
Abstract
Background
Vision loss in patients with wet/exudative age-related macular degeneration (AMD) is associated with choroidal neovascularization (CNV), and AMD is the leading cause of irreversible vision impairment in older adults. Interleukin-17A (IL-17A) is a component of the microenvironment associated with some autoimmune diseases. Previous studies have indicated that wet AMD patients have elevated serum IL-17A levels. However, the effect of IL-17A on AMD progression needs to be better understood. We aimed to investigate the role of IL-17A in a laser-induced CNV mouse model.
Methods
We established a laser-induced CNV mouse model in wild-type (WT) and IL-17A-deficient mice and then evaluated the disease severity of these mice by using fluorescence angiography. We performed enzyme-linked immunosorbent assay (ELISA) and fluorescence-activated cell sorting (FACS) to analyze the levels of IL-17A and to investigate the immune cell populations in the eyes of WT and IL-17A-deficient mice. We used ARPE-19 cells to clarify the effect of IL-17A under oxidative stress.
Results
In the laser-induced CNV model, the CNV lesions were larger in IL-17A-deficient mice than in WT mice. The numbers of γδ T cells, CD3+CD4+RORγt+ T cells, Treg cells, and neutrophils were decreased and the number of macrophages was increased in the eyes of IL-17A-deficient mice compared with WT mice. In WT mice, IL-17A-producing γδ T-cell numbers increased in a time-dependent manner from day 7 to 28 after laser injury. IL-6 levels increased and IL-10, IL-24, IL-17F, and GM-CSF levels decreased in the eyes of IL-17A-deficient mice after laser injury. In vitro, IL-17A inhibited apoptosis and induced the expression of the antioxidant protein HO-1 in ARPE-19 cells under oxidative stress conditions. IL-17A facilitated the repair of oxidative stress-induced barrier dysfunction in ARPE-19 cells.
Conclusions
Our findings provide new insight into the protective effect of IL-17A in a laser-induced CNV model and reveal a novel regulatory role of IL-17A-producing γδ T cells in the ocular microenvironment in wet AMD.
Similar content being viewed by others

Background
Age-related macular degeneration (AMD) is a retinal disease that causes damage to the macula and leads to loss of central vision. AMD is usually frequent in elderly people, and there are two types of macular degeneration associated with age: dry (atrophic) and wet (exudative) AMD [1]. Most AMD cases start as the dry type, and approximately 10–20% of individuals with dry AMD tend to progress to the wet type [2, 3]. AMD occurs in both eyes but does not necessarily progress at the same pace in both eyes. Wet AMD is associated with choroidal neovascularization (CNV), which is characterized by the proliferation of blood vessels behind the retina that begin to grow toward the macula and tend to leak fluid into the macula, causing macular damage that leads to severe central vision loss [4]. The reduction in tissue oxygenation causes the production of vascular endothelial growth factor (VEGF), a critical factor involved in the development of wet AMD [5].
The clinical efficacy of intravitreal anti-VEGF drugs has been widely demonstrated in wet AMD [6]. However, long-term anti-VEGF therapy for some patients with wet AMD may have a poor outcome [7, 8]. Under physiological conditions, VEGF can provide critical trophic support necessary for retinal function [9]. In addition, VEGFA inhibition might lead to retinal pigment epithelium (RPE) and photoreceptor cell death and vision loss [10]. An improved understanding of CNV pathogenesis is still crucial for the prevention and treatment of AMD.
Reactive oxygen species (ROS) formation is a byproduct of the normal metabolism of oxygen and plays important roles in cell signaling and homeostasis [11]. ROS levels dramatically increase, and ROS accumulate during a process called oxidative stress, which may damage cell structures [11]. Recent studies have investigated the role of excess ROS in the pathogenesis and development of AMD [12, 13]. The antioxidant capacity of retinal cells decreases with age, increasing oxidative stress, which causes irreversible damage to photoreceptors and RPE cells [14]. The upregulation of Nrf2 activity and the expression of HO-1 can suppress retinal cell apoptosis [15]. Therefore, improving the endogenous defense system against ROS damage in the retina is important.
Cytokines are critical mediators of communication for the immune system. TNF-α stimulates the expression of proangiogenic VEGF [16] and facilitates pathologic angiogenesis in AMD [17]. IL-6 is significantly associated with the volume of macular edema in patients with CNV [18]. IL-10 regulates macrophage function and promotes pathological angiogenesis [19]. Therefore, cytokine dysregulation is believed to play a critical role in the pathogenesis of AMD.
IL-17A is a pleiotropic cytokine and is mainly produced by T helper 17 cells (Th17 cells), type three innate lymphoid cells (ILC3s), and gamma delta T cells (γδ T cells). IL-17A binds to a heterodimeric receptor complex, which consists of IL-17RA and IL-17RC, to activate the MAPK and NF-κB signaling pathways and further induce the production of chemokines. These chemokines further recruit immune cells, such as monocytes and neutrophils, to sites of local inflammation. Therefore, IL-17A is critical for host defense against microbes because it promotes the recruitment of neutrophils and induces the production of antimicrobial peptides [20,21,22]. IL-17-driven inflammation is normally controlled by regulatory T (Treg) cells and the anti-inflammatory cytokines IL-10, TGF-β, and IL-35 [23].
There is much evidence indicating that AMD is associated with the immune system, which includes complement, macrophages, and T cells [24, 25]. Th17 cells are the main source of IL-17A and are involved in the pathogenesis of autoimmune diseases, including multiple sclerosis [26, 27], systemic lupus erythematosus [28], and inflammatory bowel disease [29, 30]. Treg and Th17 cells are two described lymphocyte subsets with opposing actions. Th17 cells represent a proinflammatory subset, whereas Treg cells have an anti-inflammatory effect. The Th17/Treg balance plays a critical role in the development of autoimmune diseases [31, 32]. In addition, γδ T cells are an early cellular source of IL-17A, which promotes neutrophil recruitment [33].
Previous studies reported that serum IL-17A levels were elevated in patients with AMD [34, 35]. However, the functional roles of IL-17A in AMD progression remain incompletely understood. In this study, we aimed to investigate the role of IL-17A in CNV development and verify the effects of IL-17A in a laser-induced CNV mouse model.
Materials and methods
Animals
Wild-type (WT) C57BL/6J mice were purchased from the National Laboratory Animal Center (Taipei, Taiwan). IL-17A-deficient mice on a C57BL/6J genetic background were maintained in the animal center of National Cheng Kung University. Both IL-17A-deficient and WT mice (6–8 weeks old) were used and kept on a 12-h light–dark cycle at 22 ± 2 °C. The research procedures were approved by the Animal Ethics Committee of National Cheng Kung University (IACUC Approval no. 111266). The methods were carried out in accordance with the approved guidelines.
Laser-induced CNV mouse model and fluorescence angiography
Mice were anesthetized by intraperitoneal injection of Zoletil (8 mg/kg; VIRBAC, Carros, France) and Rompun (0.32 mg/kg; Bayer, Leverkusen, Germany), and the pupils were dilated with one drop of 1% tropicamide ophthalmic solution (Santen Pharmaceutical Co., Ltd, Kita-ku, Osaka, Japan). After anesthesia and dilation, CNV was induced with a 532-nm laser photocoagulator (250 mW, 50 μm spot size, duration 80 ms, four spots per eye) with a microscopic delivery system (Micron IV, Phoenix Research Laboratories, Pleasanton, CA, USA). In all eyes, four spots were applied around the optic disc and between retinal vessels four times. A bubble formed immediately at the site of laser application, indicating successful rupture of Bruch’s membrane. Spots with subretinal hemorrhage following laser photocoagulation were excluded. Fundus fluorescein angiography (FFA) was used to evaluate CNV leakage by performing with the retinal imaging microscopy (Micron IV, Phoenix Research Laboratories) 7, 14, 21, and 28 days after laser photocoagulation. Mice were anesthetized, and their pupils were dilated and intraperitoneally injected with 2% sodium fluorescein (Sigma‒Aldrich, St. Louis, MO, USA) at 20 μg/gram body weight. The FFA images were taken with a retinal imaging microscope at 5 min after fluorescein injection. The fluorescein leakage area of each lesion was contoured and evaluated quantitatively with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Isolation of ocular-infiltrating cells
To isolate the immune cells in the retina and choroid, the tip of angled scissors was inserted between the skin and the eyeball, and then the mouse eyes were enucleated. To avoid contamination, the eye was briefly dipped in 70% ethanol and washed in PBS. The anterior segment (cornea, iris, and lens) was removed. Both eyes were pooled for each animal and enzymatically digested in DMEM-F12 serum-free medium (GeneDireX, Taiwan) supplemented with 2 mg/ml dispase II (Sigma‒Aldrich) at 37 °C for 40 min. The posterior segment of the eyes, including the sclera, choroid, and retina, was then disrupted in the digestion medium. The sample was filtered with a 70 μm cell strainer (CORNING, Tewksbury, MA, USA) and then centrifuged at 4 °C (1800 rpm/610 g for 5 min). The supernatant was removed, and the cells were resuspended in medium for further flow cytometry analysis.
