
Abstract
Background
The HIV-1 envelope glycoprotein (Env) undergoes conformational changes that mediate fusion between virus and host cell membranes. These changes involve transient exposure of two heptad-repeat domains (HR1 and HR2) in the gp41 subunit and their subsequent self-assembly into a six-helix bundle (6HB) that drives fusion. Env residues and features that influence conformational changes and the rate of virus entry, however, are poorly understood. Peptides corresponding to HR1 and HR2 (N and C peptides, respectively) interrupt formation of the 6HB by binding to the heptad repeats of a fusion-intermediate conformation of Env, making the peptides valuable probes for studying Env conformational changes.
Results
Using a panel of Envs that are resistant to N-peptide fusion inhibitors, we investigated relationships between virus entry kinetics, 6HB stability, and resistance to peptide fusion inhibitors to elucidate how HR1 and HR2 mutations affect Env conformational changes and virus entry. We found that gp41 resistance mutations increased 6HB stability without increasing entry kinetics. Similarly, we show that increased 6HB thermodynamic stability does not correlate with increased entry kinetics. Thus, N-peptide fusion inhibitors do not necessarily select for Envs with faster entry kinetics, nor does faster entry kinetics predict decreased potency of peptide fusion inhibitors.
Conclusions
These findings provide new insights into the relationship between 6HB stability and viral entry kinetics and mechanisms of resistance to inhibitors targeting fusion-intermediate conformations of Env. These studies further highlight how residues in HR1 and HR2 can influence virus entry by altering stability of the 6HB and possibly other conformations of Env that affect rate-limiting steps in HIV entry.
Similar content being viewed by others


Background
The HIV-1 envelope protein (Env) mediates virus entry into cells in a multi-step process, presenting many opportunities for blocking HIV infection [1]-[3]. The gp120 surface subunit of Env initiates the fusion process by interacting with CD4 and chemokine receptors. Subsequent conformational changes allow the transmembrane subunit (gp41) of Env to insert its hydrophobic N-terminus into the target membrane, forming the pre-hairpin intermediate. Self-assembly of the gp41 heptad-repeat regions (HR1 and HR2) then form a thermostable six-helix bundle (6HB). This energetically favorable step facilitates merger of target and virus membranes and stabilizes a fusion pore that enlarges to allow the viral capsid to pass into the target cell cytoplasm [2]. Neutralizing antibodies and fusion inhibitors can interfere with this process, but HIV rapidly mutates and evolves over the course of infection and during treatment to evade these inhibitors [4],[5]. The capacity of HIV to mutate hinders efforts to develop broadly effective vaccines and entry inhibitors. Furthermore, escape mutations may alter the functional attributes of Env.
The first entry inhibitor licensed for clinical use, Enfuvirtide (also referred to as T20), is a peptide that mimics the HR2 segment of gp41 (C peptide) [6]. T20 and other similar C peptides bind to the HR1 region of the pre-hairpin intermediate, interrupting 6HB formation in a dominant-negative manner to inhibit fusion [7]-[16]. However, T20 has a low genetic barrier to resistance, and consequently, resistance develops quickly in patients [6],[17]-[23]. While more potent, new generation C peptides are being developed, they are still susceptible to resistance in vitro[24]-[33]. The common mechanism for escape from C peptides involves mutations within HR1 that destabilize binding of the C peptide to a hydrophobic groove of the HR1 trimeric, coiled-coil core of the 6HB [23],[34]-[39]. Although these mutations necessarily diminish the stability of the 6HB, additional mutations in HR2 can compensate for the fitness cost, and in some cases, can enhance resistance [23],[40]-[43].
Peptides that mimic HR1 (N peptides) are also potent inhibitors, but they are generally less soluble and not yet in clinical use. Their inhibitory mechanism remains unclear, but current models suggest that N peptides can interfere with HR1 coiled-coil formation, and, especially if stabilized as a trimer, can sequester the HR2 region of the pre-hairpin intermediate [44]-[46]. In either case, as with C peptides, formation of the 6HB is interrupted. HIV can also develop resistance to N peptides, but unlike C peptides, the resistance mutations stabilize the 6HB [46]-[49]. This finding presents a conundrum because some resistance mutations that increase 6HB stability might also increase peptide inhibitor affinity for gp41 and therefore enhance peptide potency.
However, N-peptide resistance mutations that increase 6HB stability might also increase the rate of 6HB formation relative to peptide inhibition. Indeed, Envs with faster entry kinetics have been reported to be less sensitive to peptide fusion inhibitors [50]-[52]. Many have attributed this finding to a shorter window of opportunity for peptide accessibility to the pre-hairpin intermediate [50]-[52]. However, C-peptide fusion inhibitors have thus far not been reported to select for Envs that have faster entry kinetics. Rather, some T20-resistant Envs tended to have overall slower entry kinetics, and only after additional compensatory mutations did entry kinetics reach wild-type levels [40],[53]. Irrespective of the resistance mechanism of C-peptides, N peptides select for different resistance mutations, and their effect on Env function is unclear.
In this study, we investigated relationships between virus entry kinetics, 6HB stability, and resistance to peptide fusion inhibitors to gain insights into how residues in HR1 and HR2 can affect Env conformational changes and virus entry. Among the sixteen independent resistant cultures previously selected with one of three different N-peptide inhibitors, two resistance pathways emerged that were defined by having either a glutamic acid to lysine substitution at residue 560 (E560K, HXB2 numbering) in HR1 or a glutamic acid to lysine substitution at residue 648 (E648K, HXB2 numbering) in HR2 [46],[48]. Using pseudovirus infectivity and entry assays, we now report that increased 6HB stability, but not faster entry kinetics, correlates with resistance. We also show that increasing 6HB stability is not sufficient to increase the rate of entry. Thus, N-peptide fusion inhibitors do not necessarily select for Envs with faster entry kinetics, nor does faster entry kinetics predict decreased potency of peptide fusion inhibitors. These studies highlight an important role for HR1 and HR2 residues in influencing the relationship between stability of the final fusion-active conformation and other conformations of Env that regulates the rate of virus entry into cells.
