, Sharp JM, Palmarini M. An accessory open reading frame (orf-x) of jaagsiekte sheep retrovirus is conserved between different virus isolates. Virus Res. 2000;66:109–16.
Escalera-Zamudio M, Mendoza ML, Heeger F, Loza-Rubio E, Rojas-Anaya E, Méndez-Ojeda ML, Taboada B, Mazzoni CJ, Arias CF, Greenwood AD. A novel endogenous betaretrovirus in the common vampire bat (Desmodus rotundus) suggests multiple independent infection and cross-species transmission events. J Virol. 2015;89:5180–4.
Malicorne S, Vernochet C, Cornelis G, Mulot B, Delsuc F, Heidmann O, Heidmann T, Dupressoir A. Genome-wide screening of retroviral envelope genes in the nine-banded Armadillo (Dasypus novemcinctus, Xenarthra) reveals an unfixed chimeric endogenous betaretrovirus using the ASCT2 receptor. J Virol. 2016;90:8132–49.
Hizi A, Herzig E. dUTPase: the frequently overlooked enzyme encoded by many retroviruses. Retrovirology. 2015;12:70.
Zhu H, Gifford RJ, Murcia PR. Distribution, diversity, and evolution of endogenous retroviruses in perissodactyl genomes. J Virol 2018;92.
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12:7–8.
Bodem J, Lochelt M, Delius H, Flugel RM. Detection of subgenomic cDNAs and mapping of feline foamy virus mRNAs reveals complex patterns of transcription. Virology. 1998;244:417–26.
Pinter A, Kopelman R, Li Z, Kayman SC, Sanders DA. Localization of the labile disulfide bond between SU and TM of the murine leukemia virus envelope protein complex to a highly conserved CWLC motif in SU that resembles the active-site sequence of thiol-disulfide exchange enzymes. J Virol. 1997;71:8073–7.
Henzy JE, Johnson WE. Pushing the endogenous envelope. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120506.
Henzy JE, Gifford RJ, Kenaley CP, Johnson WE. An intact retroviral gene conserved in spiny-rayed fishes for over 100 My. Mol Biol Evol. 2017;34:634–9.
Cianciolo GJ, Copeland TD, Oroszlan S, Snyderman R. Inhibition of lymphocyte proliferation by a synthetic peptide homologous to retroviral envelope proteins. Science. 1985;230:453–5.
Tedbury PR, Freed EO. Chapter nine: The cytoplasmic tail of retroviral envelope glycoproteins. In: Klasse PJ, editor. Progress in molecular biology and translational science. Vol. 129. Academic Press; 2015: 253–284.
Dietrich MH, Ogden KM, Long JM, Ebenhoch R, Thor A, Dermody TS, Stehle T. Structural and functional features of the reovirus σ1 tail. J Virol 2018;92.
Koellhoffer JF, Dai Z, Malashkevich VN, Stenglein MD, Liu Y, Toro R, Harrison SJ, Chandran K, DeRisi JL, Almo SC, Lai JR. Structural characterization of the glycoprotein GP2 core domain from the CAS virus, a novel arenavirus-like species. J Mol Biol. 2014;426:1452–68.
Harrison JS, Koellhoffer JF, Chandran K, Lai JR. Marburg virus glycoprotein GP2: pH-dependent stability of the ectodomain α-helical bundle. Biochemistry. 2012;51:2515–25.
Higgins CD, Malashkevich VN, Almo SC, Lai JR. Influence of a heptad repeat stutter on the pH-dependent conformational behavior of the central coiled-coil from influenza hemagglutinin HA2. Proteins Struct Funct Bioinformat. 2014;82:2220–8.
Kumar S, Stecher G, Suleski M, Hedges SB. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 2017;34:1812–9.
Henzy JE, Gifford RJ, Johnson WE, Coffin JM. A novel recombinant retrovirus in the genomes of modern birds combines features of avian and mammalian retroviruses. J Virol. 2014;88:2398–405.
Huder JB, Boni J, Hatt JM, Soldati G, Lutz H, Schupbach J. Identification and characterization of two closely related unclassifiable endogenous retroviruses in pythons (Python molurus and Python curtus). J Virol. 2002;76:7607–15.