Fluorescence-activated cell sorting (FACS) analysis and detection of cytokine-expressing lymphocytes
The cells were suspended in FACS buffer (7.7 mM NaN3, 2 mM EDTA and 2% FBS dissolved in PBS) to produce a single-cell suspension. Myeloid cells were stained with surface marker antibodies, including BV 421-conjugated anti-CD45 (BD Pharmingen™, San Diego, CA, USA), 7-AAD-conjugated anti-CD11b (BD Pharmingen™), Alexa 647-conjugated anti-F4/80 (eBioscience, San Diego, CA, USA), Alexa 488-conjugated anti-Ly6G (BD Pharmingen™), and Alexa 647-conjugated anti-Ly6C (BioLegend, San Diego, CA, USA). Lymphoid cells were stained with surface marker antibodies including PE-conjugated anti-CD45 (BD Pharmingen™), BV 421-conjugated anti-TCRγδ (BD Pharmingen™), and Alexa 488-conjugated anti-CD4 (BioLegend), in the dark for 30 min at 4 °C. For transcription factor staining, cells were fixed and permeabilized for 30 min at 4 °C using BD Cytofix/Cytoperm buffer (BD Biosciences, La Jolla, CA, USA), washed twice with BD Perm/Wash buffer and then stained with transcription factor marker antibodies, including 7-AAD-conjugated RORγt (BD Pharmingen™) and Alexa 647-conjugated Foxp3 (BioLegend), in the dark for 30 min at 4 °C. For intracellular cytokine staining, cells isolated from the retina were activated with 20 ng/ml PMA (Sigma‒Aldrich), 1 μM ionomycin (Sigma‒Aldrich), and BFA (1:1000, BD Pharmingen™) and cultured for 5 h in 5% CO2 at 37 °C. Cells were stained with surface marker antibodies including APC-Cy7-conjugated anti-CD45 (BD Pharmingen™) and PE-conjugated anti-CD4 (BD Pharmingen™). Then, the cells were fixed and permeabilized for 30 min at 4 °C using BD Cytofix/Cytoperm buffer (BD Biosciences), washed twice with BD Perm/Wash buffer and then stained with Alexa 488-conjugated anti-IL-17A (BD Pharmingen™) in the dark for 30 min at 4 °C. The cell samples were acquired using a BD FACS Canto II Flow Cytometer (BD Biosciences) using BDFACS DIVA software (BD Biosciences) and analyzed with FlowJo software (v.10, FlowJo™).
Histological analysis and immunofluorescence staining
Eyes from mice were harvested and fixed in neutral buffered 4% paraformaldehyde. After paraffin embedding, retinal cross-sections were cut perpendicularly (5 μm). Hematoxylin and eosin (H&E) staining and immunofluorescence staining were performed following standard protocols. For immunofluorescence staining, the primary antibodies used were anti-IL-17A (diluted to 2 μg/ml; Genetex, Alton Pkwy Irvine, CA, USA) and anti-ZO-1 (1:50 dilution; Abcam, Cambridge, UK). Isotype-matched control antibodies were used as a negative control. The samples were washed with PBS and stained with fluorescence-conjugated secondary antibody at room temperature for 1 h. The slides were mounted with ProLong® Gold antifade reagent with DAPI (Invitrogen, Waltham, MA, USA). Images were taken using a scanning confocal laser microscope (Olympus FV1000, Waltham, MA, USA) to visualize the stained cells.
Cytokine analysis
IL-17A concentrations in serum from mice with laser-induced CNV and healthy mice were measured using a mouse IL-17A ELISA Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. To analyze dynamic changes in cytokine levels in healthy mice and mice with laser-induced CNV on day 7, day 14, day 21, and day 28, serum samples were collected and evaluated using a customized LEGENDplex™ kit according to the manufacturer’s instructions (BioLegend). The bead-based multiplex assay panel allowed simultaneous quantification of IL-17F, IL-17A, IL-22, IL-23, TNF-α, IFN-γ, IL-5, IL-13, and IL-6 using a sandwich immunoassay. Data were acquired on a BD FACSCanto II (BD Biosciences). The data were analyzed using BioLegend’s LEGENDplex™ data analysis software.
Real-time quantitative polymerase chain reaction (RT‒qPCR)
Total RNA was extracted from total mouse ocular cells or cultured cells with QIAzol Lysis Reagent (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. RNA samples (1 μg) were then subsequently used for reverse transcription using the PrimeScript™ RT Reagent Kit (Perfect Real Time) (Takara, Japan), and the cDNA samples were further diluted 10 times in diethylpyrocarbonate (DEPC)-treated water. cDNA was then amplified on a thermocycler (Roter-Gene Q detection system; QIAGEN) with gene-specific primers. The PCR mixture contained 2 µl of diluted cDNA samples, 5 µl of SYBR Green Master Mix (QIAGEN), 1.6 μl of RNase-free water (QIAGEN), and 0.7 µl of each primer at 10 pmol/µl in a final reaction volume of 10 μl. The amplification conditions were as follows: activation at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 10 sec and extension at 60 °C for 20 sec. mRNA quantification analysis results were normalized to β-actin or GAPDH, which was used as the internal control. Relative fold changes in mRNA expression were determined by calculating 2–ΔΔCt. All sequences of primers used in this study are listed in Additional file 1: Table S1.
Cell culture
The ARPE-19 human retinal pigment epithelial cell line was obtained from the American Type Culture Collection (ATCC). ARPE-19 cells were cultured in a 1:1 mixture of DMEM/Ham’s F-12 medium with L-glutamine and 15 mM HEPES (GeneDireX) and were supplemented with 2.48 g/L sodium bicarbonate (Avantor, Radnor, PA, USA), 10% FBS (HyClone™, Logan, UT, USA), and 1% antibiotic–antimycotic (GeneDireX). Cells were incubated at 37 °C with 95% air and 5% CO2.
Cell viability assay
ARPE-19 cells were cultured in 96-well plates and incubated in DMEM/F12 containing IL-17A (10–100 ng/ml; GenScript, Piscataway, NJ, USA) or 300 μM H2O2 (Honeywell Fluka™, Seelze, Germany) for 24 h. After treatment, the cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; VWR, Radnor, PA, USA) at a final concentration of 0.2 mg/ml for 3 h. The MTT solution was removed, and 150 μl of DMSO (Sigma‒Aldrich) was added to each well. The optical densities at 570 nm were read on a microplate spectrophotometer.
Apoptosis assay
ARPE-19 cells were grown overnight in a 12-well plate at a density of 8 × 104 cells/well and then treated with 300 μM H2O2 (Honeywell Fluka™) and IL-17A (100 ng/ml; GenScript) in DMEM/F12 with 2% FBS for 24 h. Cells were harvested and stained with a FITC Annexin V apoptosis detection kit (BD Pharmingen™ 556,547) according to the manufacturer’s instructions. The cells were subjected to apoptosis analysis using a BD FACSCalibur (BD Biosciences).
Permeability assay
ARPE-19 cells were seeded on 24-mm diameter, 0.4-μm pore size, polyester Transwell filters (Falcon, Tewksbury, MA, USA) at a density of 1 × 104 cells/well overnight. Cells were cultured in DMEM/F12 with 1% FBS for 7 days until they reached the appropriate cell confluence. The cells were then treated with 300 μM H2O2 and IL-17A (100 ng/ml) for 16 h. After treatment, 10 kDa FITC-dextran (100 μg/ml; Sigma–Aldrich) was added to the upper chamber of the Transwell, and then collected 50 μl aliquots from the lower chamber at 10–16 h to detect and measure the fluorescence signal at 485/538 nm excitation/emission wavelengths using a luminometer.
