
Abstract
Amyotrophic lateral sclerosis (ALS) is an adult-onset progressive neurodegenerative disorder characterized by the loss of upper and lower motor neurons in the brain and spinal cord. Accumulating evidence suggests that ALS is not solely a neuronal cell- or brain tissue-autonomous disease and that neuroinflammation plays a key role in disease progression. Furthermore, whereas both CD4 and CD8 T cells were observed in spinal cords of ALS patients and in mouse models of the disease, their role in the neuroinflammatory process, especially considering their functional changes with age, is not fully explored. In this study, we revealed the structure of the CD4 T-cell compartment during disease progression of early-onset SOD1G93A and late-onset SOD1G37R mouse models of ALS. We show age-related changes in the CD4 T-cell subset organization between these mutant SOD1 mouse models towards increased frequency of effector T cells in spleens of SOD1G37R mice and robust infiltration of CD4 T cells expressing activation markers and the checkpoint molecule PD1 into the spinal cord. The frequency of infiltrating CD4 T cells correlated with the frequency of infiltrating CD8 T cells which displayed a more exhausted phenotype. Moreover, RNA-Seq and immunohistochemistry analyses of spinal cords from SOD1G37R mice with early clinical symptoms demonstrated immunological trajectories reminiscent of a neurotoxic inflammatory response which involved proinflammatory T cells and antigen presentation related pathways. Overall, our findings suggest that age-related changes of the CD4 T cell landscape is indicative of a chronic inflammatory response, which aggravates the disease process and can be therapeutically targeted.
Similar content being viewed by others


Background
ALS is a fatal neurodegenerative disease characterized by the progressive loss of upper and lower motor neurons in the brain and spinal cord (SC) [1]. A number of risk factors for ALS have been hypothesized; however, the only recognized risk factors to date are older age, male sex, and a family history [2]. About 90% of ALS cases are sporadic, occurring without a known genetic cause. However, the remaining 10% are familial cases that are usually inherited in a dominant manner [1]. The SOD1 gene, which encodes for Cu/Zn superoxide dismutase, was the first gene found to be associated with ALS [3]. Mutations in this gene are responsible for approximately 20% of familial ALS cases and about 2% of sporadic ALS cases, making SOD1 one of the most studied genes in this disease [4, 5]. Different mutant SOD1 transgenic mice develop a disease with a similar pathogenesis to ALS, which includes motor neuron degeneration, neuroinflammation, paralysis and early death [4].
The pathology of ALS is not limited to neuronal cell-autonomous mechanisms [6]. For example, it has been shown that mice overexpressing human mutant SOD1 protein only in neurons, did not develop motor impairment [7] or had a late disease onset [8]. Further experiments showed that specific deletion of mutant SOD1 from motor neurons delayed disease onset and early phase of disease progression with no effect on the late phase of the disease [9, 10]. In contrast, deletion of mutant SOD1 from astrocytes or microglia strongly delayed the late phase of the disease [9, 11]. Therefore, non-neuronal cells play an important role in motor neuron loss and disease progression [10, 12].
Neuroinflammation is an essential host defense mechanism to protect the brain from infection or injury and restore normal structure and function [13, 14]. However, chronic inflammation can induce cytotoxicity and worsen severity of different neurodegenerative diseases such as Parkinson’s disease (PD) [15], Alzheimer’s disease (AD) [16], multiple sclerosis (MS) [17] and ALS [14, 18]. Neuroinflammation is characterized by increased microglial and astrocytic activation, leukocyte infiltration into the brain and SC and elevated levels of proinflammatory cytokines [18, 19]. In the context of ALS, neuroinflammation plays a critical role in the disease progression [18]. The dysregulated inflammatory responses, characterized by aberrant microglial activation and excessive proinflammatory cytokines, contribute to the neurodegeneration observed in ALS [20].
Microglia, as the resident macrophages of the central nervous system (CNS), play a pivotal role in maintaining brain homeostasis [21]. However, under pathological conditions, these cells can transition from their surveillant homeostatic state to various activation states including subsets which exhibit antigen presenting cell (APC) phenotypes [22, 23]. Through their MHCII-based interactions with various CD4 T-cell subsets (e.g., proinflammatory Th1 or Th17 cells [24] or anti-inflammatory Tregs [25]), microglia can contribute to inflammation-driven neuronal damage or repair. However, in the context of ALS, the nature and consequences of such interactions remain largely unknown.
In mutant SOD1G93A mice, CD4 T helper cells were observed in lumbar SC at early disease stage, whereas at end stage, both CD4 and CD8 T lymphocytes were present [18, 26, 27]. Genetic deletion of functional T cells or CD4 T cells resulted in accelerated disease progression, increased mRNA levels of NOX2 (NADPH oxidase isoform which catalyze the production of superoxide molecules) and proinflammatory cytokines, and decreased levels of the neurotrophic factors IGF-1, BDNF and GDNF [27]. Reconstitution of T cells by bone marrow transplantation, prolonged the survival, reduced proinflammatory responses, restored neuroprotective factors and inhibited M1 microglial activation [18, 27]. In ALS patients, peripheral blood cell counts of CD3, CD4, CD8 and CD3+CD56+ T cells, natural killer cells and granulocytes were increased [28]. Moreover, whereas a higher frequency of effector CD4 T cells in both blood and CSF was linked to decreased survival, increased frequencies of activated regulatory T (Treg) cells in the blood was associated with improved survival [29].
While the SOD1G93A mouse model has been widely used in ALS research primarily due to rapid progression of symptoms [30], such early onset of disease may not represent the impact of age-related immunological changes as they appear in humans. The CD4 T-cell subsets are critical mediators of immune responses, and their phenotypic and functional diversity have been shown to be modulated by age [31,32,33].
In this research, we hypothesized that there is a significant age-related CD4 T-cell component in ALS which impacts the neuroinflammatory response in the CNS. We observed distinct organization of CD4 T-cell subsets between early-onset and late-onset SOD1 mouse models. Moreover, our findings establish a positive correlation between effector memory (EM) and cytotoxic CD4 T cells with disease progression. In addition, we show upregulation in expression of genes and proteins involved in formation of immunological synapse. Taken together, these findings emphasize the significant influence of effector CD4 populations on the progression of the disease in late-onset mutant SOD1G37R mice.
Methods
Mice
Transgenic mice expressing human SOD1G93A [30] and loxSOD1G37R [9] were bred with C57BL6 mice and genotyped by PCR of DNA extracted from tails. All mice were maintained in the animal facility of Ben-Gurion University of the Negev using standard protocols. All procedures involving animals were consistent with the requirements of the Animal Care and Use Committees of Ben-Gurion University of the Negev.
Tissue collection and processing
Spleen tissue collection. SOD1G93A and SOD1G37R mice were killed using overdose of isoflurane. Immediately after, spleens were harvested, and blood was collected following incision of the mouse right atrium. Next, perfusion was performed using 20 ml of ice-cold Dulbecco’s phosphate buffered saline (PBS) and SC was collected. Both spleens and SCs were weighed and kept in HBSS solution on ice.
Tissue processing—spleens. Spleens were mashed into a 70-µm cell strainer and 300 µl of ammonium-chloride-potassium (ACK) buffer (Lonza, Switzerland) was added for 1.5 min to lyse red blood cells. For flow cytometry and splenocyte culture, cells were diluted into 2,500,000 cell/ml in RPMI 1640 media (Thermo Fisher Scientific, MA, United States), supplemented with of 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, MA, United States), HEPES buffer solution (10 mM) (Sartorius, Germany), 1 mM sodium pyruvate solution (Sartorius, Germany), 10 mM MEM non-essential amino acids solution (Sartorius, Germany), 1% penicillin–streptomycin solution (Sartorius, Germany), and 50 μM 2-mercaptoethanol, Molecular Biology Grade (Sigma-Aldrich, MO, United States).