Bamunusinghe D, Liu Q, Plishka R, Dolan MA, Skorski M, Oler AJ, Yedavalli VRK, Buckler-White A, Hartley JW, Kozak CA. Recombinant origins of pathogenic and nonpathogenic mouse gammaretroviruses with polytropic host range. J Virol 2017;91.
Sinzelle L, Carradec Q, Paillard E, Bronchain OJ, Pollet N. Characterization of a Xenopus tropicalis endogenous retrovirus with developmental and stress-dependent expression. J Virol. 2011;85:2167–79.
Kambol R, Kabat P, Tristem M. Complete nucleotide sequence of an endogenous retrovirus from the amphibian, Xenopus laevis. Virology. 2003;311:1–6.
Stake MS, Bann DV, Kaddis RJ, Parent LJ. Nuclear trafficking of retroviral RNAs and Gag proteins during late steps of replication. Viruses. 2013;5:2767–95.
Ribet D, Louvet-Vallée S, Harper F, de Parseval N, Dewannieux M, Heidmann O, Pierron G, Maro B, Heidmann T. Murine endogenous retrovirus MuERV-L is the progenitor of the “orphan” epsilon viruslike particles of the early mouse embryo. J Virol. 2008;82:1622–5.
Tan MH, Au KF, Yablonovitch AL, Wills AE, Chuang J, Baker JC, Wong WH, Li JB. RNA sequencing reveals a diverse and dynamic repertoire of the Xenopus tropicalis transcriptome over development. Genome Res. 2013;23:201–16.
Paranjpe SS, Jacobi UG, van Heeringen SJ. C Veenstra GJ: a genome-wide survey of maternal and embryonic transcripts during Xenopus tropicalis development. BMC Genomics. 2013;14:762.
Wang Q, Chung YG, deVries WN, Struwe M, Latham KE. Role of protein synthesis in the development of a transcriptionally permissive state in one-cell stage mouse embryos. Biol Reprod. 2001;65:748–54.
Grau JH, Poustka AJ, Meixner M, Plotner J. LTR retroelements are intrinsic components of transcriptional networks in frogs. BMC Genomics. 2014;15:626.
Hayward JA, Tachedjian M, Cui J, Field H, Holmes EC, Wang LF, Tachedjian G. Identification of diverse full-length endogenous betaretroviruses in megabats and microbats. Retrovirology. 2013;10:35.
Howard TM, Sheng Z, Wang M, Wu Y, Rasheed S. Molecular and phylogenetic analyses of a new amphotropic murine leukemia virus (MuLV-1313). Virol J. 2006;3:101.
Bamunusinghe D, Naghashfar Z, Buckler-White A, Plishka R, Baliji S, Liu Q, Kassner J, Oler AJ, Hartley J, Kozak CA. Sequence diversity, intersubgroup relationships, and origins of the mouse leukemia gammaretroviruses of laboratory and wild mice. J Virol. 2016;90:4186–98.
Dutch RE, Jardetzky TS, Lamb RA. Virus membrane fusion proteins: biological machines that undergo a metamorphosis. Biosci Rep. 2000;20:597–612.
White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol. 2008;43:189–219.
Barrett CT, Dutch RE. Viral membrane fusion and the transmembrane domain. Viruses. 2020;12:693.
Bénit L, De Parseval N, Casella JF, Callebaut I, Cordonnier A, Heidmann T. Cloning of a new murine endogenous retrovirus, MuERV-L, with strong similarity to the human HERV-L element and with a gag coding sequence closely related to the Fv1 restriction gene. J Virol. 1997;71:5652.
Tristem M. Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J Virol. 2000;74:3715–30.
Han G-Z, Worobey M. An endogenous foamy-like viral element in the Coelacanth genome. PLOS Pathog. 2012;8:e1002790.
Kehl T, Tan J, Materniak M. Non-simian foamy viruses: molecular virology, tropism and prevalence and zoonotic/interspecies transmission. Viruses. 2013;5:2169–209.
Igonet S, Vaney MC, Vonrhein C, Bricogne G, Stura EA, Hengartner H, Eschli B, Rey FA. X-ray structure of the arenavirus glycoprotein GP2 in its postfusion hairpin conformation. Proc Natl Acad Sci USA. 2011;108:19967–72.
Shen CH, Steiner LA. Genome structure and thymic expression of an endogenous retrovirus in zebrafish. J Virol. 2004;78:899–911.