Western blotting
Mouse ocular tissues or ARPE-19 cells were lysed with commercial Cell Lysis Buffer (#9803, Cell Signaling, Beverly, MA, USA). Proteins were separated by SDS‒PAGE and transferred electrophoretically to 0.45 μm PVDF membranes (Millipore, Burlington, MA, USA). The membranes were blocked with 5% (w/v) nonfat milk in TBST for 1 h at room temperature and then incubated overnight at 4 °C with primary antibodies: anti-NQO-1 (1:5000; Cell Signaling), anti-HO-1 (1:10,000; Cell Signaling), anti-Nrf2 (1:500; Santa Cruz, Dallas, TX, USA), anti-GAPDH (1:5000; Genetex) and anti-tubulin (1:5000; Abcam). After binding of primary antibodies, the membranes were washed four times with TBST and incubated for 1 h at room temperature with species-specific horseradish peroxidase-labeled secondary antibodies. After binding of secondary antibodies, the membranes were washed five times with TBST. The binding of secondary antibodies was detected with SuperSignal West Pico Chemiluminescent Substrate (Millipore), and the chemiluminescence signals were visualized and imaged on a luminescence imaging system following exposure and development of Hyperfilm ECL molecules (Invitrogen).
Statistical analysis
Prism 9.0 (GraphPad Software; San Diego, CA, USA) was used for the statistical analysis. GraphPad Prism software was employed to process the initial data and plot graphs. Normality tests were performed before statistical tests. In this study, the data were normally distributed, and variances were homogeneous in all groups; parametric analysis (t test for the comparison of two groups or one-way ANOVA with Tukey's multiple comparisons test for the comparison of three or more groups) was used, and data are presented as the mean ± SEM. Two-way ANOVA with Sidak's multiple comparisons test was used to analyze multiple groups. The specific test that was used is indicated in the figure legends. P < 0.05 was considered to indicate statistical significance.
Results
IL-17A deficiency exacerbated the severity of laser-induced CNV in a mouse model
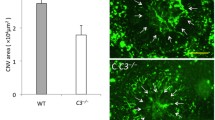
A previous study [35] reported that AMD patients had higher IL-22 and IL-17 levels than healthy individuals. IL-17 indirectly enhances vascular endothelial cell growth [36]. However, the precise role of IL-17A in AMD in vivo is still incompletely understood. To clarify the effect of IL-17A on neovascular AMD, we established a laser-induced CNV mouse model in WT and IL-17A-deficient mice and used fluorescein to observe the severity of CNV. Based on available knowledge, we hypothesized that the severity of CNV would decrease in IL-17A-deficient mice. However, we unexpectedly observed that the lesions in laser-induced CNV in IL-17A-deficient mice were more extensive than those in WT mice (Fig. 1A). Quantitative image analysis showed that there was no significant change between groups on day 7; however, the CNV area was significantly increased in IL-17A-deficient mice compared with WT mice at 14, 21, and 28 days after laser injury (Fig. 1B). H&E staining showed that the damage to retinal structure was more severe in IL-17A-deficient mice than in WT mice 28 days after laser injury (Fig. 1C). We also observed that the protein level of VEGFA was not significantly different in WT and IL-17A-deficient mice in the laser-induced CNV mouse model 28 days after laser injury; however, VEGFA mRNA was upregulated in WT mice compared with IL-17A-deficient mice 28 days after laser injury (Additional file 1: Fig. S1A–D).

IL-17A deficiency exacerbated disease severity in a laser-induced CNV mouse model. A Representative fundus fluorescein angiography (FFA) images from WT and IL-17A−/− mice at 7, 14, 21, and 28 days after laser photocoagulation. CNV lesions were analyzed by fluorescence angiography. B Quantification of the CNV area after laser photocoagulation at 7, 14, 21, and 28 days. Statistical differences were determined by unpaired t test. Data are presented as the mean ± SEM (*P < 0.05, ** < 0.01, *** < 0.001). N = 18 mice per group. C H&E staining of the retinal structure in healthy mice and WT and IL-17A−/− mice with laser-induced CNV at 28 days after laser injury. Magnification: 200X. Scale bar = 100 μm. D–J Representative flow cytometry data for the CD4+ T cell, γδ T cell, CD3+CD4+RORγt+ T cell, Treg cell, macrophage, neutrophil, and MDSC populations in the eyes of WT and IL-17A−/− mice 28 days after laser injury. N = 5 mice per group. Statistical differences were determined by two-way ANOVA and Sidak’s multiple comparisons test. Data are presented as the mean ± SEM (*P < 0.05, **P < 0.01)
IL-17A deficiency influenced ocular Treg cells, CD3+CD4+RORγt+ T cells, γδ T cells, macrophages, and neutrophils in a laser-induced CNV mouse model
Ocular immunity is involved in both maintaining visual homeostasis and driving AMD pathogenesis. We investigated whether distinct immune cell populations infiltrate the retina in WT and IL-17A-deficient mice after laser injury. We collected cervical lymph nodes from WT and IL-17A-deficient mice at 28 days after laser injury to analyze CD3+CD4+RORγt+ T cells, Treg cells (CD3+CD4+Foxp3+), and γδ T cells (CD3+TCRγδ+). FACS analysis showed no significant differences in these immune cell populations in the cervical lymph nodes of WT and IL-17A-deficient mice after laser injury (Additional file 1: Fig. S2A–C). At the same time, we also collected retina and choroidal tissue to analyze immune cell populations in the eyes (Additional file 1: Fig. S3). FACS analysis showed that the percentages of ocular CD3+CD4+RORγt+ T cells, Treg cells, and γδ T cells were lower in IL-17A-deficient mice than in WT mice at 28 days after laser injury (Fig. 1D–G). We further investigated whether IL-17A plays a role in recruiting the infiltration of innate immune cells (macrophages, MDSCs, and neutrophils) into the eyes and subsequently affects the extent of choroidal neovascularization. We collected retina and choroidal tissue to analyze the cell populations of macrophages (CD11b+F4/80+), MDSCs (CD11b+Ly6G−Ly6Chi), and neutrophils (CD11b+Ly6G+Ly6C+) in WT and IL-17A-deficient mice after laser injury. FACS analysis showed that IL-17A deficiency reduced neutrophil recruitment but increased macrophage infiltration into the eyes at 28 days after laser injury (Fig. 1H–J).
Th17 cells and γδ T cells were the main cellular sources of ocular IL-17A in mice with laser-induced CNV
To investigate the involvement of IL-17A in the laser-induced CNV mouse model, we performed immunofluorescence staining to detect the localization of IL-17A in healthy mice and mice with laser-induced CNV. IL-17A was expressed in the inner retinal layer in both healthy mice and mice with laser-induced CNV. Interestingly, IL-17A was also expressed in laser-induced CNV lesions in mice (Fig. 2A). ELISA showed that the serum level of IL-17A was not significantly different between healthy mice and mice with laser-induced CNV (Fig. 2B), which suggested that IL-17A may play an important regulatory role in local laser-induced eye lesions in mice. FACS analysis showed that the number of IL-17A-expressing cells in the mouse retina was increased after laser injury (Fig. 2C). To verify the possible cellular source of IL-17A in the laser-induced CNV mouse model, we collected retina and choroidal tissue from healthy mice and mice with laser-induced CNV on days 7, 14, 21, and 28 (Additional file 1: Fig. S4). FACS analysis showed that the percentage of CD45+IL-17A+ immune cells increased and that of CD45−IL-17A+ nonimmune cells decreased from day 7 to 28 after laser injury (Fig. 2D, E). We found that Th17 cells (CD3+CD4+RORγt+IL-17A+) and γδ T cells (CD3+TCRγδ+IL-17A+) were the two major cellular sources of IL-17A in the eyes of mice with laser-induced CNV (Fig. 2F–K). In addition, we analyzed the dynamic changes in the proportion of IL-17A-expressing immune cells and found that the percentage of γδ T cells increased in a time-dependent manner from day 7 to 28 after laser injury (Fig. 2L).