Spinal cord tissue collection. SOD1G93A and SOD1G37R mice were killed using overdose of isoflurane perfused with cold PBS. Immediately after, SCs were collected by hydraulic extrusion. The procedure involved isolating the spinal column by decapitating the head and cutting proximal to the pelvic bone. Next, a syringe filled with PBS was carefully positioned at the distal end of the spinal column and steady pressure was applied to the syringe facilitating the extrusion of the SC.
Tissue processing—spinal cord. Upon extrusion of the SCs, they were placed in Hanks' Balanced Salt Solution (HBSS) (Sartorius, Germany) and kept on ice. Then, to dissociate the tissue, the Neural Tissue Dissociation Kit (Miltenyi Biotec, Germany) was used following the manufacturer’s instructions, with slight adjustments to the protocol. Specifically, we excluded the papain incubation step, as it was found to impact the integrity of specific cell surface epitopes. Instead, to ensure tissue dissociation, we added physical dissociation step using a glass pipette, in combination with passing the tissue through a 70-μm cell strainer to eliminate larger particles.
Flow cytometry
For extracellular staining, cells were washed 3 times with FACS Buffer (2% fetal bovine serum and 1 mM EDTA in PBS). Then, cells were incubated with TruStain FcX (Biolegend, CA, United States) for 5 min in 4°C. Next, cells were incubated with antibody mix for 20 min in 4 °C. For SC cells, the antibodies that were used for extracellular membranal antigen staining are detailed in Additional file 2: Table S1. After staining with extracellular antibodies, cells were washed 3 times and were run using CytoFLEX LX instrument (Beckman Coulter, CA, United States).
For splenocytes, the antibodies that were used for extracellular membranal antigen staining are detailed in Additional file 2: Table S1. After staining with extracellular antibodies, splenocytes were washed 3 times, fixed and permeabilized using Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, MA, United States) following manufacturer’s instructions. The next day, cells were washed 3 times with permeabilization buffer and blocked using rat serum (2 µl for 90 µl permeabilization buffer) for 5 min in room temperature. Then, cells were incubated with intracellular antibody mix for 20 min at room temperature. The antibodies that were used for intracellular staining are detailed in Additional file 2: Table S1. Next, cells were washed 3 times with permeabilization buffer and FACS staining buffer was added in the last wash and samples were run using CytoFLEX LX instrument (Beckman Coulter, CA, United States). Data were analyzed using CytExpert Acquisition and Analysis Software 2.4 (Beckman Coulter, CA, United States) and FlowJo software v10.7 (BD, NJ, United States). Gating strategies excluded dead cells, clumps, and debris and were set based on fluorescence minus one (FMO) and unstained samples for splenocytes and unstained samples for SC cells. All CD4 T-cell populations were gated from lymphocytes (SSC-A vs FSC-A), live cells (Fixable Viability Dye vs FSC-A), single cells (SSC-A vs SSC-H) and were calculated as percentage from CD3+CD4+ or from effector memory (EM) (CD44+CD62L−) populations (Additional file 1: Fig. S1A).
Bulk RNA sequencing
Mice were euthanized with an overdose of isoflurane and perfused with cold PBS. Then, SCs were extracted as detailed in tissue collection and processing section, transferred into RNase-free tubes and immediately cryopreserved using liquid nitrogen. RNA was extracted from lumbar region of SC using RNeasy plus mini kit (Qiagen, Germany), according to manufacturer’s instructions. RNA was quantified using Qubit (Thermo Fisher Scientific, MA, United States) and its integrity was assessed using Bioanalyzer (Agilent Technologies, CA, United States). RNA was diluted to a concentration of 5–10 ng/µl and MARS-Seq protocol was used to generate RNA-seq libraries [34, 35].
RNA-Seq data analysis
Analysis was performed on combined data from 2 independent experiments with overall 10 SOD1G37R mice and 10 littermate control mice. One experiment included 3 female SOD1G37R mice, 3 female littermate control mice, 3 male SOD1G37R mice and 3 male littermate control mice. The other experiment included 4 male SOD1G37R mice and 4 male littermate control mice.
Initial analysis of the raw sequence reads was carried out using the NeatSeq-Flow platform [36]. The sequences were quality trimmed and filtered using Trim Galore (v0.4.5) and cutadapt (v1.15). Alignment of the reads to the mouse genome (GRCm38) was done using STAR (v2.5.2a) [37]. The number of reads per gene per sample was counted using RSEM (v1.2.28) [38]. Quality assessment (QA) of the process was carried out using FASTQC (v0.11.8) and MultiQC (v1.0.dev0) [39]. After trimming, each sample had on average 79M reads with an average sequence length of 74.4 bp.
Statistical testing for identification of differentially expressed genes, batch correction, gene annotation, clustering and enrichment analysis were performed with the DESeq2 module within the NeatSeq-Flow platform [36]. Batch correction was done using the SVA/Combat R package. Gene annotation was done using the "AnnotationHub" R package (snapshot: 2020–04-27). The statistical analysis was performed using the DESeq2 [40] R package. For comparison (Contrast) between groups, the statistical model considered two effects: the treatment group and the batch (experiment). The analysis produced P-adjusted, and fold of change (FC) per gene. Genes with P-adjusted < 0.05 were considered Differentially Expressed (DE). KEGG Enrichment analysis was performed using the clusterProfiler [41] (v3.16.0) R package.
Immunohistochemistry
Mice were euthanized with an overdose of isoflurane and perfused with cold PBS. Then, SCs were extracted as detailed in tissue collection and processing, immersed in 4% paraformaldehyde solution at 4 °C overnight, transferred to a freshly prepared 30% sucrose solution at 4 °C for 24 h and fixed in OCT (Tissue-Tek, Torrance, CA). Transverse sections (40 µm) of SCs were produced using a Leica CM3050S Research Cryostat (Leica Biosystems, Germany) with object and chamber temperature of − 25 °C. Sections kept at − 20 °C in cryopreservation solution (25% glycerol, 25% ethylene glycol, and 50% PBS). Sections were rinsed twice in 0.05% Tween 20 washing solution and permeabilized in a 0.5% Triton X-100 solution for 30 min. Prior to staining, sections were blocked with 10% serum of secondary antibody and 1% BSA solution. The primary antibodies that were used for immunohistochemistry staining are detailed in Additional file 3: Table S2. The antibodies used for secondary staining are detailed in Additional file 3: Table S2.
Confocal image analysis
Confocal images were generated with a 4-channel Olympus XI81-ZDC confocal microscope (Olympus, Hamburg, Germany) at a 1024 × 1024pixel resolution with ×20, or ×60 objectives. To generate 3D images, z-stack of at least 40 μm thickness, with serial images taken every 1 μm, was imaged using confocal microscope and reconstructed using IMARIS 9.8 software (Oxford Instruments, United Kingdom).
Statistical analysis
Data are presented as mean ± SEM. Statistical evaluations were performed using the non-parametric Mann–Whitney U test, Spearman's rank correlation test, and Wilcoxon matched-pairs signed rank test Further details of the RNA-Seq statistical analysis are detailed in RNA-Seq data analysis section. All analyses were executed with a P < 0.05 threshold for statistical significance.