Results
Effect of different combinations of resistance mutations on Env function
We previously generated escape-mutant viruses selected with peptides corresponding to either 44 (N44) or 36 residues (N36 or the trimer-stabilized IZN36 [54]) in gp41 HR1 and identified two genetic resistance pathways, each defined by a key mutation in either HR1 (E560K) or HR2 (E648K) [46],[48]. Each pathway was frequently associated with additional mutations in either the CD4 binding site (E560K pathway) or the V3 loop of gp120 (E648K pathway). To determine whether there were functional relationships between these gp120 and gp41 mutations, we made several chimeric Envs and Envs with site-directed mutations (Table 1). In one set of chimeras, we paired gp41 resistance mutations from one pathway with gp120 mutations from the other pathway. In addition, we made dual-pathway Envs containing the most common HR1 mutations (E560K and Q577R) and the HR2 mutation E648K (WT-KKR, C2-KKR, and C4-KKR) because these pathway-defining mutations were never isolated together despite their high frequency of selection. Finally, we also made single-site mutations that either added or removed C-terminal cytoplasmic tail (C-tail) mutations seen in cultures 4 and 5 to see whether those mutations contribute to the functional properties of Env.
We first assessed Env function in the pseudotype infectivity assay. Notably, all chimeric Envs (C4-C1, C3-C2, C2-C3, C1-C4), including those Envs with gp41 mutations from both (dual) pathways (WT-KKR, C2-KKR, and C4-KKR), were highly functional and exhibited only modest differences compared with their respective parental (selected) Envs (Figure 1A; Table 2). Reversion of the C-tail mutations (C4-C4-T and C5-C5-T) and the C-tail mutations only (WT-V833I and WT-P714L) also had little effect on infectivity levels. Overall, Envs containing E560K, including both the chimeric Envs (C4-C1, C3-C2) and the selected Envs derived from resistance cultures C1 and C2 (C1-C1, C2-C2, C6-C6), generally conferred lower infectivity than WT Env. In four of five pseudotypes with Envs containing E648K, including the chimeric Envs (C2-C3 and C1-C4) and selected Envs derived from the resistance cultures C3 and C4 (C3-C3 and C4-C4), infectivity was more similar to WT. The exception was the pseudotype with the Env from culture 5 (C5-C5), which had reduced infectivity.

Chimeric Env pseudotypes containing HR1 and HR2 mutations are functional and confer resistance to N44. (A) Relative pseudovirus infectivity titered on U87-CD4-CCR5 cells in the absence of selection. Constructs are named by donor gp120-donor gp41. For example, C1-C4 represents the gp120 from C1 and the gp41 from C4. Relative N44 resistance of pseudoviruses bearing Envs with (B) E560K pathway mutations (checkered bars), (C) E648K pathway mutations (diagonally-hatched bars), or (D) Envs with gp120 mutations only (open bars), with both pathway (dual) mutations (vertically-hatched bars), or with mutations to the C-terminal tail (horizontal-hatched bars). Results are normalized to wild type (WT-WT) and averaged across two independent experiments using at least two different pseudovirus lots (±S.E.M). A full description of Env constructs are presented in Table 1.
We next tested the chimeric Envs for susceptibility to inhibition by the N44 N-peptide and extended the analysis to include chimeras containing wild-type gp120 or gp41 sequences (Figure 1B-D). Most chimeras displayed resistance levels similar to their respective parental Envs, including the Envs with dual mutations from both pathways. These findings confirm and extend our prior work showing that the gp41 mutations were responsible for resistance. In two cases (C3-C2 compared to C2-C2 and C1-C4 compared to C4-C4 and WT-C4), however, there were modest differences in resistance between the chimera and parental Envs, suggesting that cross-talk between gp120 and gp41 may modulate resistance levels (Figure 1B-C; Table 2).
Relationship between gp41 6HB stability and resistance
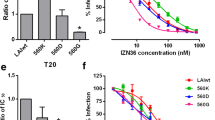
Using this extended panel of resistant Envs, we next evaluated whether changes in 6HB stability among both selected and chimeric Envs contributed to resistance. We estimated the relative stability of different 6HBs by determining their transition mid-point temperature (T m ) in thermal denaturation studies using mixtures of N and C peptides to model 6HB interactions. Using our previous T m values [46],[48], as well as additional data obtained in the current study (Table 3), we found a strong correlation between increased resistance and increased 6HB stability (Figure 2A). For example, when the T m increased 20°C, resistance increased five-fold. Additionally, using 50% inhibition concentrations and T m data from our previous study [46], we found an even stronger relationship between increased 6HB stability and resistance to the other HR1 peptides N36 and IZN36 (Figure 2B and C). Indeed, for IZN36, T m increases of 20°C led to a nearly 100-fold increase in resistance to IZN36.

6HB thermostablity increases with N-peptide resistance. Scatter plots show correlation between transition mid-point temperature (Tm) and peptide resistance of pseudoviruses relative to wild type (WT) for (A) N44, (B) N36, and (C) IZN36. Each dot in (A) represents the mean resistance for each Env depicted in Figure 1B and C. Results in (B-C) were derived from Envs reported previously. Linear regressions and statistics for correlation are indicated for each panel. rs, Spearman correlation coefficient.
Effect of resistance mutations on entry kinetics
We next assessed whether increased 6HB stability affected the rate of virus entry, since entry kinetics has been linked to susceptibility to inhibition by peptide fusion inhibitors [50]-[52]. To measure entry kinetics, we recorded fusion in real time using pseudoviruses that incorporated β-lactamase-Vpr enzyme, which cleaves a fluorescent substrate in the cytoplasm of target cells after virus entry [55]-[58]. This assay showed that five of six Envs selected in the resistance cultures conferred significantly slower (C1-C1, C2-C2, C6-C6, C4-C4, C5-C5) entry kinetics relative to WT (Figure 3A-B; Table 2). The Env from culture 3 (C3-C3), which had only a single gp41 and gp120 mutation, was more similar to WT. Envs containing HR1 pathway mutations generally showed the slowest entry kinetics, with an entry time to half maximal fusion (T1/2) that was ~20-30% slower than WT. The same pattern was seen for Envs containing only the gp41 mutations (Figure 3B-C).