Cellular source of IL-17A in WT mice with the laser-induced CNV. A Immunofluorescence staining of ocular tissue with an anti-IL-17A antibody in vivo. Rabbit IgG isotype was used as the negative control. Magnification: 200X. Scale bar = 50 μm. B Serum IL-17A levels were measured by using ELISA at 7, 14, 21 and 28 days after laser photocoagulation. N = 5 mice per group. C–E Flow cytometry analysis of IL-17A-expressing cells among ocular cells and further gating of immune cells (CD45+) and nonimmune cells (CD45−). Statistical differences were determined by one-way ANOVA and Tukey’s multiple comparisons test. Data are presented as the mean ± SEM (*P < 0.05, **P < 0.01). N = 8 mice per group. F–K Representative flow cytometry data for the CD3+IL-17A+ T cell, CD3+CD4+IL-17A+ T cell, CD3+CD8+IL-17A+ T cell, γδ T cell, Th17 cell, and Treg cell populations in the eyes of WT mice at 7, 14, 21, and 28 days after laser injury in comparison with healthy control mice. N = 8 mice per group. Statistical differences were determined by one-way ANOVA and Tukey’s multiple comparisons test. Data are presented as the mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001). The data shown are representative of three independent experiments with similar results. L The dynamic changes in the proportion of IL-17A-expressing immune cells and the percentages of nonimmune cells, Treg cells, Th17 cells, and γδ T cells were analyzed at 7, 14, 21, and 28 days after laser injury. N = 8 mice per group
IL-17A deficiency influenced the expression of ocular proinflammatory molecules in a laser-induced CNV mouse model
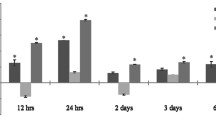
We further analyzed the inflammatory response in the eyes after laser injury. We performed RT‒qPCR to investigate the expression of proinflammatory cytokines (TNF-α, IL-1β, IL-6, and MCP-1), anti-inflammatory cytokines (IL-10 and TGF-β), and Th17-associated cytokines (IL-17F, GM-CSF, and IL-24). There was no significant change in TNF-α, IL-1β, and TGF-β expression between IL-17A-deficient and WT mice (Fig. 3A, B and F). IL-17A deficiency increased IL-6 and decreased MCP-1 and IL-10 expression in the eyes after laser injury (Fig. 3C–E). IL-17F, GM-CSF, and IL-24 levels were significantly reduced in IL-17A-deficient mice compared with WT mice at 28 days after laser injury (Fig. 3G–I). In addition, we performed a cytometric bead assay to further measure dynamic changes in systemic cytokine levels (Th1-, Th2-, and Th17-associated cytokines) in a laser-induced CNV mouse model. We observed an increasing trend for IFN-γ and IL-6 levels and a decreasing trend for IL-22 levels in IL-17A-deficient mice compared with WT mice (Additional file 1: Fig. S5A–I).

IL-17A deficiency influenced ocular cytokine expression in a laser-induced CNV mouse model. Eyes were harvested from healthy mice and WT and IL-17A−/− mice with laser-induced CNV at 28 days after laser injury (N = 8). Both eyes were pooled for each animal. A–I The mRNA expression of IL-10, MCP-1, IL-1β, TNF-α, IL-6, TGF-β, IL-24, IL-17F, and GM-CSF in the eyes was analyzed by using RT‒qPCR with specific primers. Statistical differences were determined by two-way ANOVA and Sidak’s multiple comparisons test. Data are presented as the mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). The data shown are representative of three independent experiments with similar results
IL-17A inhibited H2O2-induced apoptosis in ARPE-19 cells
Previous studies [12, 13, 37, 38] have indicated that oxidative stress in the RPE is a crucial contributor to the development of AMD. Based on our in vivo findings which raised the possibility that IL-17A might have a protective effect, we further clarified whether IL-17A can protect against oxidative stress in RPE cells. To confirm this hypothesis, we examined the viability of human ARPE-19 cells after IL-17A treatment by using an MTT assay. We found that IL-17A did not affect the viability of ARPE-19 cells (Fig. 4A). To determine the appropriate concentration of H2O2 to test the protective effect of IL-17A on H2O2-induced cytotoxicity, the viability of ARPE-19 cells was examined after H2O2 (0–2 mM) exposure for 24 h. We found that the IC50 value of H2O2 after stimulation for 24 h was 300 μM (data not shown). Therefore, we selected this experimental condition for the following experiments. Next, ARPE-19 cells were cotreated with IL-17A and H2O2, and then cell viability was analyzed. MTT assays showed that IL-17A did not affect ARPE-19 cell viability during exposure to H2O2-induced oxidative stress (Fig. 4B). We used the Annexin V/PI apoptosis assay to clarify the effect of IL-17A during exposure to oxidative stress. FACS analysis showed that IL-17A inhibited cell apoptosis during exposure to H2O2-induced oxidative stress in ARPE-19 cells (Fig. 4C, D).

IL-17A inhibited H2O2-induced apoptosis through HO-1 in ARPE-19 cells. A, B ARPE-19 cells were treated with IL-17A (10-100 ng/ml), H2O2 (300 μM), and IL-17A (100 ng/ml) plus H2O2 (300 μM) for 24 h. Cell viability was analyzed using the MTT assay. Statistical differences were determined by one-way ANOVA and Tukey’s multiple comparisons test. Data are presented as the mean ± SEM (*P < 0.05, **P < 0.01). C ARPE-19 cells were treated with or without IL-17A (100 ng/ml) for 24 h and then treated with H2O2 (300 μM) for 24 h, and cell apoptosis was analyzed using FACS. D Quantification of Annexin V+/ PI+ ratios in ARPE-19 cells after H2O2 and IL-17A treatment. Statistical differences were determined by one-way ANOVA and Tukey’s multiple comparisons test. Data are presented as the mean ± SEM (**P < 0.01, ****P < 0.0001). The data shown are representative of three independent experiments with similar results. E, F ARPE-19 cells were cotreated with H2O2 and IL-17A for 8 h, and the mRNA levels of HO-1 and NQO-1 were analyzed by RT‒qPCR with specific primers. N = 3. Statistical differences were determined by one-way ANOVA and Tukey’s multiple comparisons test. Data are presented as the mean ± SEM (*P < 0.05, **P < 0.01, ****P < 0.0001). The data shown are representative of three independent experiments with similar results. G, H ARPE-19 cells were cotreated with H2O2 and IL-17A for 24 h. The cell lysates were collected and analyzed using immunoblotting with specific antibodies against Nrf2, HO-1, and NQO-1. Tubulin was used as an internal control. Statistical differences were determined by one-way ANOVA and Tukey’s multiple comparisons test. Data are presented as the mean ± SEM of three independent experiments. (*P < 0.05, **P < 0.01, ****P < 0.0001)
IL-17A induced the expression of the antioxidant protein HO-1 during exposure to H2O2-induced oxidative stress
Previous studies [39, 40] reported the essential role of the Nrf2/Keap1/ARE pathway in redox homeostasis; this pathway leads to the expression of cytoprotective enzymes, such as NAD(P)H: quinone oxidoreductase 1 (NQO1) and heme oxygenase 1 (HO-1), to counteract oxidative stress. The Nrf2 pathway and its downstream antioxidant enzyme HO-1 are crucial for cellular defense against H2O2-induced oxidative damage [41]. HO-1 upregulation inhibited H2O2-induced apoptosis in ARPE-19 cells [42]. RT‒qPCR showed that H2O2 treatment induced the expression of HO-1 and NQO1. H2O2 combined with IL-17A treatment induced more HO-1 but no NQO1 expression compared with H2O2 treatment alone in ARPE-19 cells (Fig. 4E, F). Western blotting showed that IL-17A treatment increased HO-1 protein levels during exposure to H2O2-induced oxidative stress (Fig. 4G, H).