Results
CD4 T-cell subsets in spleens of symptomatic late-onset SOD1G37R mice exhibit enhanced effector functions compared with symptomatic early-onset SOD1G93A mice
Splenocytes were isolated from spleens of symptomatic SOD1G37R mice (stage 2, approximately 370 days of age) and SOD1G93A (stage 2, approximately 130 days of age), during which foot-dragging or toe-curling is observed during walking [42] (Fig. 1A).


Increased frequencies of effector CD4 T-cell subsets in spleens of symptomatic, late-onset, SOD1G37R mice. A Splenocytes from mutant SOD1G37R and SOD1G93A mice were mechanically dissociated, immunostained for CD4 T-cell markers, and analyzed with flow cytometry. CD4 T-cell populations were gated from lymphocytes (SSC-A vs FSC-A), live cells (Fixable Viability Dye vs FSC-A), and single cells (SSC-A vs SSC-H). B CD4 T-cell subsets in late-onset SOD1G37R (upper panels) and early-onset SOD1G93A (lower panels) mice (gating strategy is shown in Additional file 1: Fig. S1A). C tSNE plot of CD4 T-cell subset distribution in symptomatic late-onset SOD1G37R (n = 3) and in early-onset SOD1G93A mice (n = 3). D–O CD4 T-cell subset frequency in spleens of NT mice (n = 15, D–F and H–M; n = 10, G, N and O) and SOD1G37R mice (n = 23, D–F and H–M; n = 16, G, N and O), shown as percentage of CD4 or EM CD4 T cells (mean ± SEM). *P ≤ 0.05, two-tailed Mann–Whitney U-test
To assess general differences in CD4 T-cell distribution between late-onset SOD1G37R and early-onset SOD1G93A mice, tSNE analysis of flow cytometry data was performed. tSNE plot indicates differences in CD4 T-cell subset distribution between both models (Fig. 1C). Compared with early-onset SOD1G93A mice, splenocytes from late-onset SOD1G37R mice exhibited increased frequencies of EM CD4 T cells, Tregs and cytotoxic CD4 T cells (CTLs) along with reduced frequencies of the naïve population (Fig. 1C and Additional file 4: Table S3), similar to littermate controls (Additional file 1: Fig. S1B and Additional file 4: Table S3). When comparing to littermate controls, late-onset SOD1G37R mice exhibited no difference in frequencies of CD4 (CD3+CD4+), EM (CD3+CD4+CD44+CD62L−), naïve (CD3+CD4+CD44+CD62L−), Tregs (CD3+CD4+FOXP3+), Th17 (CD3+CD4+RORγ+), and EM Th17 (CD3+CD4+CD44+CD62L−RORγ+) cells (Fig. 1D–I). Notably, splenocytes from the late-onset SOD1G37R mice exhibited increased frequencies of proinflammatory Th1 (CD3+CD4+Tbethigh), EM Th1 (CD3+CD4+ CD44+CD62L−Tbethigh), anti-inflammatory Th2 (CD3+CD4+ GATA3high), but not EM Th2 (CD3+CD4+ +CD44+CD62L−GATA3high) cells (Fig. 1J–M); differences which were not observed in SOD1G93A mice (Additional file 1: Fig. S1C-N and Additional file 4: Table S3). Interestingly, we also observed reduced frequencies of CD4 CTLs (CD3+CD4+CCL5+EOMES+) and EM CD4 CTLs (CD3+CD4+CD44+CD62L−CCL5+EOMES+) (Fig. 1N–O), cells which were recently implicated in aging and ALS [29, 31]. Together, our results reveal a shift with age and disease towards effector phenotypes of CD4 T cells in symptomatic late-onset SOD1G37R which may aggravate disease progression.
Effector phenotypes of CD4 T cells correlate with disease severity in SOD1G37R mice
To determine whether alterations in frequencies of CD4 T-cell subsets correlate with disease progression, SOD1G37R mice were subjected to weekly weight assessment from disease onset. Upon reaching stage 2, mice were killed, and spleens were excised for flow cytometry analysis (Fig. 2A). As shown in Fig. 2B, weight plotted over time for both SOD1G37R mice and age- and sex-matched littermate controls shows that individual SOD1G37R mice variably lose weight during disease progression (Fig. 2B). To determine whether age- and disease-related CD4 T cell subsets contribute to disease severity, we correlated their frequency with weight ratio, defined as the weight at stage 2 (approximately 370 days old), divided by weight at 240 days old. Lower weight ratio values indicate enhanced disease progression in SOD1G37R mice [42]. Notably, whereas the frequency of CD3+, CD4+, naïve CD4 T cells, Tregs, and Th2/Th1 ratio did not correlate with weight loss (Fig. 2C–G), frequencies of EM CD4 T cells and CD4 CTLs correlated with weight loss (Fig. 2H, I). In addition, there was a strong trend for the correlation between frequencies of exhausted CD4 T cells and weight loss (Fig. 2J). These findings highlight a potential role of circulating effector T cells and CD4 CTLs in shaping the disease course in SOD1G37R mice.

Cytotoxic and effector phenotypes of CD4 T-cell populations correlate with disease progression of mutant SOD1G37R mice. A Mice were weighted from disease onset until symptomatic stage 2. Spleens were mechanically dissociated, immunostained for CD4 T-cell markers, and flow cytometry was performed. B Weight loss curves of SOD1G37R mice starting from disease onset (approximately 240 days). C–J Correlations between CD4 T-cell populations and weight ratio in SOD1G37R mice (n = 21, C–G and I–J; n = 17, H). *P ≤ 0.05, Spearman's rank correlation
CD4 T cells infiltrate the spinal cord of SOD1G37R mice exhibiting effector and exhausted phenotypes
To determine whether the correlation of effector CD4 T cells with disease severity parallel the inflammatory process in the SC, we characterized the infiltrating T cells with flow cytometry. To that end, whole SCs were isolated from symptomatic mutant SOD1G37R and littermate controls (stage 2, approximately 370 days of age) and symptomatic mutant SOD1G93A (stage 2, approximately 130 days of age) and age-matched littermate controls, mechanically dissociated, and stained for surface receptors. All CD4 T-cell subsets were calculated as event number or percentage from CD3+ CD4+ or CD3+ CD8+ T cells (Fig. 3A, B). Whereas the event number of CD4 and CD8 T cells in whole SCs was greater in SOD1G37R mice as compared with littermate controls (Fig. 3C, D), there was only a trend increase in SCs of SOD1G93A mice (Additional file 1: Fig. S2). Furthermore, frequencies of infiltrating CD4 and CD8 T cells were strongly correlated in SOD1G37R mice (Fig. 3E). Within the CD4 T-cell population of SOD1G37R mice, event number of CXCR3+CCR6− (Th1), CD81 (induced in formation of immunologic synapse) [43], PD1 and LAG-3 (inhibitory receptors) [44] and CD69 (activation marker) [43] were all increased compared with littermate controls (Fig. 3F). Similarly, within CD8 T cells, Lag3, CD81, PD1 and CD69 were highly expressed compared with controls (Fig. 3G). Notably, whereas most of the activated CD4+ CD69+ T cells (77.77% ± 2.34% of the infiltrating CD4 T cells) in the SC of SOD1G37R mice were CD81+ (38.37% ± 2.65%), most of the activated CD8 T cells (91.74% ± 1.05% of the infiltrating CD8 T cells) were Lag3+ (46.38% ± 4.33) and PD1+ Lag3+ (48.44% ± 4.44%) (Fig. 3H), indicating a pronounced T-cell response with greater exhaustion of the CD8 subset.