Pseudoviruses with Envs from resistance cultures have slower entry kinetics than WT. (A) Entry of β-Lactamase-Vpr pseudoviruses inoculated onto HeLa cells with high CD4 and high CCR5 receptor levels (JC53), normalized to the fusion plateau. Each time point is the average of at least three separate pseudovirus lots tested in independent experiments (±S.E.M). (B-C) The time to half maximal (T1/2) fusion in target cells comparing (B) parental resistant Envs to WT, or (C) Env containing only gp41mutations to WT. *unpaired t-test indicates P values <0.05.
The kinetics results were confirmed in a second assay involving the addition of peptide to cultures at different time points to measure the time during entry when Env is susceptible to peptide inhibition. The peptide prevents fusion after Env binds to CD4 to expose the peptide-sensitive, pre-hairpin, fusion-intermediate conformation of Env and before formation of the 6HB. Therefore, the longer Env remains sensitive to the peptide, the slower it forms the 6HB and fusion pores that drive fusion. Results of these studies showed that pseudotypes with Envs from resistant viruses were not sensitive to inhibition by peptide for shorter periods of time (Figure 4A-B). In particular, pseudotypes with Envs from the E560K pathway (C1-C1, C2-C2) were sensitive to N44 significantly longer than pseudotypes with WT Env (T1/2 ~ 75% longer), while the T1/2’s of N44 susceptibility for pseudotypes with Envs from the 648 K pathway (C3-C3, and C4-C4) were similar to WT. Thus, both assays indicate that the mechanism of resistance does not require faster entry into cells, and the two assays were strongly correlated with each other (Figure 4C).

Resistant Envs do not lose susceptibility to inhibition by N44 faster than WT. (A) Percent infectivity of pseudovirus on U87-CD4-CCR5 cells after addition of N44 at different time points. (B) Calculated T1/2 of N44 susceptibility from time of addition experiments in (A). (C) Scatter plot correlating the T1/2 of N44 susceptibility and the T1/2 of fusion (Figure 3). *unpaired t-test indicates P values <0.05 showing longer time of susceptibility compared to WT. Linear regression and statistics for correlation is indicated. rs, Spearman correlation coefficient.
Relationship between entry kinetics and resistance
Despite prior reports about associations between faster entry kinetics and decreased sensitivity to peptide fusion inhibitors [50]-[52], we did not find that overall entry kinetics correlated with resistance to the N44 peptide (Figure 5A). Rather, pseudotyped Envs that fused the slowest were still two to three-fold more resistant than WT Envs (Figure 1B and C). 6HB stability and entry kinetics were also not correlated (Figure 5B). This finding was initially surprising because 6HB formation is a major driving force in the fusion process. Nevertheless, this result is consistent with our data showing that N-peptide resistance is correlated with 6HB stability (Figure 2) but not entry kinetics (Figure 5A). Clearly, other Env interactions are more important for controlling the rate-limiting step for entry than the energy contributed by 6HB formation. Mutations that increase bundle stability and confer resistance to N peptides do not necessarily confer inherently faster entry kinetics.

Pseudovirus entry kinetics does not correlate with resistance or thermodynamic stability of the 6HB. (A) Scatter plots of the T1/2 of fusion and resistance for each pseudovirus described in Table 1. (B) Scatter plots of the T1/2 fusion value and T m of each Env.
Discussion
In this study we analyzed relationships between N-peptide resistance, 6HB stability, and entry kinetics to gain insights into how HR1 and HR2 residues affect conformational changes and provide new information on the Env entry and peptide resistance mechanisms. Among our large panel of resistant Envs, we found a correlation between increased 6HB stability and increased resistance, but not between N44 resistance and entry kinetics or time to escape from N44 inhibition. Furthermore, while providing the energy to drive fusion and escape from peptide, the stability of the 6HB was not sufficient to increase the rate of entry kinetics.
Our current study with a large panel of Envs extends our previous work to establish the link between bundle stability and peptide resistance. Previously, we and others have showed that N-peptide resistance mutations that stabilize the 6HB can confer cross-resistance to C peptides (T20 and C34), indicating that N-peptide resistance mutations provide a more universal mechanism of resistance to fusion inhibitors compared to the C-peptide resistance mutations [46],[48],[49]. Escape mutations that develop after C-peptide selection generally destabilize interactions between Env and C peptides. Perhaps such a direct mechanism of escape is not selected by N peptides because mutations that would disrupt N peptide binding to Env could disrupt both the conserved inner core of HR1 helices, as well as interactions between HR2 and HR1 in the endogenous 6HB needed for virus entry. N peptides have the potential to bind both HR1 and HR2 in the fusion intermediate, so more than one mutation may be needed to directly disrupt N peptide binding to Env [59]. Therefore, the more universal mechanism of improving the stability of the endogenous 6HB bundle to favor the virus might be a more efficient resistance mechanism, especially for an inhibitor that can bind to two different sites of Env.
By contrast, C peptides may not select for such an escape mechanism because resistance mutations in the exposed surface of the HR1 coiled coil that confer high-level resistance to C peptides may not be overly detrimental to 6HB formation. Nonetheless, secondary C-peptide resistance mutations in HR2 do increase 6HB stability and may directly contribute to resistance, but these mutations generally occur after the primary resistance mutations [23],[40]-[43]. Indeed, a more potent C peptide inhibitor with increased affinity for the 6HB selects a more complex profile of resistance mutations, suggesting that resistance mutations that are needed to directly compete for the higher affinity binding of the inhibitor may impose a fitness cost on Env function [26]. These examples highlight the complex relationship between mutations that directly confer resistance and mutations that affect the inherent kinetics and efficiency of Env entry. The interplay between these attributes determines the overall fitness of the resistant virus.