IL-17A facilitates repair of oxidative stress-induced barrier dysfunction
The outer blood‒retina barrier (BRB) formed by the tight junctions between RPE cells is critical for maintaining retinal homeostasis. The integrity of the outer BRB prevents choroidal neovascularization from invading the retina [43, 44]. The components of the BRB are tight junction proteins, including zonula occludens 1 (ZO-1), occludin (OCLN), and the claudin (CLDN) family [45]. A previous study [46] indicated that the HO-1-dependent MAPK pathway regulated intestinal barrier disruption. Our in vitro findings showed that IL-17A treatment increased HO-1 protein levels during exposure to H2O2-induced oxidative stress, suggesting that IL-17A may play a role in maintaining barrier function in the retina. To confirm this hypothesis, we used ARPE-19 cells to establish an in vitro model to evaluate the effect of IL-17A on retina barrier function. Immunofluorescence staining of the tight junction marker ZO-1 showed that the ARPE-19 monolayer successfully formed a tight junction in the untreated control group (Fig. 5A). We observed that H2O2 treatment destroyed the ARPE-19 monolayer, which suggested that H2O2 can damage the RPE by disrupting tight junctions. Compared with H2O2 treatment alone, IL-17A and H2O2 cotreatment preserved the integrity of the whole monolayer (Fig. 5A). Additionally, we set up a FITC-dextran permeability assay to evaluate oxidative stress-induced barrier dysfunction in ARPE-19 cells. We found that the FITC-dextran concentration in the H2O2-treated group increased in a time-dependent manner compared to that in the untreated control group. The concentration of FITC-dextran was significantly reduced in the IL-17A and H2O2 cotreated group compared with the H2O2-treated group (Fig. 5B). To further verify that IL-17A has a beneficial effect on the restoration of oxidative stress-induced barrier dysfunction, we analyzed tight junction proteins, including ZO-1, OCLN, and the CLDN family, in the eyes of mice with laser-induced CNV. RT‒qPCR showed that the expression of ZO-1, OCLN, CLDN-3, CLDN-10, and CLDN-19 in the eyes was decreased in IL-17A-deficient mice compared with WT mice at 28 days after laser injury (Fig. 5C–G). These data suggested that IL-17A plays a role in maintaining retinal homeostasis and impacts the repair of oxidative stress-induced barrier dysfunction in the laser-induced CNV mouse model.

IL-17A facilitated repair of oxidative stress-induced barrier dysfunction. ARPE-19 cells were cultured in DMEM/F12 with 1% FBS for 7 days until they reached the appropriate cell confluence. A ARPE-19 cells were cotreated with 300 μM H2O2 and IL-17A (100 ng/ml) in the Transwell insert for 16 h, and immunofluorescence staining was performed with an anti-ZO-1-specific antibody in ARPE-19 monolayers in vitro. Scale bar = 75 μm. B To analyze the permeability function, ARPE-19 cells were cotreated with 300 μM H2O2 and IL-17A (100 ng/ml) in the Transwell insert for 16 h. FITC-dextran (100 μg/ml) was added after treatment, and the fluorescent content was measured at 485/538 nm excitation/emission wavelengths using a luminometer at 10-16 h. Statistical differences were determined by one-way ANOVA and Tukey’s multiple comparisons test. Data are presented as the mean ± SEM (****P < 0.0001). The data shown are representative of three independent experiments with similar results. C–G The mRNA expression of ZO-1, OCLN, CLDN-3, CLDN-10, and CLDN-19 in eyes isolated from WT and IL-17A−/− mice (N = 6) at 28 days after laser injury was analyzed using RT‒qPCR with specific primers. Statistical differences were determined by two-way ANOVA and Sidak's multiple comparisons test. Data are presented as the mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). The data shown are representative of three independent experiments with similar results
Discussion
Oxidative stress is a critical inducer of RPE cell dysregulation in wet AMD; this process causes RPE barrier dysfunction and might result from disruption of intercellular tight junctions. We demonstrated that IL-17A deficiency exacerbated disease severity in a laser-induced CNV mouse model. IL-17A deficiency influenced ocular Treg cells, CD3+CD4+RORγt+ T cells, γδ T cells, macrophages, and neutrophils and affected the expression of ocular proinflammatory molecules. IL-17A inhibited cell apoptosis and induced the expression of the antioxidant protein HO-1 in ARPE-19 cells under oxidative stress. IL-17A has the ability to repair oxidative stress-induced barrier dysfunction in ARPE-19 cells. These findings suggest a protective effect of IL-17A in the laser-induced CNV model.
To confirm the elevated serum IL-17A levels in AMD patients, we established a laser-induced CNV mouse model and found that IL-17A was expressed mainly in CNV lesions. We did not observe a significant change in serum IL-17A levels in mice after laser injury. However, we found that the number of IL-17A-expressing cells in the eyes increased after laser injury. IL-17A deficiency influenced Treg, Th17, and γδ T-cell populations in the eyes but not in the cervical lymph nodes. In addition, IL-17A deficiency decreased neutrophil infiltration and increased macrophage infiltration into the retina. Therefore, the laser-induced CNV mouse model might involve a local inflammatory response and disruption of the ocular microenvironment but not the systemic microenvironment 28 days after laser injury. In addition, we analyzed the dynamic changes in the proportions of IL-17A-expressing immune cells and observed that the number of γδ T cells significantly increased from day 7 to 28 after laser injury; this change occurred in a time-dependent manner. These data suggested that IL-17A-secreting γδ T cells might play a critical role in maintaining ocular homeostasis at later time points after laser-induced injury.
Previous studies [47,48,49] reported that CNV lesions trigger local ocular inflammation via the infiltration of IL-17-producing γδ T cells, which are presumably recruited to the eye in a manner dependent on complement factor 5a. IL-17 generates a proinflammatory environment in the RPE by affecting barrier function. Gene transfer of a soluble IL-17 receptor prevents retinotoxicity in DKO/rd8 mice [50]. In addition, IL-17A deficiency in mice was reported to inhibit CNV development in the early phase after laser injury [51]. These findings provide evidence for a pathogenic role of IL-17A in AMD. Since AMD is an age-related progressive chronic disease, we speculated that the dynamic change in the laser-induced CNV model needs to be observed for a longer time. We observed no significant change in CNV lesions between WT and IL-17A-deficient mice at 7 days after laser injury. However, unexpectedly, the size of CNV lesions was significantly increased in IL-17A-deficient mice at 14, 21, and 28 days after laser injury. IL-17A deficiency caused a larger area of damage to the retinal structure. These data suggested that IL-17A might play a protective role in the laser-induced CNV mouse model.
Clinical trials targeting IL-17A in rheumatoid arthritis [52] and uveitis [53] have reported disappointing results. A previous study [54] reported that an IL-17A negative feedback loop limited Th17 pathogenicity in experimental autoimmune uveitis (EAU). The authors revealed that IL-17A could bind its receptor and further trigger the Th17 cell-intrinsic autocrine loop, which activates the transcription factor NF-κB and induces IL-24; IL-24 can then further suppress the expression of the Th17-lineage cytokines IL-17F and GM-CSF. A recent study [55] reported that IL-24 suppressed the production of proinflammatory cytokines and chemokines in ocular-infiltrating Th1 and Th17 cells and RPE cells in the EAU mouse model. IL-24 significantly suppressed the expression of RORγt, the master regulator of Th17 cells, and inhibited IL-17A production by Th17 cells. In our in vivo data, we found that IL-17F, GM-CSF, and IL-24 levels were significantly reduced in IL-17A-deficient mice compared with WT mice at 28 days after laser injury. These data indicated that the IL-17A-IL-24 circuit functions through a negative feedback mechanism and is critical for regulating ocular inflammation.
Previous studies [23, 33, 56, 57] have demonstrated that γδ T-cell-produced IL-17 can activate neutrophils and initiate an antibacterial immune response against infections occurring in several tissues. However, it is also important to note that excessive IL-17 production and the resultant neutrophilia can lead to immunopathology, further contributing to the onset or exacerbation of inflammatory diseases. These results support the idea that IL-17A may be a double-edged sword in terms of its effects during the early and late stages of laser-induced CNV in mice. On the other hand, a previous study [58] showed that in the absence of γδ T cells, bleomycin-stimulated mice showed more severe pulmonary interstitial inflammation and a significantly lower level of IL-17 production. The deficiency of IL-17 secretion resulted in a notable delay in epithelial regeneration following bleomycin instillation. Consistently, our study showed that the laser-induced inflammation and epithelial destruction in IL-17A-deficient CNV mice were more severe than those in WT CNV mice. These results implied that the specific tissue environment dictates the effects of γδ T-cell-produced IL-17A, allowing the immune system to respond to different disease-induced injuries. Our study suggested that the protective function of IL-17A produced by γδ T cells in the eyes involves more than just regulating ocular inflammation; it also involves antioxidant activities that promote the restoration of epithelial integrity.
