
Abstract
Neuroinflammation is instigated by the misfiring of immune cells in the central nervous system (CNS) involving microglia and astrocytes as key cell-types. Neuroinflammation is a consequence of CNS injury, infection, toxicity, or autoimmunity. It is favorable as well as a detrimental process for neurodevelopment and associated processes. Transient activation of inflammatory response involving release of cytokines and growth factors positively affects the development and post-injury tissue. However, chronic or uncontrolled inflammatory responses may lead to various neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis, and multiple sclerosis. These diseases have variable clinical and pathological features, but are underlaid by the aggregation of misfolded proteins with a cytotoxic effect. Notably, abnormal activation of glial cells could mediate neuroinflammation, leading to the neurodegenerative condition. Microglia, a type of glial cell, a resident immune cell, form the forefront defense of the CNS immune system. Dysfunctional microglia and astrocyte, a different kind of glial cell with homeostatic function, impairs the protein aggregate (amyloid-beta plaque) clearance in AD. Studies have shown that microglia and astrocytes undergo alterations in their genetic profile, cellular and molecular responses, and thus promote dysfunctional immune cross-talk in AD. Hence, targeting microglia and astrocytes-driven molecular pathways could resolve the particular layers of neuroinflammation and set a reliable therapeutic intervention in AD progression.
Similar content being viewed by others

Introduction
Alzheimer's disease is a neurodegenerative disorder characterized by cognitive deficit and memory loss. The pathological characteristic of the disease is the extracellular accumulation of amyloid-beta (Aβ) plaque and intracellular neurofibrillary tangles (NFTs). NFTs are composed of the hyperphosphorylated microtubule-binding protein tau, which regulates the cytoskeleton range. Aβ plaque constitutes Aβ oligomer and fibrils generated by the catalytic cleavage of amyloid precursor protein (APP) by the enzyme gamma (γ) and beta (β)-secretase. Mutation of APP and γ secretase cause rare familial AD, emphasizing that Aβ may contribute to AD pathogenesis [1, 2]. The pathogenic role of Aβ is known, but the mechanism of pathogenesis remains unknown. Aβ aggregates affect neuronal development along with the cellular and molecular components of the immune system. Glial cells, including microglia and astrocytes, are known to propagate Aβ toxicity. Microglia acts as a sentinel that surveys and senses the changes within the brain. Aβ aggregates induce microglial activation, leading to the release of nitric oxide (NO), reactive oxygen species (ROS), pro-inflammatory cytokines, chemokine, which may contribute to neuronal death [3, 4]. Aβ aggregates engage with cellular receptors such as Toll-like receptors (TLRs) and receptors for advanced glycoxidation end-products (RAGE) on microglia. This engagement induces the transcriptional activation of downstream inflammatory response genes [5,6,7]. The binding of Aβ aggregates to TLRs and RAGE have also recognized on astrocytes. The activation of astrocytes referred as reactive astrocytes produces toxic factors due to the induction of downstream target genes [8]. The inflammation becomes detrimental when inflammatory responses establish feed-forward loops with the Aβ formation, which overwhelms the standard resolution mechanism of Aβ aggregates [3]. Moreover, the runaway inflammatory response imbalances the immune cross-talk between microglia, astrocytes, and neurons, affecting regulatory mechanisms of synaptic plasticity, neuronal survival, and cognition [9].
Furthermore, in contrast to a rare cause of familial AD, the altered expression of microglia-associated genes may promote the late onset of AD (LOAD) [10]. An allelic variant of the apolipoprotein E (APOE) gene is significantly associated with an increased risk of AD [11]. Microglia and astrocytes encoded APOE4 display an immunomodulatory effect; therefore, they actively participate in neuroinflammation. Besides APOE4, several other gene variants expressed on microglia such as Triggering Receptor Expressed on Myeloid Cells (TREM2), PLCG2, CD33, transient receptor potential melastatin 2 (TRPM2), etc., linked with increased risk of AD (10). Although genetic, biochemical studies have shown a potential role of neuroinflammation in AD pathogenesis, but are not solely considered the causative factor. However, there is always a pursuing question of whether resolving neuroinflammation would regress the AD progression. Thus, to effectively address this question, it will be vital to understand the role of microglia and astrocytes and the causative mechanism in AD pathology. Here, we review the microglia and astrocytes-mediated neuroinflammation, therapeutic interventions, and further prospects of neuroinflammation biology in AD pathology.