Notably, increasing 6HB stability confers resistance without increasing the rate of virus entry. This finding was initially unexpected because Envs that confer faster entry have been reported to be less sensitive to peptide fusion inhibitors than Envs that confer slower entry [50]-[52]. In addition, increased stability of the 6HB should facilitate formation of the final fusion-active conformation of Env. Two considerations may help to address the issue of why increasing 6HB stability doesn’t increase entry kinetics. First, high 6HB stability may increase the likelihood of premature 6HB formation and inactivation of Env, resulting in fewer functional Env trimers. This could slow entry kinetics if several functional Env trimers need to coalesce to form a fusion pore. Second, most kinetic experiments, including our own, report overall entry kinetics rather than the half-life of discreet, peptide-sensitive fusion intermediates. In fact, the rate constant for early steps in the fusion process, and the relative competition between peptide association rates and intermediate lifetimes, are both important for defining peptide sensitivity [60],[61]. It therefore remains possible that the 6HB-stabilizing mutations shorten the lifetime of peptide-sensitive conformational intermediates without speeding up the overall fusion process.
Our experiments measuring time of escape from N44 inhibition also indicate that resistant Envs do not lose susceptibility to N44 inhibition faster than WT Env. Two alternate explanations might account for this observation. In one case, the peptide-sensitive, pre-hairpin intermediate might indeed have a longer lifetime, but resistance can be explained by an equilibrium between the peptide-sensitive, pre-hairpin intermediate and a later, pre-bundle conformation that is insensitive to peptide. Increased bundle stability would favor the later peptide-insensitive conformation and thus fusion in the presence of peptide. Alternatively, increasing bundle stability could increase the rate of 6HB formation and shorten the lifetime of the sensitive intermediate, but the bundle-stabilizing mutations could additionally affect a different conformation of Env, resulting in slower entry kinetics. For example, the resistance mutations might also impair receptor interactions or subsequent conformational changes and delay the start of the window of susceptibility to peptide, without changing the length of time of the window of susceptibility. The recent pre-fusion structure of trimeric Env supports such a possibility because gp41, and especially HR1, makes intimate contact with the inner domain of gp120 [62]-[65]. The time of escape from peptide inhibition experiments therefore could reflect the rate-limiting steps prior to peptide inhibition and make the window of peptide susceptibility look longer in that it ends later, even if the length of the window is actually unchanged or shorter.
Conclusions
In summary, we show that mutations in HR1 and HR2 that confer resistance to peptide fusion inhibitors increase 6HB stability without conferring increased entry kinetics. Our data indicate that increased 6HB stability does not necessarily enhance entry kinetics and that N peptide inhibitors do not select for Envs with faster entry kinetics. These studies highlight an important role for HR1 and HR2 residues in influencing the relationship between stability of the final fusion-active conformation and other conformations of Env that regulates the rate of virus entry into cells.
Methods
Cells
293 T cells and U87 cells expressing high levels of CD4 and CCR5 (U87-CD4-CCR5) were provided by Dan Littman (New York University, New York, NY). HeLa cells expressing high levels of CD4 and CCR5 (JC53) were provided by David Kabat (Oregon Health and Science University, Portland, OR). 293 T and HeLa cells were passaged in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech, Inc, Manassas, VA) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, Lawrenceville, GA), 1× penicillin, 1× streptomycin, 2 mM glutamine, 1 mM HEPES buffer, and 1× non-essential amino acids (DMEM+). U87-CD4-CCR5 cells were passaged in DMEM+ supplemented with 300 μg/ml G418 Sulfate (Mediatech, Inc, Manassas, VA) and 1 μg/ml puromycin (Sigma, St. Louis, MO).
Plasmids/constructs
Env-deficient HIV-1 genome (gag/pol) plasmid pCMVΔR8.2, luciferase (Luc) reporter plasmid pHR’CMV-Luc, and expression vector pCMV/R were obtained from Gary J. Nabel (NIH, Bethesda, MD) and described previously [48]. pMM310-β-lactamase-Vpr reporter plasmid was obtained from Gregory B. Melikyan (Emory Children's Center, Atlanta, GA). JR-CSF wild-type (WT) and resistant Envs were cloned into the pCMV/R vector as previously described [46],[48]. Chimeras between the gp120 and gp41 of select Envs were constructed by swapping segments using Mfe1 and Not1 restriction sites within the construct. C-terminal tail mutations were incorporated using standard site-direct mutagenesis techniques. The entire env gene was sequenced to confirm that the desired mutations were made.
Pseudovirus production
293 T cells were seeded at 3.25 × 105cells/mL in a 10-cm dish approximately 20 h prior to transfection. Pseudovirus stocks were obtained by transfecting cells with 0.5 μg pCMV/R-Env, and either 4 μg each of pCMVΔR8.2 and pHR’CMV-Luc, or 10 μg of pCMVΔR8.2 and 5 μg of pMM310-b-lactamase-Vpr, together with Fugene 6 according to the manufacturer’s recommendations (Promega, Madison, WI). Media was changed at 18–20 h and at 48 h post-transfection, supernatants were harvested and filtered through a 0.45-μm pore-size low protein binding filter. Virus stocks were quantified by HIV-1 p24 Gag ELISA (AIDS Vaccine Program, NCI-Frederick Cancer Research and Development Center, Frederick, MD) and stored at −80°C.
Infectivity assay
U87-CD4-CCR5 cells were plated at 2 × 104 cells per well in white-walled, 96-well plates (Nunclon Delta Surface, Thermo Scientific, Denmark) and spun at 200 × g in a Sorvall RT 6000D centrifuge (Dupont, Wilmington, DE) for 3 min at 25°C the day prior to infection. Seven steps of two-fold serial dilutions of pseudoviruses containing 8 μg/ml polybrene (Sigma-Aldrich, St. Louis, MO) were applied to cells for infection. After 24 h, an additional 100 μl of warm DMEM+ was added to each well. At ~48 h post-infection, cells were lysed with buffer (Luciferase Cell Culture Lysis 5x reagent, Promega, Madison, WI), and luciferase activity was assayed using kit reagents (Luciferase Assay System, Promega, Madison, WI) according to the manufacturer’s instructions. To determine the infectivity titer, luciferase activity for replicate means of each dilution within the linear range were normalized by p24 Gag content and averaged across dilutions. The infectivity of at least two different pseudovirus lots was determined from separate experiments and normalized to the average WT titer.