A previous study [59] reported that macrophages activated under conditions of oxidative stress could enhance the inflammatory response but impair the phagocytic response, leading to ineffective clearance of apoptotic cells. Therefore, we speculated that IL-17A deficiency might cause a high level of oxidative stress and further direct macrophage activation to promote a robust inflammatory response that causes larger CNV lesions. Whether IL-17A-mediated oxidative stress promotes macrophage infiltration/activation and causes larger CNV lesions awaits further investigation. Future studies are needed to clarify these mechanisms by evaluating the effect of ROS inhibitor treatment in IL-17A-deficient mice with laser-induced CNV.
IL-10 was upregulated in ocular macrophages of aged mice after laser injury. In addition, IL-10 regulates macrophage function and promotes angiogenesis [19]. IL-6 was shown to be associated with the volume of macular edema in patients with CNV [18]. In our in vivo data, IL-10 expression was upregulated in the eyes of IL-17A-deficient mice 7 days after laser injury. IL-6 expression was increased in the eyes and serum of IL-17A-deficient mice 28 days after laser injury. We speculated that IL-10 might influence macrophage clearance of apoptotic cells in IL-17A-deficient mice at 7 days and further increase macrophage infiltration into the retina at 28 days after laser injury. Therefore, IL-10 may directly cause CNV lesion expansion at 14, 21, and 28 days.
Th17 cells, ILC3s, and γδ T cells mainly produce IL-17A. Neutrophils and macrophages also secrete IL-17A. In addition, a previous study [60] reported that intestinal Paneth cells produce IL-17A in an IL-23-independent fashion, which is one of the causes of systemic inflammatory response syndrome. IL-17A and IL-17F are expressed in colonic [61] and lung epithelial cells [62]. Our study also showed that nonimmune cells can secrete IL-17A in the retina. However, the role of IL-17A-expressing nonimmune cells in the development of AMD requires further investigation.
IL-17A acts as a pathogenic cytokine in a variety of inflammatory diseases, including multiple sclerosis [26, 27], inflammatory bowel disease [29, 30], and uveitis [63,64,65,66]. A previous study [29] reported that IL-17A-producing resident γδ T cells are essential for maintaining and protecting the epithelial barriers in the intestinal mucosa. The data from that study demonstrated one possible mechanism by which IL-17A signaling through Act-1 maintains OCLN localization to support barrier function during DSS-induced injury. Within the RPE, CLDN and OCLN traverse the plasma membrane, where ZO proteins anchor them. This anchoring enables their binding with signaling molecules and the actin cytoskeleton, thereby promoting the formation and stabilization of tight junctions [45]. Although the CLDN family includes at least 27 members, their expression patterns vary in different organs and tissues, contributing to the tissue-specific characteristics of tight junctions [67]. CLDN-19 is predominantly expressed in the human RPE and is essential for maintaining RPE tight junction formation and barrier function [68]. A previous study showed that patients harboring mutations in the CLDN-19 gene in the RPE suffer severe ocular impairment [69]. In addition, there is detectable, albeit lower, expression of CLDN-3 and CLDN-10 in RPE cells [70]. Therefore, we examined the expression of OCLN, ZO-1, CLDN-3, CLDN-10, and CLDN-19 in our laser-induced CNV model, and we found that these tight junction genes were expressed at lower levels in IL-17A-deficient mice than in WT mice. The laser injured the RPE and Bruch’s membrane and caused RPE barrier and membrane rupture [71], which may explain why the levels of tight junction genes were decreased at 28 days after laser injury.
Previous studies reported that IL-17A promoted VEGF production in corneal neovascularization [72], osteoarthritis [73], and rheumatoid arthritis [74, 75]. In our in vivo data, IL-17A deficiency did not influence VEGF expression in the laser-induced CNV mouse model. The retina resides in an environment that is primed for the generation of ROS and resultant oxidative damage. Oxidative stress contributes to the production of proangiogenic factors in the retina [76]. Previous studies have reported that many chemical compounds can induce the production of antioxidant proteins, including HO-1 and NQO-1, through the Nrf2 pathway to inhibit ROS production and further protect against oxidative stress [77,78,79,80]. According to our in vitro data, IL-17A-induced HO-1 expression might occur via the Nrf2 pathway. Therefore, we speculated that IL-17A might play a critical role in activating the oxidative stress-associated pathway without directly influencing the VEGF-associated signaling pathway in the laser-induced CNV mouse model. In addition, a previous study showed that VEGF expression peaked 7 days after laser injury [81]. Our data showed that VEGF expression could be detected at 28 days after laser injury. However, we cannot exclude the possibility that the use of different observation time points led us to miss a difference in VEGF expression between WT and IL-17A-deficient mice.
Intravitreal injections of anti-VEGF agents have become a first-line treatment for wet AMD. However, some patients still have persistent fluid or recurrent exudation, which is called refractory neovascular AMD [82]. A previous study also reported many obvious markers of oxidative damage in AMD, and antioxidant therapy is a potential treatment for AMD [83]. However, whether refractory neovascular AMD patients have high levels of oxidative stress in the retina is still unknown. According to our in vitro data, IL-17A treatment induced a high level of antioxidant proteins in APRE-19 cells. Future studies are needed to clarify the possible effect of combination treatment with IL-17A and anti-VEGF in animal models.
In summary, our findings demonstrate a possible effect of IL-17A-producing γδ T cells on the progression of laser-induced CNV in mice. IL-17A can exert a protective effect against laser-induced CNV injury. The underlying mechanism is that IL-17A induces the expression of the antioxidant protein HO-1, which might inhibit RPE cell apoptosis and prevent redundant macrophage infiltration into the retina and the subsequent generation of an excessive immune response. IL-17A repaired oxidative stress-induced barrier dysfunction. Our findings provide insight into the protective mechanisms of IL-17A in the ocular microenvironment, which may provide a new direction for understanding wet AMD.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- AMD:
-
Age-related macular degeneration
- CNV:
-
Choroidal neovascularization
- IL-17A:
-
Interleukin-17A
- WT:
-
Wild-type
- FACS:
-
Fluorescence-activated cell sorting
- VEGF:
-
Vascular endothelial growth factor
- RPE:
-
Retinal pigment epithelium
- Th17 cells:
-
T helper 17 cells
- ILC3:
-
Type three innate lymphoid cells
- γδ T cells:
-
Gamma delta T cells
- IBD:
-
Inflammatory bowel diseases
- DR:
-
Diabetic retinopathy
- EAU:
-
Experimental autoimmune uveitis
- ELISA:
-
Enzyme-linked immunosorbent assay
- RT‒PCR:
-
Reverse transcription polymerase chain reaction
- RT‒qPCR:
-
Real time quantitative polymerase chain reaction
- PBS:
-
Phosphate-buffered saline
- HRP:
-
Horseradish peroxidase
- TNF-α:
-
Tumor necrosis factor-α
- IHC:
-
Immunohistochemistry
- IF:
-
Immunofluorescence
- ZO-1:
-
Zonula occludens-1
- OCLN:
-
Occludin
- NQO-1:
-
NAD(P)H quinone dehydrogenase-1
- HO-1:
-
Heme oxygenase-1
References
Mehta S. Age-related macular degeneration. Prim Care. 2015;42:377–91.
Friedman DS, O’Colmain BJ, Muñoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–72.
Tielsch JM, Javitt JC, Coleman A, Katz J, Sommer A. The prevalence of blindness and visual impairment among nursing home residents in Baltimore. N Engl J Med. 1995;332:1205–9.
Ferris FL 3rd, Fine SL, Hyman L. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch Ophthalmol. 1984;102:1640–2.
Caprara C, Grimm C. From oxygen to erythropoietin: relevance of hypoxia for retinal development, health and disease. Prog Retin Eye Res. 2012;31:89–119.
Fogli S, Del Re M, Rofi E, Posarelli C, Figus M, Danesi R. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye (Lond). 2018;32:1010–20.
Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology. 2013;120:2292–9.
Scott AW, Bressler SB. Long-term follow-up of vascular endothelial growth factor inhibitor therapy for neovascular age-related macular degeneration. Curr Opin Ophthalmol. 2013;24:190–6.
Long D, Kanan Y, Shen J, Hackett SF, Liu Y, Hafiz Z, Khan M, Lu L, Campochiaro PA. VEGF/VEGFR2 blockade does not cause retinal atrophy in AMD-relevant models. JCI Insight. 2018;3: e120231.
Kurihara T, Westenskow PD, Bravo S, Aguilar E, Friedlander M. Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest. 2012;122:4213–7.
Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–90.