Carre-Eusebe D, Coudouel N, Magre S. OVEX1, a novel chicken endogenous retrovirus with sex-specific and left-right asymmetrical expression in gonads. Retrovirology. 2009;6:59.
Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41:563–71.
Dennis S, Sheth U, Feldman JL, English KA, Priess JR. C. elegans germ cells show temperature and age-dependent expression of Cer1, a Gypsy/Ty3-related retrotransposon. PLoS Pathog. 2012;8:e1002591.
Schlesinger S, Goff SP. Retroviral transcriptional regulation and embryonic stem cells: war and peace. Mol Cell Biol. 2015;35:770–7.
Göke J, Lu X, Chan Y-S, Ng H-H, Ly L-H, Sachs F, Szczerbinska I. Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell Stem Cell. 2015;16:135–41.
Crowell RC, Kiessling AA. Endogenous retrovirus expression in testis and epididymis. Biochem Soc Trans. 2007;35:629–33.
Robbez-Masson L, Rowe HM. Retrotransposons shape species-specific embryonic stem cell gene expression. Retrovirology. 2015;12:45.
Magiorkinis G, Katzourakis A, Lagiou P. Roles of endogenous retroviruses in early life events. Trends Microbiol. 2017;25:876–7.
Colicelli J, Goff SP. Sequence and spacing requirements of a retrovirus integration site. J Mol Biol. 1988;199:47–59.
Yedavalli VS, Benkirane M, Jeang KT. Tat and trans-activation-responsive (TAR) RNA-independent induction of HIV-1 long terminal repeat by human and murine cyclin T1 requires Sp1. J Biol Chem. 2003;278:6404–10.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25.
Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8:275–82.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome project data processing S: the sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
https://amphibiaweb.org. Accessed 9 Mar 2021.
Acknowledgements
We thank Dr. Frank L. Conlon (Department of Biology and Genetics, McAllister Heart Institute, UNC-Chapel Hill, Chapel Hill, NC 27599, USA) for providing us with genomic DNA of X. tropicalis and X. laevis.
Funding
This work was funded by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (https://www.niaid.nih.gov/about/dir) under Grant Number AI000300-38 to C.A.K.
Author information
Authors and Affiliations
Contributions
VY conceived the concept and developed it under supervision of CK. VY and CK conducted the sequence analysis. VY cloned the XtERV-S. AP and VY conducted the EST and RNAseq analysis. AP and JP conducted cell transfections and reporter assays. CK and VY and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Sequence similarity of the RTs of XtERV-S and ERV-L. The amino acid sequences of the RTs of XtERV-S and other families of RVs were aligned using MUSCLE. Asterisks indicate conserved amino acids. The RT catalytic domain YIDD is shaded in grey.
Additional file 3
: Figure S3. 19 deleted XtERV-S like sequences in the X. tropicalis genome.
Additional file 4: Figure S4.
Dot-plot and alignments compare the XtERV-S and XlERV-S genomes. Arrows indicate the site of insertions in the XlERV-S proviral sequence. Dots in the alignment represent identities. Shaded portions represent different regions of the proviruses – LTR (grey), PBS (red), gag (yellow), pol (green) and env (blue).
Additional file 5
: Figure S5. XtERV-S like sequences in the African bull frog (P. adspersus) genome.
Additional file 6: Figure S6.
Alignment of RV MHR sequences including XtERV-S and mouse ERV-L. MHR is defined by three conserved residues and a fourth site occupied by a hydrophobic residue, all with conserved spacing as shown in the consensus sequence.
Additional file 7: Figure S7.
Alignment of the TM regions of the ERV and RV envelopes. Some of TMenv genes in the Fig. 6C tree have a heptad repeat stutter marked by N-linked glycosylation site (shaded in grey). The ISD region is shown in red and the CX6CC motif is highlighted in green.
Additional file 8: Table S1
. Genome positions and age of the XtERV-S like elements in Xenopus tropicalis.
Additional file 9
: Table S2. Accession numbers for sequences used for phylogenetic analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yedavalli, V.R.K., Patil, A., Parrish, J. et al. A novel class III endogenous retrovirus with a class I envelope gene in African frogs with an intact genome and developmentally regulated transcripts in Xenopus tropicalis. Retrovirology 18, 20 (2021). https://doi.org/10.1186/s12977-021-00564-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-021-00564-2