Peptide inhibition assay
The N44 peptide dose–response assay was performed as described above except 300 RLU of pseudovirus was used and peptide was added prior to adding to cells. The N44 peptide was synthesized using standard 9-fluorenylmethoxy carbonyl chemistry and purified by high-pressure liquid chromatography (CBER Facility for Biotechnology Resources, Bethesda, MD). The sequence is based on residues 547 to 590 (HXB2 numbering), except for an isoleucine to glycine change at position 548 (GGVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARILAVERYLKDQ). The 50% inhibitory concentration (IC50) of N44 was determined from dose–response curves using non-linear regression analysis in Graph Pad Prism 6 (Graph Pad Prism Software, San Diego, California). Experiments were performed in duplicate and at least two different pseudovirus lots were used in independent experiments to determine the sensitivity to N44. The IC50values were normalized against WT for each individual experiment before averaging across experiments to give the mean relative resistance.
Kinetic assay
JC53 cells, frequently used for beta-lactamase kinetic assays, were plated at 2 × 104 per well in clear-bottom, black-walled, 96-well plates (Costar, Corning Inc., Corning, NY) the day prior to the assay [55]-[57],[61]. Beta-lactamase loading solution (Life Technologies, Madison, WI) was prepared at room temperature using DMEM (minus phenol red) supplemented with 25 mM HEPES, 2 μM CCF2-AM Dye (Solution A), 10 μL per mL of Solution B, 157 μL per mL of Solution C, and 3.1 mM probenecid (Sigma-Aldrich, St. Louis, MO). Cells were washed with Hanks Buffered Saline Solution (HBSS; Mediatech, Inc, Manassas, VA), prior to adding 50 μl of loading substrate to each well. Cells were allowed to take up substrate for 1 h at room temperature in the dark prior to placing on ice. Meanwhile, β-lactamase-containing pseudoviruses were first thawed at room temperature, then placed on ice and diluted into ice-cold DMEM+ supplemented with probenecid and polybrene for final concentrations of 2.5 mM and 8 μg per mL, respectively. After this initial 1.3-fold dilution, three additional 1.4-fold serial dilutions of each pseudovirus were applied in duplicate to cells that had been washed twice with HBSS containing 2.5 mM probenecid. These dilutions spanned the entire dynamic range of the assay for most pseudoviruses tested (Additional file 1: Figure S1). All manipulations were performed on ice. The 96-well plate was placed on a thermo-conductive aluminum block covered with a wet paper towel and immersed in ice to keep the cells cold throughout washing and pseudovirus application. Pseudoviruses were spinoculated in the Sorvall RT 6000D centrifuge for 30 min at 4°C and 2500 × g and then immediately placed into a Spectra Max Gemini EM (Molecular Devices, Sunnyvale, CA) fluorescence plate reader at 37°C. Florescence changes were monitored over time using a 409 nm excitation and both a 460 nm (blue) and 528 nm (green) emission detection. We measured fusion for 100 minutes to capture maximum fusion, which was comparable to time points reported by others using similar assays [56],[58]. We also noticed that control pseudoviruses without Env produced a time-dependent increase in background signal as incubation periods approach 100 min (Additional file 1: Figure S1).
To obtain the working fusion signal in florescence units, the blue:green ratio for wells mock treated only with DMEM+ was subtracted from the blue:green ratio of wells treated with pseudovirus. The signal at each time point was normalized to the signal at 100 min. The normalized signal for each replicate was averaged prior to averaging across the four dilutions to give the final entry kinetic curve. This trace was fitted to a 4-parameter logistic equation to obtain the T1/2 (min) for each pseudovirus using Graph Pad Prism 6. Three independent measurements of time to half-maximal fusion (T1/2) were determined for each Env using at least three different pseudovirus lots.
Time to escape N44 inhibition
U87-CD4-CCR5 cells were plated as described above for the infectivity assay. The following day the cells were cooled on ice, and media was removed. 100 μl of pseudovirus containing 8 μg/mL of polybrene were spinoculated in the Sorvall RT 6000D centrifuge for 30 min at 4°C and 2500 × g. Pseudovirus was then removed and 75 μl of DMEM+ warmed to 37°C was added to start infectivity and placed into a 37°C incubator. 25 μl of 20 μM (excess) N44 was then added to the cells at times corresponding to 0, 5, 10, 15, 20, 25, 35, 55, 75, 100, and 150 min incubations at 37°C. Additionally, 25 μl of diluent without N44 was added at 150 min to serve as a control to normalize for total infectivity. 24 h later, DMEM+ warmed to 37°C was added, and the assay plate was allowed to incubate for another 24 hr. Luciferease activity was then assayed as described above and normalized to the infectivity of the 150 min time point without peptide. Samples were run in duplicate, and at least two independent experiments were performed using different pseudovirus lots. The time course was fit to a 4-parameter logistic equation with a constraint that the bottom asymptote must be equal to zero to determine the T1/2 (min) using Graph Pad Prism 6. All independent measures of T1/2 (min) were then averaged.
Circular dichroism experiments
6HB stability studies were performed as previously described [46],[48]. In this study, 10 μM of each HR1 and HR2 peptide (KKR- and C1-mutant, respectively) were mixed in sodium phosphate buffer (PBS) at room temperature. A Jasco spectropolarimeter (model J-810, Jasco Inc., Easton, MD) was used to collect circular dichroism (CD) spectra of this peptide mixture. Thermal denaturation was monitored at 220 nm between 4° and 95°C. The transition mid-point temperature (T m ) was estimated from the first-derivative of the denaturation curves using Jasco software utilities.
Statistical analysis
Spearman correlation coefficients were used to determine correlations, and t-tests were performed using GraphPad Prism software. P values < 0.05 were considered significant. Comparisons were made using mean values from different pseudovirus lots for a particular Env. Unpaired, parametric two-tailed t-tests were used as indicated in the figure captions.