Beatty S, Koh HH, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–34.
Liang F-Q, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403.
Sachdeva MM, Cano M, Handa JT. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp Eye Res. 2014;119:111–4.
Fang Y, Su T, Qiu X, Mao P, Xu Y, Hu Z, Zhang Y, Zheng X, Xie P, Liu Q. Protective effect of alpha-mangostin against oxidative stress induced-retinal cell death. Sci Rep. 2016;6:21018.
Thichanpiang P, Harper SJ, Wongprasert K, Bates DO. TNF-α-induced ICAM-1 expression and monocyte adhesion in human RPE cells is mediated in part through autocrine VEGF stimulation. Mol Vis. 2014;20:781–9.
Wang H, Han X, Wittchen ES, Hartnett ME. TNF-α mediates choroidal neovascularization by upregulating VEGF expression in RPE through ROS-dependent β-catenin activation. Mol Vis. 2016;22:116–28.
Miao H, Tao Y, Li XX. Inflammatory cytokines in aqueous humor of patients with choroidal neovascularization. Mol Vis. 2012;18:574–80.
Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. 2007;117:3421–6.
Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79.
Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect. 2013;2: e60.
Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11:763–76.
Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. 2023;23:38–54.
Whitcup SM, Sodhi A, Atkinson JP, Holers VM, Sinha D, Rohrer B, Dick AD. The role of the immune response in age-related macular degeneration. Int J Inflam. 2013;2013: 348092.
DeMaio A, Mehrotra S, Sambamurti K, Husain S. The role of the adaptive immune system and T cell dysfunction in neurodegenerative diseases. J Neuroinflammation. 2022;19:251.
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8.
Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–4.
Martin JC, Baeten DL, Josien R. Emerging role of IL-17 and Th17 cells in systemic lupus erythematosus. Clin Immunol. 2014;154:1–12.
Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, Blumenschein WM, Judo M, Ayanoglu G, McClanahan TK, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. 2015;43:727–38.
Sarra M, Pallone F, Macdonald TT, Monteleone G. IL-23/IL-17 axis in IBD. Inflamm Bowel Dis. 2010;16:1808–13.
Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev. 2014;13:668–77.
Eisenstein EM, Williams CB. The T(reg)/Th17 cell balance: a new paradigm for autoimmunity. Pediatr Res. 2009;65:26r–31r.
Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. 2007;178:4466–72.
Chen J, Wang W, Li Q. Increased Th1/Th17 responses contribute to low-grade inflammation in age-related macular degeneration. Cell Physiol Biochem. 2017;44:357–67.
Liu B, Wei L, Meyerle C, Tuo J, Sen HN, Li Z, Chakrabarty S, Agron E, Chan CC, Klein ML, et al. Complement component C5a promotes expression of IL-22 and IL-17 from human T cells and its implication in age-related macular degeneration. J Transl Med. 2011;9:1–12.
Takahashi H, Numasaki M, Lotze MT, Sasaki H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol Lett. 2005;98:189–93.
Cai JY, Nelson KC, Wu M, Sternberg P, Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–21.
Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32.
Jian Z, Li K, Liu L, Zhang Y, Zhou Z, Li C, Gao T. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J Invest Dermatol. 2011;131:1420–7.
Papaiahgari S, Kleeberger SR, Cho HY, Kalvakolanu DV, Reddy SP. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J Biol Chem. 2004;279:42302–12.
Li L, Dong H, Song E, Xu X, Liu L, Song Y. Nrf2/ARE pathway activation, HO-1 and NQO1 induction by polychlorinated biphenyl quinone is associated with reactive oxygen species and PI3K/AKT signaling. Chem Biol Interact. 2014;209:56–67.
Hong Y, Liang Y-P, Chen W-Q, You L-X, Ni Q-F, Gao X-Y, Lin X-R. Protective effects of upregulated HO-1 gene against the apoptosis of human retinal pigment epithelial cells in vitro. Int J Ophthalmol. 2021;14:649–55.
Cunha-Vaz J, Bernardes R, Lobo C. Blood-retinal barrier. Eur J Ophthalmol. 2011;21(Suppl 6):S3-9.
Runkle EA, Antonetti DA. The blood-retinal barrier: structure and functional significance. Methods Mol Biol. 2011;686:133–48.
Naylor A, Hopkins A, Hudson N, Campbell M. Tight junctions of the outer blood retina barrier. Int J Mol Sci. 2019;21:211.
Zhang Z, Zhang Q, Li F, Xin Y, Duan Z. Contributions of HO-1-dependent MAPK to regulating intestinal barrier disruption. Biomol Ther. 2021;29:175–83.
Chen Y, Yang P, Li F, Kijlstra A. The effects of Th17 cytokines on the inflammatory mediator production and barrier function of ARPE-19 cells. PLoS ONE. 2011;6: e18139.
Chan CC, Ardeljan D. Molecular pathology of macrophages and interleukin-17 in age-related macular degeneration. Adv Exp Med Biol. 2014;801:193–8.
Coughlin B, Schnabolk G, Joseph K, Raikwar H, Kunchithapautham K, Johnson K, Moore K, Wang Y, Rohrer B. Connecting the innate and adaptive immune responses in mouse choroidal neovascularization via the anaphylatoxin C5a and gammadeltaT-cells. Sci Rep. 2016;6:23794.
Ardeljan D, Wang Y, Park S, Shen D, Chu XK, Yu CR, Abu-Asab M, Tuo J, Eberhart CG, Olsen TW, et al. Interleukin-17 retinotoxicity is prevented by gene transfer of a soluble interleukin-17 receptor acting as a cytokine blocker: implications for age-related macular degeneration. PLoS ONE. 2014;9: e95900.
Hasegawa E, Sonoda K-H, Shichita T, Morita R, Sekiya T, Kimura A, Oshima Y, Takeda A, Yoshimura T, Yoshida S, et al. IL-23–independent induction of IL-17 from γδT cells and innate lymphoid cells promotes experimental intraocular neovascularization. J Immunol. 2013;190:1778.
Kunwar S, Dahal K, Sharma S. Anti-IL-17 therapy in treatment of rheumatoid arthritis: a systematic literature review and meta-analysis of randomized controlled trials. Rheumatol Int. 2016;36:1065–75.
Letko E, Yeh S, Foster CS, Pleyer U, Brigell M, Grosskreutz CL. Efficacy and safety of intravenous secukinumab in noninfectious uveitis requiring steroid-sparing immunosuppressive therapy. Ophthalmology. 2015;122:939–48.
Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, Wang W, Chen Z, Silver PB, Jittayasothorn Y, et al. The cytokine IL-17A limits Th17 pathogenicity via a negative feedback loop driven by autocrine induction of IL-24. Immunity. 2020;53(384–397): e385.
Zhang X, Hu C, Zhong Y, Qiao D, Chi W, Shen H, Chong W. Multifunctional interleukin-24 resolves neuroretina autoimmunity via diverse mechanisms. Int J Mol Sci. 2022;23:11988.
Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, Oshiro K, Okamoto Y, Watanabe H, Kawakami K, et al. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against Listeria monocytogenes infection in the liver. J Immunol. 2008;181:3456–63.
Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest. 2010;120:1762–73.
Braun RK, Ferrick C, Neubauer P, Sjoding M, Sterner-Kock A, Kock M, Putney L, Ferrick DA, Hyde DM, Love RB. IL-17 producing gammadelta T cells are required for a controlled inflammatory response after bleomycin-induced lung injury. Inflammation. 2008;31:167–79.
Kirkham P. Oxidative stress and macrophage function: a failure to resolve the inflammatory response. Biochem Soc Trans. 2007;35:284–7.
Takahashi N, Vanlaere I, de Rycke R, Cauwels A, Joosten LAB, Lubberts E, van den Berg WB, Libert C. IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med. 2008;205:1755–61.
Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–19.
Suzuki S, Kokubu F, Kawaguchi M, Homma T, Odaka M, Watanabe S, Ieki K, Matsukura S, Kurokawa M, Takeuchi H, et al. Expression of interleukin-17F in a mouse model of allergic asthma. Int Arch Allergy Immunol. 2007;143(suppl 1):89–94.
Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. 2007;13:711–8.
Guedes MCE, Borrego LM, Proença RD. Roles of interleukin-17 in uveitis. Indian J Ophthalmol. 2016;64:628–34.