Activated and exhausted CD4 T cells infiltrate the spinal cord of mutant SOD1G37R mice. A SCs from symptomatic mutant SOD1G37R mice were mechanically dissociated and immunostained for flow cytometry. B Gating strategy for flow cytometry analysis; all T-cell populations were gated from lymphocytes (SSC-A vs FSC-A), live cells (Fixable Viability Dye vs FSC-A), single cells (SSC-A vs SSC-H). C, D Event number of CD4 and CD8 T cells in the SC of NT (n = 11) and symptomatic SOD1G37R (n = 19) mice. E Correlation of event number of CD4 T cells and CD8 T cells. ****P ≤ 0.0001, Spearman's rank correlation. F Event numbers representing cells expressing CD4 T-cell receptors in SCs of NT (n = 11) and SOD1G37R (n = 19) mice. G Event numbers representing cells expressing CD8 T-cell receptors in SCs of NT (n = 11) and SOD1G37R (n = 19) mice. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, Mann–Whitney U test. H Percentage of CD69highCD4+ and CD69highCD8+ T cells expressing activation and exhaustion receptors (mean ± SEM) in SC of symptomatic SOD1G37R mice. ****P ≤ 0.0001, Wilcoxon matched-pairs signed rank test.
Neuroinflammation and T cell-mediated pathways are upregulated in spinal cords of mutant SOD1G37R mice
To explore the genes involved in disease progression of mutant SOD1G37R mice, RNA was extracted from lumbar SCs of 10 SOD1G37R and 10 littermate control mice and was subjected to bulk RNA-seq (Fig. 4A and Additional file 5: Table S4). A principal component analysis (PCA) was conducted and demonstrated a clear separation between SOD1G37R mice and controls (Fig. 4B). A volcano plot prominently highlighted upregulated genes related to immune cell activation (e.g., IL-2rg, CD44), complement system (e.g., C1qa, C1qc, C4b), and leukocyte migration (e.g., ICAM-1) (Additional file 1: Fig. S3A). Accordingly, Gene Ontology (GO) analysis highlights various processes related to immune system activation and inflammation (Fig. 4C and Additional file 6: Table S5) including an upregulation of genes related to immune system process (gene ratio: 0.2438, Padj = 8.278E−63) and cytokine production (gene ratio: 0.1003, Padj = 3.770E−38) (Fig. 4C, D). Furthermore, there was upregulation of genes associated with CD4 T-cell activation pathway (gene ratio: 0.0144, Padj = 9.505E−07); for instance, CD44 which contributes to the activation, migration, and effector functions of T cells; CD81 which plays a role in various aspects of CD4 T-cell activation and function, CD3 signaling, immune synapse formation and migration [43]; IL2RG which is a critical component of IL-2 receptor complexes, and its presence is essential for various CD4 T-cell functions, including activation, proliferation, differentiation, and immune regulation (Fig. 4C, D; Additional file 6: Table S5).

Immune and antigen presentation related pathways are upregulated in the spinal cord of mutant SOD1G37R mice. A The lumbar region of the SC was extracted and cryopreserved, RNA was extracted and quantified, library was prepared and bulk RNA-seq was performed. B PCA analysis representing the gene expression profiles of symptomatic mutant SOD1G37R mice (n = 10) compared with NT mice (n = 10). C Plot showing the ten most upregulated GO biological processes in the SC of symptomatic mutant SOD1G37R mice compared with NT mice. D Heatmap of differentially expressed genes (see arrows) in the SC of symptomatic mutant SOD1G37R and NT mice that are involved in CD4 T-cell activation biological processes (GO:0035710). E Upregulated immune-related KEGG pathways. F Genes upregulated (red) or unchanged (grey) in KEGG pathway for antigen processing and presentation
KEGG pathway analysis provides further insight into mechanisms that support CD4 T-cell migration and activity. These include upregulation of the leukocyte transendothelial migration pathway (gene ratio: 0.0417, Padj = 7.332E−11) [45], NF-kappa B signaling pathway (gene ratio: 0.0359 Padj = 6.816E−08), Th17 cell differentiation (gene ratio: 0.0281, Padj = 9.489E−05), and antigen processing and presentation (gene ratio: 0.0204, Padj = 0.00126) (Fig. 4E; Additional file 7: Table S6) [46, 47]. As indicated in GO analysis (Additional file 1: Fig. S3B), Fig. 4F depicts a detailed representation of the antigen processing and presentation pathway, emphasizing genes central to enzymatic processing of antigens in the endosome (i.e., AEP and CTSB) and genes related to MHCII complex assembly (i.e., Ii, MHCII and CTSB/L/S), which were notably enhanced in SOD1G37R mice (red boxes). Together, these results strongly suggest that CD4 T cells play a crucial role in the neuroinflammation observed in SOD1G37R mice, likely through their increased activation, altered trafficking, and potential skewing towards a more inflammatory phenotype.
Infiltrating CD4 T cells interact with antigen presenting cells in spinal cords of mutant SOD1G37R mice
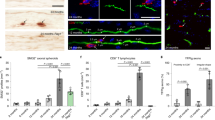
To investigate the potential activation of effector CD4 T cells by microglia, we performed immunostaining on lumbar SC sections from SOD1G37R mice, targeting both CD4 T cells and the microglial marker, Iba1. In line with our flow cytometry analysis (Fig. 3C), confocal image analysis revealed increased numbers of CD4 T cells in SC sections from SOD1G37R mice as compared with age-matched littermate controls (Fig. 5A–E). In addition, CD4 T cells were in a close association with Iba1+ microglia (Fig. 5F–H). Notably, Iba1 MFI was found to be positively correlated with the number of infiltrating CD4 T cells. However, when considering the volume of Iba1-positive areas, there was no significant correlation with the number of infiltrating CD4 T cells (Fig. 5I). Concomitantly, there was an increase in the volume of Iba-1+ and CD86+ cells (Fig. 5J–N and Additional file 1: Fig. S4A, B), along with increased volume of MHCII+ cells in regions of aggregated misfolded SOD1 (Fig. 5O–S and Additional file 1: Fig. S4C, D). Collectively, our data suggest that active immunological synapse occurs between microglia and infiltrating effector CD4 T cells potentially modulating the neuroinflammatory process in SOD1G37R mice.