Additional file
Abbreviations
- Env:
-
Envelope protein
- gp41:
-
HIV envelope transmembrane subunit
- gp120:
-
HIV envelope surface subunit
- HR1 and HR2:
-
Heptad-repeat regions one and two
- 6HB:
-
Thermostable six-helix bundle
- T20:
-
Enfuvirtide
- C peptide:
-
HR2 segment of gp41
- N peptide:
-
HR1 segment of gp41
- N44:
-
N Peptide 44 residue long
- N36:
-
N peptide 36 residues long
- IZN36:
-
Trimer stabilized N peptide
- C-tail:
-
C-terminal cytoplasmic tail
- T m :
-
Transition mid-point temperature
- T1/2:
-
Time to half maximal fusion
References
Eckert DM, Kim PS: Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001, 70: 777-810. 10.1146/annurev.biochem.70.1.777.
Melikyan GB: Membrane fusion mediated by human immunodeficiency virus envelope glycoprotein. Curr Top Membr. 2011, 68: 81-106. 10.1016/B978-0-12-385891-7.00004-0.
Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, Rawat SS, Puri A, Durell S, Blumenthal R: The HIV Env-mediated fusion reaction. Biochim Biophys Acta. 2003, 1614 (1): 36-50. 10.1016/S0005-2736(03)00161-5.
De Feo CJ, Weiss CD: Escape from human immunodeficiency virus type 1 (HIV-1) entry inhibitors. Viruses. 2012, 4 (12): 3859-3911. 10.3390/v4123859.
Walker LM, Burton DR: Rational antibody-based HIV-1 vaccine design: current approaches and future directions. Curr Opin Immunol. 2010, 22 (3): 358-366. 10.1016/j.coi.2010.02.012.
Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS: Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med. 1998, 4 (11): 1302-1307. 10.1038/3293.
Wild C, Greenwell T, Shugars D, Rimsky-Clarke L, Matthews T: The inhibitory activity of an HIV type 1 peptide correlates with its ability to interact with a leucine zipper structure. AIDS Res Hum Retroviruses. 1995, 11 (3): 323-325. 10.1089/aid.1995.11.323.
Chen CH, Matthews TJ, McDanal CB, Bolognesi DP, Greenberg ML: A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: implication for viral fusion. J Virol. 1995, 69 (6): 3771-3777.
Tan K, Liu J, Wang J, Shen S, Lu M: Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A. 1997, 94 (23): 12303-12308. 10.1073/pnas.94.23.12303.
Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC: Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997, 387 (6631): 426-430. 10.1038/387426a0.
Chan DC, Kim PS: HIV entry and its inhibition. Cell. 1998, 93 (5): 681-684. 10.1016/S0092-8674(00)81430-0.
Chan DC, Fass D, Berger JM, Kim PS: Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997, 89 (2): 263-273. 10.1016/S0092-8674(00)80205-6.
He Y, Vassell R, Zaitseva M, Nguyen N, Yang Z, Weng Y, Weiss CD: Peptides trap the human immunodeficiency virus type 1 envelope glycoprotein fusion intermediate at two sites. J Virol. 2003, 77 (3): 1666-1671. 10.1128/JVI.77.3.1666-1671.2003.
Kilgore NR, Salzwedel K, Reddick M, Allaway GP, Wild CT: Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J Virol. 2003, 77 (13): 7669-7672. 10.1128/JVI.77.13.7669-7672.2003.
Chan DC, Chutkowski CT, Kim PS: Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A. 1998, 95 (26): 15613-15617. 10.1073/pnas.95.26.15613.
Furuta RA, Wild CT, Weng Y, Weiss CD: Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol. 1998, 5 (4): 276-279. 10.1038/nsb0498-276.
Lalezari JP, Henry K, O'Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M: Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med. 2003, 348 (22): 2175-2185. 10.1056/NEJMoa035026.
Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ: Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci U S A. 1994, 91 (21): 9770-9774. 10.1073/pnas.91.21.9770.
Greenberg ML, Cammack N: Resistance to enfuvirtide, the first HIV fusion inhibitor. J Antimicrob Chemother. 2004, 54 (2): 333-340. 10.1093/jac/dkh330.
Lu J, Deeks SG, Hoh R, Beatty G, Kuritzkes BA, Martin JN, Kuritzkes DR: Rapid emergence of enfuvirtide resistance in HIV-1-infected patients: results of a clonal analysis. J Acquir Immune Defic Syndr. 2006, 43 (1): 60-64. 10.1097/01.qai.0000234083.34161.55.
Sista PR, Melby T, Davison D, Jin L, Mosier S, Mink M, Nelson EL, DeMasi R, Cammack N, Salgo MP, Matthews TJ, Greenberg ML: Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS. 2004, 18 (13): 1787-1794. 10.1097/00002030-200409030-00007.
Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D: Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov. 2004, 3 (3): 215-225. 10.1038/nrd1331.
Baldwin CE, Sanders RW, Deng Y, Jurriaans S, Lange JM, Lu M, Berkhout B: Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J Virol. 2004, 78 (22): 12428-12437. 10.1128/JVI.78.22.12428-12437.2004.
Cai L, Jiang S: Development of peptide and small-molecule HIV-1 fusion inhibitors that target gp41. ChemMedChem. 2010, 5 (11): 1813-1824. 10.1002/cmdc.201000289.
Yu X, Lu L, Cai L, Tong P, Tan S, Zou P, Meng F, Chen YH, Jiang S: Mutations of Gln64 in the HIV-1 gp41 N-terminal heptad repeat render viruses resistant to peptide HIV fusion inhibitors targeting the gp41 pocket. J Virol. 2012, 86 (1): 589-593. 10.1128/JVI.05066-11.
Eggink D, Bontjer I, Langedijk JP, Berkhout B, Sanders RW: Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J Virol. 2011, 85 (20): 10785-10797. 10.1128/JVI.05331-11.
Hermann FG, Egerer L, Brauer F, Gerum C, Schwalbe H, Dietrich U, Von Laer D: Mutations in gp120 contribute to the resistance of human immunodeficiency virus type 1 to membrane-anchored C-peptide maC46. J Virol. 2009, 83 (10): 4844-4853. 10.1128/JVI.00666-08.
Lohrengel S, Hermann F, Hagmann I, Oberwinkler H, Scrivano L, Hoffmann C, von Laer D, Dittmar MT: Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J Virol. 2005, 79 (16): 10237-10246. 10.1128/JVI.79.16.10237-10246.2005.