The role of microglia in neuroinflammation
Microglia are the brain resident macrophages derived from monocyte precursor cells during neurodevelopment. The two significant functional aspects of microglia are immune defense and homeostasis maintenance. Besides, they have an essential role in neurogenesis, the formation of the neuronal circuit, and maintaining neuron health [12,13,14]. Microglia undergo dynamic changes during the physiological and pathological conditions, exemplified by a change in its morphology from ramified (resting) to amoeboid (active) form [15]. Under normal physiological conditions, the microglia patrol the brain's microenvironment to provide immune surveillance and neuronal survival (Fig. 1A). Studies have shown that Aβ aggregates induce microglia to acquire a full spectrum of activation by acquiring morphological and molecular changes. This activated form of microglia is referred to as disease-associated microglia (DAM) and has overexpression of particular receptors, chemokines, cytokines, and other factors [16]. The pattern recognition receptors (PRRs) such as TLRs, RAGE, and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) detect Aβ in the vicinity. The binding of Aβ, specifically to TLR4, has been suggested to activate microglia. Studies in the mouse model of AD have shown the role of TLR4 microglia signaling in mediating the increase of inflammatory cytokines production, phagocytosis inhibition, and higher plaque accumulation [17, 18]. Likewise, the binding of Aβ to RAGE promotes microglia activation-mediated neurotoxicity [7]. Besides, Aβ peptide activates NLRs, specifically the NLRP3, which triggers an inflammatory response by facilitating inflammasome complex formation and maturation of IL-1β [19]. Microglia express different purinergic receptors (e.g., P2Y12, P2Y6) that binds to extracellular nucleotides released by damaged cells. In turn, activated microglia have enhanced phagocytic potential [20, 21]. The PRRs ligation activates multiple transcription factors, such as NF-kB, AP-1, cAMP response element-binding protein, and interferon regulator families. These factors work in a combinatorial manner to regulate the expression of numerous downstream inflammatory response genes [22] (Fig. 1B).

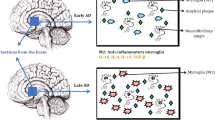
Schematic showing the modes of neuroinflammation in healthy and AD brain. A The healthy brain has minimal Aβ aggregates. Under normal physiological function microglia and astrocytes maintain neuronal homeostasis by clearing Aβ aggregates and providing neurotrophic factor to the brain. B The Alzheimer’s brain is associated with a large number of Aβ aggregates. The inhibited phagocytosis of Aβ aggregates and abrupt inflammatory response by microglia and astrocytes lead to the Aβ aggregates accumulation. Aβ aggregates bind to the pattern recognition receptors (PRRs) of microglia and stimulate downstream target genes NF-κB and AP-1. Subsequently, activated microglia produces cytokines. Cytokines contribute to astrocytes activation (referred to as reactive astrocytes) and affect neuronal health by causing neurotoxicity. The binding of Aβ aggregates to the microglia induces the NADPH oxidase and inducible nitric oxide synthase to produce ROS and NO leading to neurotoxicity. Likewise, Aβ aggregates bind to astrocyte receptors leading to the activation of downstream target genes NF-κB and AP-1 that subsequently produce cytokines. Cytokines affect neuronal health and cause neurotoxicity. The Aβ aggregates induce NADPH oxidase and inducible nitric oxide to produce ROS and NO by reactive astrocytes. The abrupt cross-talk between neurons, astrocytes, and microglia involving inflammatory molecules shown in the picture causes an imbalance in brain homeostasis and promotes neuronal death
Clinical and preclinical studies have shown the prominent role of activated microglia in AD progression [23]. Researchers have found microglia associated with Aβ plaque in AD patient's brains [3, 24, 25]. Microglia surrounding Aβ plaque has shown high immunoreactivity toward activation markers (e.g., MHCII and COX2), cytokines (e.g., IL-1, MCP-1, MIP-1α, IL-1β, TNFα, and IL-6), and chemokine receptors (e.g., CCR3, CCR5) [3]. MCP-1 induced chemotaxis of astrocytes and recruitment around Aβ plaque is reported [26]. Moreover, Aβ-induced microglia activation produces ROS through the NADPH oxidase system and reactive nitrogen species (RNS) by induction of inducible nitric oxide synthase (iNOS). These toxic factors stimulate amyloidogenesis, inhibition of Aβ clearance, and neurotoxicity [27] (Fig. 1B). In contrast, few studies have shown a neuroprotective effect of activated microglia. The research study has shown that the overexpression of IL-1β in the APP/PS1 mouse model reduces a load of Aβ plaque by increasing the number of activated microglia [28]. Another study has shown the benefit of IL-6 overexpression in APP transgenic mice against Aβ plaque accumulation by inducing gliosis and microglial activation [29]. Therefore, a discrepancy between studies suggests that microglia-mediated inflammation could be neurotoxic or neuroprotective. This may depend on the timing, duration, and amplitude of microglia activation.