Infiltrating CD4 T cells interact with antigen presenting cells in spinal cords of mutant SOD1G37R mice. Mice underwent euthanasia followed by perfusion with cold PBS. Thereafter, SCs were harvested, fixed, and transversely sectioned. For 3D visualization, a z-stack encompassing a minimum of 40 μm thickness was captured using a confocal microscope, with images acquired every 1 μm. A–D Lumbar SC sections of NT (A, C) and SOD1G37R (B, D) mice, immunolabeled with anti-Iba1 (red), anti-NeuN (white), anti-CD4 (green), and DAPI (blue) (n = 3). Scale bars represent ×10 (A, B) and ×40 magnification, Z-stack (C, D). E 3D projection of z-sections from panel D using Imaris (Methods). F Lumbar SC section of symptomatic mutant SOD1G37R mice (×60 magnification, Z-stack), immunolabeled with anti-Iba1 (red) and anti-CD4 (green) (n = 3). G, H 3D reconstruction of z-sections (from F) using Imaris. I Correlation between Iba-1 fluorescence intensity (left) or Iba-1 fluorescence volume (right) and frequencies of colocalized CD4 T cells (n = 6, 3 sections from different regions within the lumbar SC). *P ≤ 0.05, Spearman's rank correlation. J–L Lumbar SC sections of NT (n = 4) (J) and symptomatic mutant SOD1G37R mice (n = 5) (K, L), immunolabeled with anti-Iba1 (red), anti-CD86 (white), and DAPI (blue) [×10 (J, K) and ×20 (L) magnification, z-stack). M 3D reconstruction of z-sections (from L) using Imaris. N Quantification of Iba-1 (left) and CD86 (right) fluorescence volume in SC sections from NT (n = 4) and mutant SOD1G37R (n = 5) mice. *P ≤ 0.05, Mann–Whitney U test. O–Q Lumbar SC sections from NT (n = 6) (O) and symptomatic mutant SOD1G37R mice (n = 8) (P, Q) [×10 magnification (O, P) and ×20 magnification, z-stack (Q)] immunolabeled with anti-MHCII (white), anti-B8H10 (green) and DAPI (blue). R 3D reconstruction of z-sections (from Q) using Imaris. S Quantification of misfolded aggregated SOD1 (left, B8H10) and MHCII (right) fluorescence volume in NT and mutant SOD1G37R. ***P ≤ 0.001, Mann–Whitney U test
Discussion
In the present study, we sought to elucidate the contribution of T cells in the neuroinflammatory process of ALS, primarily using the late-onset SOD1 mouse model. First, we observed a skew towards effector memory phenotypes, which correlated with disease progression. Within the SC of SOD1G37R mice, CD4 T cells showed primarily a proinflammatory phenotype which coincided with activated microglia and positively correlated with infiltrating CD8 T cells. RNA-seq analyses reinforced these observations, uncovering a robust adaptive immune response underscored by upregulated pathways related to CD4 T-cell activity and antigen presentation. These findings thus highlight an important role for age-related T-cell subsets in ALS progression.
In our work, we examined the immune compartment of mice at symptomatic disease stage, characterized by specific phenotypic markers such as foot-dragging or toe-curling during walking [42]. Specifically, SOD1G37R mice were approximately 370 days of age and SOD1G93A mice were approximately 130 days of age at the time of sample collection. This approach was intended to capture a representative similar state of disease progression in these models. Using the late-onset SOD1G37R mice, we identified a pronounced representation of effector memory phenotypes, which suggest a heightened state of T-cell effector functions. This is in contrast to the dominance of naive T cells in the early-onset and commonly used SOD1G93A mice, reflecting a landscape of reduced antigenic exposure along with increased turnover of naïve T cells [48,49,50,51]. These results are in line with the most noticeable changes observed in the overall T cell population with age; the reduced frequencies of naive T cells along with increased frequencies of exhausted and effector phenotypes [31, 48, 49] result in disrupted immune tolerance and consequently improper T cell reactions, excessive cytokine production, cytotoxic responses, and tissue damage [52, 53]. Such impact of age-related dysregulated T-cell effector functions were recently observed in patients with ALS [29] and several neurodegenerative processes such as MS [54], PD [55], and AD [56]. Together, our results suggest that late-onset mouse models more accurately reflect the complexity of immune responses occurring in ALS.
Neuroinflammation, particularly related to the activity and distribution of different T-cell subsets, is becoming increasingly recognized as a hallmark of ALS [20]. Research conducted on leukocytes and SCs from ALS patients and SOD1 mouse models has demonstrated a significant inflammatory component in disease pathobiology; peripheral blood cell counts of natural killer cells, granulocytes, CD3+, CD4+, CD8+ and CD3+CD56+T cells were all shown to increase in patients with ALS [28]. Furthermore, within the realm of CD4 T cells, a correlation between the proinflammatory Th1/Th17 phenotype and disease severity was documented [57] and Tregs were shown to be dysfunctional in both ALS patients [58] and SOD1G93A mice [59]. Similarly, an augmented presence of expanding CD4 T cells was shown in CSF samples of ALS patients, which express lineage-defining transcription factors typical to CD4 CTLs, Th1, and Th2 cells [29]. Our results in the late-onset SOD1G37R mice show a simultaneous rise in pro- and anti-inflammatory populations (Th1 and Th2) and a decrease in the CD4 CTL population. Furthermore, EM CD4 T cells and CD4 CTL populations correlated with weight loss in SOD1G37R mice and thus highlight a possible association with disease progression. This pattern hints at a chronic inflammatory response which, along with the accumulation of dysregulated T cells, exacerbate the disease process [29, 57, 60]. However, in contrast to a previous study showing a correlation between IL-17+ cells and disease progression[57], our study neither observed significant changes in the frequency of Th17 cells—immunolabeled with anti-RORγt—nor their correlation with disease severity. This may be attributed either to the use of RORγt staining for the identification of Th17 cells or to the variation in disease stage or mechanism. In any case, the effector function of CNS infiltrating proinflammatory T cells and particularly Th17 cells may be extremely pathogenic in ALS and should be further explored. In addition, we found that while the frequency of circulating CD4 CTLs decreased, they correlated with disease progression, suggesting that they may accumulate within the CNS as part of the disease process [29]. Together, these results highlight the sophisticated, multifaceted immune dynamics in ALS, underscoring the need for further research to elucidate these intricate immune mechanisms and their implications for ALS therapy.
The intricate relationship between T cell activation, their infiltration into CNS borders and parenchyma, and the subsequent neuroinflammatory process are central to unveil pathological mechanisms underpinning neurodegenerative diseases [29, 61,62,63,64]. Several studies have indicated increased T-cell frequencies within the CNS of individuals with ALS [26, 65], and mouse models [66], with both CD4+ and CD8+ T cells observed adjacent to deteriorating neurons [26]. Here, we observed a significant infiltration of T cells into the SCs of SOD1G37R mice, which was not observed to the same extent in SOD1G93A mice. This aligns with previous findings showing minimal neuroinflammatory response in early clinical stages of ALS in SOD1G93A mice [66]. Intriguingly, T cells from CSF of ALS patients were found to undergo clonal expansion, hinting at antigen-triggered activation within the CNS [67, 68]. In line with findings showing an increase in proinflammatory Th1 cells in the CSF of ALS [29] and MS patients [69], we showed that the majority of CD4 T cells in the SC of SOD1G37R mice exhibited a Th1 phenotype and expressed the activation markers CD69, CD81 and the checkpoint molecule PD1 as part of their effector function within the CNS. Although activated CD8 T cells expressed both PD1 and Lag3, indicating a more exhausted phenotype [70,71,72], the overall T-cell response in the SC of SOD1G37R mice appears extremely neurotoxic, yet to be fully explored [73,74,75].
APCs at the CNS barriers and/or parenchyma are essential for locally executing and regulating T-cell responses via MHC–TCR interactions [22, 25]. Our RNA-seq studies demonstrate upregulated genes related to CD4 T-cell activation (e.g., CD44, CD81 and Il2rg) along with upregulated genes related to antigen processing and presentation (e.g., MHCII complex related genes: H2-Aa, H2-Ab1, H2-Eb1, H2-DMa, H2-DMb). Recent studies have shown that within the CNS parenchyma, certain disease-associated microglial subsets exhibit APC phenotypes [45, 76] which either promote [64] or limit [77] pathology. In a rat model of PD, infiltration of CD4 and CD8 T cells into the brain was followed by increased frequencies of MHCII-expressing microglia along with enhanced neuronal loss [22]. Similar to ALS mice, RNA-seq analysis of immune cells in the brain of PD mice revealed enrichment in GO terms related to antigen processing and presentation and T-cell activation [78]. Targeting the expression of MHCII, resulted in reduced T-cell and monocyte infiltration into the brain along with attenuated neurodegeneration [79]. Together, these results suggest that T-cell activation within the CNS might be a central mechanism underlying the progression of neurodegenerative diseases such as PD and ALS [80].