Liu Z, Shan M, Li L, Lu L, Meng S, Chen C, He Y, Jiang S, Zhang L: In vitro selection and characterization of HIV-1 variants with increased resistance to sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem. 2011, 286 (5): 3277-3287. 10.1074/jbc.M110.199323.
Shimura K, Nameki D, Kajiwara K, Watanabe K, Sakagami Y, Oishi S, Fujii N, Matsuoka M, Sarafianos SG, Kodama EN: Resistance profiles of novel electrostatically constrained HIV-1 fusion inhibitors. J Biol Chem. 2010, 285 (50): 39471-39480. 10.1074/jbc.M110.145789.
Eggink D, Baldwin CE, Deng Y, Langedijk JP, Lu M, Sanders RW, Berkhout B: Selection of T1249-resistant human immunodeficiency virus type 1 variants. J Virol. 2008, 82 (13): 6678-6688. 10.1128/JVI.00352-08.
Berkhout B, Eggink D, Sanders RW: Is there a future for antiviral fusion inhibitors?. Curr Opin Virol. 2012, 2 (1): 50-59. 10.1016/j.coviro.2012.01.002.
Eggink D, Berkhout B, Sanders RW: Inhibition of HIV-1 by fusion inhibitors. Curr Pharm Des. 2010, 16 (33): 3716-3728. 10.2174/138161210794079218.
Rimsky LT, Shugars DC, Matthews TJ: Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J Virol. 1998, 72 (2): 986-993.
Poveda E, Rodes B, Lebel-Binay S, Faudon JL, Jimenez V, Soriano V: Dynamics of enfuvirtide resistance in HIV-infected patients during and after long-term enfuvirtide salvage therapy. J Clin Virol. 2005, 34 (4): 295-301. 10.1016/j.jcv.2005.02.004.
Eggink D, Langedijk JP, Bonvin AM, Deng Y, Lu M, Berkhout B, Sanders RW: Detailed mechanistic insights into HIV-1 sensitivity to three generations of fusion inhibitors. J Biol Chem. 2009, 284 (39): 26941-26950. 10.1074/jbc.M109.004416.
Mink M, Mosier SM, Janumpalli S, Davison D, Jin L, Melby T, Sista P, Erickson J, Lambert D, Stanfield-Oakley SA, Salgo M, Cammack N, Matthews T, Greenberg ML: Impact of human immunodeficiency virus type 1 gp41 amino acid substitutions selected during enfuvirtide treatment on gp41 binding and antiviral potency of enfuvirtide in vitro. J Virol. 2005, 79 (19): 12447-12454. 10.1128/JVI.79.19.12447-12454.2005.
Nameki D, Kodama E, Ikeuchi M, Mabuchi N, Otaka A, Tamamura H, Ohno M, Fujii N, Matsuoka M: Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp41 and Rev-responsive element functions. J Virol. 2005, 79 (2): 764-770. 10.1128/JVI.79.2.764-770.2005.
Menzo S, Castagna A, Monachetti A, Hasson H, Danise A, Carini E, Bagnarelli P, Lazzarin A, Clementi M: Genotype and phenotype patterns of human immunodeficiency virus type 1 resistance to enfuvirtide during long-term treatment. Antimicrob Agents Chemother. 2004, 48 (9): 3253-3259. 10.1128/AAC.48.9.3253-3259.2004.
Ray N, Blackburn LA, Doms RW: HR-2 mutations in human immunodeficiency virus type 1 gp41 restore fusion kinetics delayed by HR-1 mutations that cause clinical resistance to enfuvirtide. J Virol. 2009, 83 (7): 2989-2995. 10.1128/JVI.02496-08.
Tolstrup M, Selzer-Plon J, Laursen AL, Bertelsen L, Gerstoft J, Duch M, Pedersen FS, Ostergaard L: Full fusion competence rescue of the enfuvirtide resistant HIV-1 gp41 genotype (43D) by a prevalent polymorphism (137K). AIDS. 2007, 21 (4): 519-521. 10.1097/QAD.0b013e3280187558.
Bai X, Wilson KL, Seedorff JE, Ahrens D, Green J, Davison DK, Jin L, Stanfield-Oakley SA, Mosier SM, Melby TE, Cammack N, Wang Z, Greenberg ML, Dwyer JJ: Impact of the enfuvirtide resistance mutation N43D and the associated baseline polymorphism E137K on peptide sensitivity and six-helix bundle structure. Biochemistry. 2008, 47 (25): 6662-6670. 10.1021/bi702509d.
Xu L, Pozniak A, Wildfire A, Stanfield-Oakley SA, Mosier SM, Ratcliffe D, Workman J, Joall A, Myers R, Smit E, Cane PA, Greenberg ML, Pillay D: Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob Agents Chemother. 2005, 49 (3): 1113-1119. 10.1128/AAC.49.3.1113-1119.2005.
Bewley CA, Louis JM, Ghirlando R, Clore GM: Design of a novel peptide inhibitor of HIV fusion that disrupts the internal trimeric coiled-coil of gp41. J Biol Chem. 2002, 277 (16): 14238-14245. 10.1074/jbc.M201453200.
Root MJ, Kay MS, Kim PS: Protein design of an HIV-1 entry inhibitor. Science. 2001, 291 (5505): 884-888. 10.1126/science.1057453.
Zhuang M, Wang W, De Feo CJ, Vassell R, Weiss CD: Trimeric, coiled-coil extension on peptide fusion inhibitor of HIV-1 influences selection of resistance pathways. J Biol Chem. 2012, 287 (11): 8297-8309. 10.1074/jbc.M111.324483.
Desmezieres E, Gupta N, Vassell R, He Y, Peden K, Sirota L, Yang Z, Wingfield P, Weiss CD: Human immunodeficiency virus (HIV) gp41 escape mutants: cross-resistance to peptide inhibitors of HIV fusion and altered receptor activation of gp120. J Virol. 2005, 79 (8): 4774-4781. 10.1128/JVI.79.8.4774-4781.2005.