Li Y, Zhou Y. Interleukin-17: the role for pathological angiogenesis in ocular neovascular diseases. Tohoku J Exp Med. 2019;247:87–98.
Yoshimura T, Sonoda KH, Miyazaki Y, Iwakura Y, Ishibashi T, Yoshimura A, Yoshida H. Differential roles for IFN-gamma and IL-17 in experimental autoimmune uveoretinitis. Int Immunol. 2008;20:209–14.
Tokumasu R, Tamura A, Tsukita S. Time- and dose-dependent claudin contribution to biological functions: lessons from claudin-1 in skin. Tissue Barriers. 2017;5: e1336194.
Peng S, Rao VS, Adelman RA, Rizzolo LJ. Claudin-19 and the barrier properties of the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2011;52:1392–403.
Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79:949–57.
Peng S, Adelman RA, Rizzolo LJ. Minimal effects of VEGF and anti-VEGF drugs on the permeability or selectivity of RPE tight junctions. Invest Ophthalmol Vis Sci. 2010;51:3216–25.
Kim J, Park JR, Choi J, Park I, Hwang Y, Bae H, Kim Y, Choi W, Yang JM, Han S, et al. Tie2 activation promotes choriocapillary regeneration for alleviating neovascular age-related macular degeneration. Sci Adv. 2019;5:eaau6732.
Li Z, Burns AR, Han L, Rumbaut RE, Smith CW. IL-17 and VEGF are necessary for efficient corneal nerve regeneration. Am J Pathol. 2011;178:1106–16.
Honorati MC, Neri S, Cattini L, Facchini A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage. 2006;14:345–52.
Ryu S, Lee JH, Kim SI. IL-17 increased the production of vascular endothelial growth factor in rheumatoid arthritis synoviocytes. Clin Rheumatol. 2006;25:16–20.
Moran EM, Connolly M, Gao W, McCormick J, Fearon U, Veale DJ. Interleukin-17A induction of angiogenesis, cell migration, and cytoskeletal rearrangement. Arthritis Rheum. 2011;63:3263–73.
Jarrett SG, Boulton ME. Consequences of oxidative stress in age-related macular degeneration. Mol Aspects Med. 2012;33:399–417.
Dong X, Li Z, Wang W, Zhang W, Liu S, Zhang X, Fang J, Maeda H, Matsukura M. Protective effect of canolol from oxidative stress-induced cell damage in ARPE-19 cells via an ERK mediated antioxidative pathway. Mol Vis. 2011;17:2040–8.
Huang S-Y, Chang S-F, Chau S-F, Chiu S-C. The protective effect of hispidin against hydrogen peroxide-induced oxidative stress in ARPE-19 cells via Nrf2 signaling pathway. Biomolecules. 2019;9:380.
Yang JH, Shin BY, Han JY, Kim MG, Wi JE, Kim YW, Cho IJ, Kim SC, Shin SM, Ki SH. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol Appl Pharmacol. 2014;274:293–301.
Zhu C, Dong Y, Liu H, Ren H, Cui Z. Hesperetin protects against H2O2-triggered oxidative damage via upregulation of the Keap1-Nrf2/HO-1 signal pathway in ARPE-19 cells. Biomed Pharmacother. 2017;88:124–33.
Ding D, Zhu M, Liu X, Jiang L, Xu J, Chen L, Liang J, Li L, Zhou T, Wang Y, et al. Inhibition of TRAF6 alleviates choroidal neovascularization in vivo. Biochem Biophys Res Commun. 2018;503:2742–8.
Yang S, Zhao J, Sun X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: a comprehensive review. Drug Des Dev Ther. 2016;10:1857–67.
Payne AJ, Kaja S, Naumchuk Y, Kunjukunju N, Koulen P. Antioxidant drug therapy approaches for neuroprotection in chronic diseases of the retina. Int J Mol Sci. 2014;15:1865–86.
Acknowledgements
We are grateful for the support from the Human Biobank, Research Center of Clinical Medicine, National Cheng Kung University Hospital.
Funding
This work was supported by the Ministry of Science and Technology of Taiwan (MOST-111-2628-B-006-018 and MOST-111-2314-B-384-010-MY2).
Author information
Authors and Affiliations
Contributions
YHH conceived, supervised and coordinated the project. YHC, CJC and YHH designed the experiments. YHC, CJC, YRW, SMH and YHH designed and performed the experiments and analyzed the data. YHC, CHH, and YHH wrote the manuscript. All authors have reviewed and agreed upon the manuscript before submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All surgical intervention, preoperative care, and treatment of all animals were in strict accordance with Institutional Animal Care and approved by the Animal Ethics Committee.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Expression of VEGFA in the laser-induced CNV mouse model. A Immunofluorescence staining for VEGFA expression in ocular tissue in WT and IL-17A−/− mice 28 days after laser injury (N = 4). Magnification: 200X. B, C The ocular tissues from WT and IL-17A−/− mice 28 days after laser injury (N = 3) were collected and analyzed using immunoblotting with specific antibodies against VEGFA. GAPDH was used as an internal control. D The mRNA expression of VEGFA in eyes isolated from WT and IL-17A−/− mice (N = 6) 28 days after laser injury was analyzed using RT‒qPCR with specific primers. Statistical differences were determined by two-way ANOVA and Sidak’s multiple comparisons test. Data are presented as the mean ± SEM (***P < 0.001, ****P < 0.0001). Figure S2. Analysis of CD3+CD4+RORγt+ T cells, γδ T cells, and Treg cells in cervical lymph nodes of mice in a laser-induced CNV mouse model. A–C Representative flow cytometry data of CD3+CD4+RORγt+ T cells, γδ T cells, and Treg cells in cervical lymph nodes of WT and IL-17A−/− mice at 28 days after laser injury. N = 5 mice per group. Data are presented as the mean ± SEM. The data shown are representative of three independent experiments with similar results. Figure S3. Gating strategy for flow cytometry analysis for Fig. 1. In this sample gating, cells were gated in an SSC-A and FSC-A dot plot to select live cells and then gated in an FSC-H and FSC-A dot plot to eliminate doublets. The singlet gate was then gated on the CD45+ population. These were then further gated for the subsets of interest, namely, CD3+CD4+ T cells, CD3+CD4+RORγt+ T cells, Treg cells (CD3+CD4+Foxp3+), γδ T cells (CD3+TCRγδ+), macrophages (CD11b+F4/80+), MDSCs (CD11b+Ly6G−Ly6Chi), and neutrophils (CD11b+Ly6G+Ly6C+). Data were analyzed using FlowJo software, and population frequencies were expressed as percentages of the CD45+ parent population. Figure S4. Gating strategy for flow cytometry analysis for Fig. 2. In this sample gating, cells were gated in an SSC-A and FSC-A dot plot to select live cells and then gated in an FSC-H and FSC-A dot plot to eliminate doublets. The singlet gate was then gated on the IL-17A+ population. These were then further gated for the subsets of interest, namely, IL-17A+ immune cells (IL-17A+CD45+), IL-17A+ nonimmune cells (IL-17A+CD45−), CD3+IL-17A+ T cells, CD3+CD4+IL-17A+ T cells, CD3+CD8+IL-17A+ T cells, γδ T cells (CD3+TCRγδ+IL-17A+), Th17 cells (CD3+CD4+RORγt+IL-17A+), and Treg cells (CD3+CD4+Foxp3+IL-17A+). Data were analyzed using FlowJo software, and population frequencies were expressed as percentages of the IL-17A+ parent population. Figure S5. Dynamic changes in systemic cytokine levels in a laser-induced CNV mouse model. Serum samples were collected from healthy mice and WT and IL-17A−/− mice with laser-induced CNV (N = 5) at 7 days, 14 days, 21 days, and 28 days after laser injury. A–I The cytokine levels of IL-17F, IL-17A, IL-22, IL-23, TNF-α, IFN-γ, IL-5, IL-13, and IL-6 were measured by LEGENDplex™. Statistical differences were determined by two-way ANOVA and Sidak's multiple comparisons test. Data are presented as the mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001). The data shown are representative of two independent experiments with similar results. Table S1. List of primers sequences used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chang, YH., Hsing, CH., Chiu, CJ. et al. Protective role of IL-17-producing γδ T cells in a laser-induced choroidal neovascularization mouse model. J Neuroinflammation 20, 279 (2023). https://doi.org/10.1186/s12974-023-02952-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-02952-1