Providing a cellular context in SOD1G37R mice, we show that CD4 T cells were in close proximity to microglia cells along with marked elevated expression of Iba1 and MHCII. Likewise, the association of Iba-1 levels with increased frequencies of infiltrating CD4 T cells, suggests a plausible causal relationship between T-cell infiltration and neurotoxic microglia activation in the course of ALS. Figure 6 depicts a possible scenario whereby CD4 T cells in spleens of late-onset SOD1G37R show an age-related proinflammatory phenotype. Upon infiltration into the SC, these CD4 T cells are re-activated by antigen-presenting microglia cells, thereby facilitating their activation and furthering the inflammatory response, which could trigger release of neurotoxic factors leading to neuronal death. Our findings hint at a possible feedback loop, wherein the activation and interaction of CD4 T cells with microglia could lead to sustained neuroinflammation, possibly exacerbating disease progression [62].

CD4 T-cell-mediated neuroinflammation in a late-onset mouse model of ALS. Elevated frequencies of systemic age-related T-cell subsets lead to neurotoxic inflammation in the lumbar SC, a process characterized by APCs activation, infiltration of CD4 (blue) and CD8 (red) T cells, and an upsurge in the expression of genes and proteins involved in antigen presentation and T cell activation. This cascade can be both a precursor to and a consequence of neuronal death
Although the SOD1 mouse models exhibit similar neuropathology and neuroinflammation characteristics of ALS [18], the disease in human is substantially more heterogeneous due to genetic and environmental variations [81, 82]. Nonetheless, while the human disease is primarily age-related with the average age of patients around 60–70 years [2], the immunological changes occurring with age were mostly neglected in animal models. Moreover, despite the fact that these animal models remain essential for the development of optimized therapeutic drugs, in some cases, only human studies will definitely determine the real therapeutic value of these strategies. Thus, further human studies considering our findings can unveil various age-related T-cell subsets such as the increased frequencies of effector T cells and CD4 CTLs, which were also evident in the CSF of ALS patients, that can be promising targets to modify the disease process. The molecular profiles, signaling pathways, antigen specificities, and infiltration routes of such neurotoxic T cell subsets can all be valuable diagnostic and therapeutic targets. In addition, further understanding of the infiltration process and synapse formation of T cells with brain APCs can be harnessed to develop novel immunoregulatory approaches such as the use of antigen-specific Tregs or chimeric antigen receptor (CAR) T cells that target specific antigens within the CNS [83].
Conclusions
To conclude, our investigations delineate the intricate immune dynamics in the context of both early and late-onset SOD1 mouse models of ALS, emphasizing a potential pathogenic role of infiltrating T cells. The contrasting predominance of effector memory phenotype in late-onset SOD1G37R mice compared to naive T cells in early-onset SOD1G93A suggests distinct immunological trajectories and, potentially, differing disease progression pathways. Our RNA-seq data underscore an amplified adaptive immune response within the SC of SOD1G37R mice, hallmarked by an elevation in pathways related to CD4 T-cell activation and antigen presentation. This is further supported by our observations of CD4 T cells closely interacting with microglial cells, suggestive of potential antigen presentation and subsequent T cell activation. Together, these interactions hint at a self-sustaining feedback loop between CD4 T cells and microglia, driving persistent neuroinflammation and potentially accelerating ALS progression. Consequently, our insights extend the current understanding of immune involvement in ALS and offer valuable perspectives for potential therapeutic interventions targeting the immune dynamics in neurodegenerative diseases.
Availability of data and materials
The RNA-seq data have been deposited to the GEO repository with accession number GSE246397.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- APC:
-
Antigen presenting cell
- CNS:
-
Central nervous system
- EM:
-
Effector memory
- CD4 CTLs:
-
Cytotoxic CD4 T cells
- IHC:
-
Immunohistochemistry
- MHC:
-
Major histocompatibility complex
- SC:
-
Spinal cord
References
Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:162–72.
Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol. 2015;7:181.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253.
Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206.
Parker SE, Hanton AM, Stefanou SN, Noakes PG, Woodruff TM, Lee JD. Revisiting the role of the innate immune complement system in ALS. Neurobiol Dis. 2019;127:223–32.
Pramatarova A, Laganière J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci. 2001;21:3369–74.
Jaarsma D, Teuling E, Haasdijk ED, De Zeeuw CI, Hoogenraad CC. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci. 2008;28:2075–88.
Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–92.
Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253–63.
Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet. 2011;20:286–93.
Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol. 2013;8:888–99.
Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflamm. 2014;11:1–15.
Kempuraj D, Thangavel R, Selvakumar GP, Zaheer S, Ahmed ME, Raikwar SP, Zahoor H, Saeed D, Natteru PA, Iyer S, Zaheer A. Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation and neurodegeneration. Front Cell Neurosci. 2017;11:216.
Chung Y-C, Ko H-W, Bok E-G, Park E-S, Huh S-H, Nam J-H, Jin B-K. The role of neuroinflammation on the pathogenesis of Parkinson’s disease. BMB Rep. 2010;43:225–32.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405.
Matthews PM. Chronic inflammation in multiple sclerosis—seeing what was always there. Nat Rev Neurol. 2019;15:582–93.
Liu J, Wang F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front Immunol. 2017;8:1005.
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139:136.
Béland LC, Markovinovic A, Jakovac H, De Marchi F, Bilic E, Mazzini L, Kriz J, Munitic I. Immunity in amyotrophic lateral sclerosis: blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020;2: fcaa124.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–68.
Subbarayan MS, Hudson C, Moss LD, Nash KR, Bickford PC. T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an α-synuclein rat model of Parkinson’s disease. J Neuroinflamm. 2020;17:1–16.
Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun. 2020;11:6129.
Murphy ÁC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–51.
Haimon Z, Frumer GR, Kim J-S, Trzebanski S, Haffner-Krausz R, Ben-Dor S, Porat Z, Muschaweckh A, Chappell-Maor L, Boura-Halfon S. Cognate microglia–T cell interactions shape the functional regulatory T cell pool in experimental autoimmune encephalomyelitis pathology. Nat Immunol. 2022;23:1749–62.
Engelhardt JI, Tajti J, Appel SH. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol. 1993;50:30–6.
Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci USA. 2008;105:15558–63.
Gustafson MP, Staff NP, Bornschlegl S, Butler GW, Maas ML, Kazamel M, Zubair A, Gastineau DA, Windebank AJ, Dietz AB. Comprehensive immune profiling reveals substantial immune system alterations in a subset of patients with amyotrophic lateral sclerosis. PLoS ONE. 2017;12: e0182002.
Yazdani S, Seitz C, Cui C, Lovik A, Pan L, Piehl F, Pawitan Y, Kläppe U, Press R, Samuelsson K, et al. T cell responses at diagnosis of amyotrophic lateral sclerosis predict disease progression. Nat Commun. 2022;13:1–13.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–5.