Wang W, De Feo CJ, Zhuang M, Vassell R, Weiss CD: Selection with a peptide fusion inhibitor corresponding to the first heptad repeat of HIV-1 gp41 identifies two genetic pathways conferring cross-resistance to peptide fusion inhibitors corresponding to the first and second heptad repeats (HR1 and HR2) of gp41. J Virol. 2011, 85 (24): 12929-12938. 10.1128/JVI.05391-11.
Izumi K, Nakamura S, Nakano H, Shimura K, Sakagami Y, Oishi S, Uchiyama S, Ohkubo T, Kobayashi Y, Fujii N, Matsuoka M, Kodama EN: Characterization of HIV-1 resistance to a fusion inhibitor, N36, derived from the gp41 amino-terminal heptad repeat. Antiviral Res. 2010, 87 (2): 179-186. 10.1016/j.antiviral.2010.04.011.
Reeves JD, Gallo SA, Ahmad N, Miamidian JL, Harvey PE, Sharron M, Pohlmann S, Sfakianos JN, Derdeyn CA, Blumenthal R, Hunter E, Doms RW: Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc Natl Acad Sci U S A. 2002, 99 (25): 16249-16254. 10.1073/pnas.252469399.
Gallo SA, Puri A, Blumenthal R: HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry. 2001, 40 (41): 12231-12236. 10.1021/bi0155596.
Abrahamyan LG, Mkrtchyan SR, Binley J, Lu M, Melikyan GB, Cohen FS: The cytoplasmic tail slows the folding of human immunodeficiency virus type 1 Env from a late prebundle configuration into the six-helix bundle. J Virol. 2005, 79 (1): 106-115. 10.1128/JVI.79.1.106-115.2005.
Reeves JD, Lee FH, Miamidian JL, Jabara CB, Juntilla MM, Doms RW: Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J Virol. 2005, 79 (8): 4991-4999. 10.1128/JVI.79.8.4991-4999.2005.
Eckert DM, Kim PS: Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci U S A. 2001, 98 (20): 11187-11192. 10.1073/pnas.201392898.
Cavrois M, De Noronha C, Greene WC: A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002, 20 (11): 1151-1154. 10.1038/nbt745.
Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB: HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009, 137 (3): 433-444. 10.1016/j.cell.2009.02.046.
Putcharoen O, Lee SH, Henrich TJ, Hu Z, Vanichanan J, Coakley E, Greaves W, Gulick RM, Kuritzkes DR, Tsibris AM: HIV-1 clinical isolates resistant to CCR5 antagonists exhibit delayed entry kinetics that are corrected in the presence of drug. J Virol. 2012, 86 (2): 1119-1128. 10.1128/JVI.06421-11.
Tsibris AM, Hu Z, Paredes R, Leopold KE, Putcharoen O, Schure AL, Mazur N, Coakley E, Su Z, Gulick RM, Kuritzkes DR: Vicriviroc resistance decay and relative replicative fitness in HIV-1 clinical isolates under sequential drug selection pressures. J Virol. 2012, 86 (12): 6416-6426. 10.1128/JVI.00286-12.
Louis JM, Bewley CA, Clore GM: Design and properties of N (CCG)-gp41, a chimeric gp41 molecule with nanomolar HIV fusion inhibitory activity. J Biol Chem. 2001, 276 (31): 29485-29489. 10.1074/jbc.C100317200.
Kahle KM, Steger HK, Root MJ: Asymmetric deactivation of HIV-1 gp41 following fusion inhibitor binding. PLoS Pathog. 2009, 5 (11): e1000674-10.1371/journal.ppat.1000674.
Miyauchi K, Kozlov MM, Melikyan GB: Early steps of HIV-1 fusion define the sensitivity to inhibitory peptides that block 6-helix bundle formation. PLoS Pathog. 2009, 5 (9): e1000585-10.1371/journal.ppat.1000585.
Bartesaghi A, Merk A, Borgnia MJ, Milne JL, Subramaniam S: Prefusion structure of trimeric HIV-1 envelope glycoprotein determined by cryo-electron microscopy. Nat Struct Mol Biol. 2013, 20 (12): 1352-1357. 10.1038/nsmb.2711.
Mao Y, Wang L, Gu C, Herschhorn A, Xiang SH, Haim H, Yang X, Sodroski J: Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat Struct Mol Biol. 2012, 19 (9): 893-899. 10.1038/nsmb.2351.
Lyumkis D, Julien JP, De Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, Wilson IA, Ward AB: Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science. 2013, 342 (6165): 1484-1490. 10.1126/science.1245627.
Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA: Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013, 342 (6165): 1477-1483. 10.1126/science.1245625.
Acknowledgements
We thank Drs. Tzanko Stantchev, Hana Golding, and Ira Berkower (U.S. Food and Drug Administration, Bethesda, MD) for critical reading of the manuscript. This work was supported in part by institutional funds from the U.S. Food and Drug Administration and the Intramural AIDS Targeted Antiviral Program from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CD and CW designed the study and wrote the manuscript. CD, WW, and MH performed the experiments. WW, MZ and RV provided constructs and critical intellectual input. All authors read and approved the final draft.
Electronic supplementary material
12977_2014_86_MOESM1_ESM.pdf
Additional file 1: Figure S1.: Raw kinetic data. (A) Representative real time entry for four serial dilutions (1.3-fold to 3.6-fold) of the WT β-lam pseudovirus. (B) Representative real time entry for the lowest dilutions of either WT Env, C1-C1 Env, or the no HIV Env control β-lam pseudoviruses. (C) Entry kinetics for WT-WT and C1-C1 using the no HIV Env as the background control. Here the kinetic relationships are preserved using this alternate background correction. Error bars, standard deviation of mean for each time point. Inset, T1/2 of each of the curves in (±95% C.I). (PDF 411 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
De Feo, C.J., Wang, W., Hsieh, ML. et al. Resistance to N-peptide fusion inhibitors correlates with thermodynamic stability of the gp41 six-helix bundle but not HIV entry kinetics. Retrovirology 11, 86 (2014). https://doi.org/10.1186/s12977-014-0086-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-014-0086-8