Elyahu Y, Hekselman I, Eizenberg-Magar I, Berner O, Strominger I, Schiller M, Mittal K, Nemirovsky A, Eremenko E, Vital A, et al. Aging promotes reorganization of the CD4 T cell landscape toward extreme regulatory and effector phenotypes. Sci Adv. 2019;5: eaaw8330.
Lefebvre JS, Haynes L. Aging of the CD4 T cell compartment. Open Longev Sci. 2012;6:83.
Moro-García MA, Alonso-Arias R, López-Larrea C. When aging reaches CD4+ T-cells: phenotypic and functional changes. Front Immunol. 2013;4:107.
Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I. Massively parallel single cell RNA-Seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776.
Keren-Shaul H, Kenigsberg E, Jaitin DA, David E, Paul F, Tanay A, Amit I. MARS-seq2.0: an experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat Protocols. 2019;14:1841–62.
Sklarz M, Levin L, Gordon M, Chalifa-Caspi V. NeatSeq-Flow: a lightweight high-throughput sequencing workflow platform for non-programmers and programmers alike. bioRxiv. 2018:173005.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:1–16.
Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Hatzipetros T, Kidd JD, Moreno AJ, Thompson K, Gill A, Vieira FG. A quick phenotypic neurological scoring system for evaluating disease progression in the SOD1-G93A mouse model of ALS. J Vis Exp. 2015. https://doi.org/10.3791/53257.
Rocha-Perugini V, Zamai M, González-Granado JM, Barreiro O, Tejera E, Yañez-Mó M, Caiolfa VR, Sanchez-Madrid F. CD81 controls sustained T cell activation signaling and defines the maturation stages of cognate immunological synapses. Mol Cell Biol. 2013;33:3644–58.
Shah K, Al-Haidari A, Sun J, Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther. 2021;6:1–26.
Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to Tango. Front Immunol. 2017;8:25.
Karin N, Wildbaum G. The role of chemokines in shaping the balance between CD4+ T cell subsets and its therapeutic implications in autoimmune and cancer diseases. Front Immunol. 2015;6:609.
Daniels MA, Luera D, Teixeiro E. NFκB signaling in T cell memory. Front Immunol. 2023;14:1129191.
Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018;19:10–9.
Goronzy JJ, Weyand CM. Successful and maladaptive T cell aging. Immunity. 2017;46:364–78.
Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+ T cells: differentiation and functions. Clin Dev Immunol. 2012;2012: 925135.
Elyahu Y, Monsonego A. Thymus involution sets the clock of the aging T-cell landscape: implications for declined immunity and tissue repair. Ageing Res Rev. 2021;65: 101231.
Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E, Brioschi S, Shchukina I, Kerndl M, Bambouskova M. Comprehensive profiling of an aging immune system reveals clonal GZMK+ CD8+ T cells as conserved hallmark of inflammaging. Immunity. 2021;54(99–115): e112.
Carrasco E, Gómez de las Heras MM, Gabandé-Rodríguez E, Desdín-Micó G, Aranda JF, Mittelbrunn M. The role of T cells in age-related diseases. Nat Rev Immunol. 2022;22:97–111.
Zuroff L, Rezk A, Shinoda K, Espinoza DA, Elyahu Y, Zhang B, Chen AA, Shinohara RT, Jacobs D, Alcalay RN. Immune aging in multiple sclerosis is characterized by abnormal CD4 T cell activation and increased frequencies of cytotoxic CD4 T cells with advancing age. EBioMedicine. 2022;82: 104179.
Kouli A, Williams-Gray CH. Age-related adaptive immune changes in Parkinson’s disease. J Parkinsons Dis. 2022;12:S93–104.
Andronie-Cioara FL, Ardelean AI, Nistor-Cseppento CD, Jurcau A, Jurcau MC, Pascalau N, Marcu F. Molecular mechanisms of neuroinflammation in aging and Alzheimer’s disease progression. Int J Mol Sci. 1869;2023:24.
Jin M, Günther R, Akgün K, Hermann A, Ziemssen T. Peripheral proinflammatory Th1/Th17 immune cell shift is linked to disease severity in amyotrophic lateral sclerosis. Sci Rep. 2020;10:5941.
Beers DR, Zhao W, Wang J, Zhang X, Wen S, Neal D, Thonhoff JR, Alsuliman AS, Shpall EJ, Rezvani K, Appel SH. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight. 2017;2: e89530.
Banerjee R, Mosley RL, Reynolds AD, Dhar A, Jackson-Lewis V, Gordon PH, Przedborski S, Gendelman HE. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS ONE. 2008;3: e2740.
Chen Y, Qi B, Xu W, Ma B, Li L, Chen Q, Qian W, Liu X, Qu H. Clinical correlation of peripheral CD4+-cell sub-sets, their imbalance and Parkinson’s disease. Mol Med Rep. 2015;12:6105–11.
Bhatia D, Grozdanov V, Ruf WP, Kassubek J, Ludolph AC, Weishaupt JH, Danzer KM. T-cell dysregulation is associated with disease severity in Parkinson’s Disease. J Neuroinflamm. 2021;18:1–10.
González H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflamm. 2014;11:201.
Jorfi M, Park J, Hall CK, Lin CJ, Chen M, von Maydell D, Kruskop JM, Kang B, Choi Y, Prokopenko D. Infiltrating CD8+ T cells exacerbate Alzheimer’s disease pathology in a 3D human neuroimmune axis model. Nat Neurosci. 2023;26:1–16.
Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, Wang C, Bao X, Finn MB, Hu H. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615:668–77.
Kawamata T, Akiyama H, Yamada T, McGeer P. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140:691.
Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH, Carroll MC. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci USA. 2008;105:17913–8.
Panzara MA, Gussoni E, Begovich AB, Murray RS, Zang YQ, Appel SH, Steinman L, Zhang J. T cell receptor BV gene rearrangements in the spinal cords and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Neurobiol Dis. 1999;6:392–405.
Evans FL, Dittmer M, de la Fuente AG, Fitzgerald DC. Protective and regenerative roles of T cells in central nervous system disorders. Front Immunol. 2019;10:2171.
Schafflick D, Xu CA, Hartlehnert M, Cole M, Schulte-Mecklenbeck A, Lautwein T, Wolbert J, Heming M, Meuth SG, Kuhlmann T. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat Commun. 2020;11:247.
Li H, Zheng C, Han J, Zhu J, Liu S, Jin T. PD-1/PD-L1 axis as a potential therapeutic target for multiple sclerosis: AT cell perspective. Front Cell Neurosci. 2021;15: 716747.
Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. LAG-3: from molecular functions to clinical applications. J Immunother Cancer. 2020;8:1014.
Gao Z, Feng Y, Xu J, Liang J. T-cell exhaustion in immune-mediated inflammatory diseases: new implications for immunotherapy. Front Immunol. 2022;13: 977394.
Rocha-Perugini V, Zamai M, González-Granado JM, Barreiro O, Tejera E, Yañez-Mó M, Caiolfa VR, Sánchez-Madrid F. CD81 controls sustained T cell activation signaling and defines the maturation stages of cognate immunological synapses. Mol Cell Biol. 2013;33:3644–58.
Zhao J, Roberts A, Wang Z, Savage J, Ji R-R. Emerging role of PD-1 in the central nervous system and brain diseases. Neurosci Bull. 2021;37:1188–202.
Beers DR, Zhao W, Thonhoff JR, Faridar A, Thome AD, Wen S, Wang J, Appel SH. Serum programmed cell death proteins in amyotrophic lateral sclerosis. Brain Behav Immunity Health. 2021;12: 100209.
Muzio L, Viotti A, Martino G. Microglia in neuroinflammation and neurodegeneration: from understanding to therapy. Front Neurosci. 2021;15: 742065.
Mittal K, Eremenko E, Berner O, Elyahu Y, Strominger I, Apelblat D, Nemirovsky A, Spiegel I, Monsonego A. CD4 T cells induce a subset of MHCII-expressing microglia that attenuates Alzheimer pathology. iScience. 2019;16:298–311.
Karikari AA, McFleder RL, Ribechini E, Blum R, Bruttel V, Knorr S, Gehmeyr M, Volkmann J, Brotchie JM, Ahsan F. Neurodegeneration by α-synuclein-specific T cells in AAV-A53T-α-synuclein Parkinson’s disease mice. Brain Behav Immun. 2022;101:194–210.
Williams GP, Schonhoff AM, Jurkuvenaite A, Thome AD, Standaert DG, Harms AS. Targeting of the class II transactivator attenuates inflammation and neurodegeneration in an alpha-synuclein model of Parkinson’s disease. J Neuroinflamm. 2018;15:1–14.
Zang X, Chen S, Zhu J, Ma J, Zhai Y. The emerging role of central and peripheral immune systems in neurodegenerative diseases. Front Aging Neurosci. 2022;14: 872134.
Boddy SL, Giovannelli I, Sassani M, Cooper-Knock J, Snyder MP, Segal E, Elinav E, Barker LA, Shaw PJ, McDermott CJ. The gut microbiome: a key player in the complexity of amyotrophic lateral sclerosis (ALS). BMC Med. 2021;19:1–14.
Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, Feldman EL. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21:465–79.
Graber DJ, Cook WJ, Sentman M-L, Murad-Mabaera JM, Sentman CL. Human CD4+ CD25+ T cells expressing a chimeric antigen receptor against aberrant superoxide dismutase 1 trigger antigen-specific immunomodulation. Cytotherapy. 2023. https://doi.org/10.1016/j.jcyt.2023.11.007.
Acknowledgements
The authors gratefully acknowledge the expert assistance of Dr. Hadas Keren-Shaul from The Nancy and Stephen Grand Israel National Center for Personalized Medicine, Weizmann Institute of Science and Dr. Liron Levin and Dr. Vered Chalifa-Caspi from The Bioinformatics Core Facility, Ben-Gurion University.
Funding
This research was supported by the Israel Science Foundation Grant 284/19 (to A.I.), and the Ministry of Science and Technology Grant 3-16148 (to A.M).
Author information
Authors and Affiliations
Contributions
SZ designed the research, performed experiments, analyzed data, wrote the first draft of the manuscript, and edited the manuscript. AN, LL, and LA assisted with experiments. EE provided intellectual assistance in interpreting the data. AI and AM provided research supervision and reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by Ben-Gurion University Institutional Animal Care and Use Committee (IL-36-07-2018) and was conducted according to the Israeli Animal Welfare Act following the guidelines of the Guide for Care and Use of Laboratory Animal (National Research Council, 1996).
Consent for publication
Not applicative.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Flow cytometry gating strategy and immune alterations in spleens of SOD1 mice. (A) Gating strategy for spleen flow cytometry experiment for SOD1G37R and SOD1G93A mice. CD4 T-cell T populations were gated from lymphocytes (SSC-A vs FSC-A), live cells (Fixable Viability Dye vs FSC-A), and single cells (SSC-A vs SSC-H). (B) tSNE plot of CD4 T-cell population distribution of NT littermates of late-onset SOD1G37R (n = 3) and early-onset SOD1G93A mice (n = 3). (C–N) CD4 T-cell subsets frequency (mean ± SEM) of NT (n = 5) and symptomatic SOD1G93A mice (n = 5), shown as percentage. Figure S2: CD4 and CD8 T-cell infiltration into the spinal cord of SOD1G93A mice. (A) Gating strategy for SC flow cytometry experiment for SOD1G37R and SOD1G93A mice. (B, C) Event number of CD4 (B) and CD8 (C) T cells in SCs of NT mice (n = 7) and symptomatic mutant SOD1G93A mice (n = 8). Figure S3: Elevation of genes related to inflammation and antigen presentation in SOD1G37R mice. (A) Volcano plot of upregulated and downregulated genes related to inflammatory pathway. (B) Heatmap of differentially expressed genes in the SC from symptomatic mutant SOD1G37R (n = 10) and NT mice (n = 10) that are involved in antigen processing and presentation (GO:0019882). Figure S4: Microglial expression of CD86 and MHCII in SCs of non-transgenic control mice. (A) Lumbar SC sections of NT (n = 4) mice. Sections were immunolabeled with anti-Iba-1 (red), anti-CD86 (white) and DAPI (blue) (×20 magnification, z-stack). (B) 3D reconstruction of z-sections (from A) using Imaris. (C) Lumbar SC sections of NT (n = 6) mice. Sections were immunolabeled with anti-MHCII (white), anti-B8H10 (green) and DAPI (blue) (×20 magnification, z-stack). (D) 3D reconstruction of z-sections (from C) using Imaris.
Additional file 2: Table S1.
Antibodies used in flow cytometry experiments for SC cells and splenocytes. This table lists the antibodies used in the flow cytometry analysis of SCs and splenocytes. It includes details on conjugation, target antigens, the cell types for which each antibody was used, and the suppliers. This detailed categorization facilitates understanding of the specific reagents used in the experimental process.
Additional file 3: Table S2.
Antibodies used in IHC experiments. This table lists the antibodies used in the IHC analysis of SC sections. The table includes information on the target antigen, the dilution used, the source of the antibody, and the species origin.
Additional file 4: Table S3.
Frequencies of CD4 T-cell populations in SOD1 and littermate control mice. Percentages of CD4 T-cell populations derived from spleens of SOD1G93A, SOD1G37R and littermate control mice are displayed. Data for each genotype are presented in terms of Avg. and SEM.
Additional file 5: Table S4.
Differentially expressed genes in lumbar spinal cord sections of SOD1G37R and littermate control mice. Lumbar SC RNA-seq results of 10 SOD1G37R mice and 10 littermate control mice. Genes are detailed with their corresponding ENSEMBL ID and gene symbols, log2 fold change values, and adjusted p-values. Fold change values > 1 suggest upregulation whereas values < − 1 downregulation. Data analysis and organization were executed utilizing the NeatSeq Flow DeSeq2 module.
Additional file 6: Table S5.
Upregulated Immune-related GO biological processes in lumbar spinal cords of SOD1G37R mice. The table outlines the GO biological processes found to be upregulated in lumbar SC samples, as ascertained through bulk RNA-seq analysis. Columns from left to right present the biological process name, gene ratio, and p.adjust.
Additional file 7: Table S6.
Upregulated Immune-related KEGG pathways in lumbar spinal cords of SOD1G37R mice. The table delineates the KEGG pathways found to be upregulated in lumbar SC samples, as determined by bulk RNA-seq analysis. Columns from left to right represent the pathway name, gene ratio, and p.adjust.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zaccai, S., Nemirovsky, A., Lerner, L. et al. CD4 T-cell aging exacerbates neuroinflammation in a late-onset mouse model of amyotrophic lateral sclerosis. J Neuroinflammation 21, 17 (2024). https://doi.org/10.1186/s12974-023-03007-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-03007-1