The role of astrocytes in neuroinflammation
Astrocyte is the most abundant glial cell of the CNS, derived from neural stem cells (NSCs) [30] (Fig. 1A). In the coarse similarity with microglia, astrocytes maintain homeostasis and provide metabolite and growth factors to neurons. Besides, astrocyte has an imperial role in synapse formation, synaptic plasticity, and modulates the extracellular balance of ions, fluid, removal of free radicals at the blood–brain barrier functions [31]. Astrocytes are an essential component of tripartite synapses where along with the inter-neuronal communications, bidirectional communication exists between neurons and astrocytes, which maintains homeostasis and neuronal survival [32]. Under pathological conditions, astrocytes undergo morphological and functional changes characterized by cell hypertrophy and excessive release of neurotoxic factors, referred to as reactive astrocytes. The reactive astrocytes have an increased expression of prominent markers such as glial fibrillary acidic protein (GFAP), and vimentin which underlies cell hypertrophy. In the context of AD, evidence from post-mortem and rodent model studies has shown the presence of reactive astrocytes in proximity with Aβ plaque [33, 34]. The depletion of GFAP and vimentin reduces the astrocytic reaction and increases the Aβ plaque load in the APP/PS1 mouse model of AD [35]. Reactive astrocytes may have a neuroprotective role. It surrounds (forms a physical barrier called glial scar) and infiltrates Aβ plaque to reduce collateral damage due to neurotoxic Aβ species. Also, in vitro and in situ studies have shown that reactive astrocytes clear or reduce Aβ deposits by phagocytosis [26, 36]. Moreover, by improving the phagocytic potential of astrocytes, impairment in a neural circuit, inflammatory response, and Aβ-mediated pathology could be restored, as suggested by the study [37].
However, several studies have shown the neurotoxic role of reactive astrocytes. Similar to microglia, astrocytes also sense Aβ aggregates in the TLR/RAGE-dependent manner, which leads to activation of downstream target genes and subsequent production of factors [8]. Aβ induces the increased production of neurotoxic factors, including ROS, NO, cytokines (e.g., IL-1β, IL-6, TNF-α, IFN-α, granulocyte–macrophage colony-stimulating factor, chemokines (e.g., MCP-1, MIP1-α, CCL4, IL-8, IFN-γ-inducible protein-10) [3, 38]. The astrocytes’ reaction has shown to be carried by four signaling pathways including, the Janus kinase (JAK)/STAT3, the calcium/calcineurin (CN)/nuclear factor of activated T-cells (NFAT), nuclear factor kappa-B (NF-κB), and the MAPK pathway [39] (Fig. 1B). Excessive production of neurotoxic factors modulates astrocytes APP processing homeostasis, which leads to increased Aβ load and toxicity. A study has shown that the treatment of TNF-α and IFN-γ or Aβ42 oligomers and fibrils to cultured primary astrocytes increases β-secretase processing of APP and Aβ load in AD transgenic mice. The study has revealed a significant role of the feed-forward loop driven by cytokines and Aβ42 in enhancing Aβ load [40]. Also, the contribution of inflammatory mediators to synaptic degeneration, cognitive impairment, and neuronal death has been reported [41, 42]. Particularly IL-1-mediated signaling is recognized for its pathogenic role in AD [43]. The study has shown abrogation of IL-1 signaling ameliorates impaired inflammatory response, improves cognitive deficit, reduces tau and Aβ pathology in a transgenic mouse model of AD [44]. Thus, existing evidence has shown the imperial role of astrocytes in neuroinflammation. Ongoing research studies are focusing on astrocytes-mediated neuroinflammatory pathways to resolve AD progression.
Dysfunctional immune cross-talk cause neuroinflammation
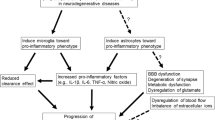
Communication between immune cells and neurons is an essential process in CNS. The cross-talk between microglia, astrocyte, and neurons is necessary to provide homeostasis and neuronal survival. Dysregulated cross-talk is known to underlay neuroinflammation in AD. Soluble factors such as cytokines, chemokines, ATP, complement proteins, and growth factors are the players of immune cross-talk.
Cellular and animal studies have shown that the impaired activation of astrocytes alters microglia status in AD. Impaired astrocytes induce an increase in the number of microglia around Aβ plaque in GFAP − / − Vim − / − APP/PS1 mice which can be a compensatory mechanism to resolve the inefficient role of astrocytes as suggested by the study [34]. In the Aβ-induced mixed glial culture (microglia and astrocyte), the loss of the neurotoxic potential of microglia-conditioned media is because astrocytes modulate microglia activation, as shown by study [45]. Also, the inhibition of the CN/NFAT signaling pathway in activated astrocytes showed to modulate microglial activation and Aβ load in APP/PS1 AD mouse model [42]. Moreover, studies have reported the impact of complement proteins on the Aβ dynamics and microglia status in AD. The Aβ produced by neurons activates NFκB-signaling in astrocytes, which in turn releases complement protein C3a extracellularly. The C3a interacts with the C3a receptor (C3aR) on microglia and neurons and aggravates Aβ pathology by facilitating the feed-forward loop. The blocking of this pathogenic cycle by employing a C3aR antagonist reduces microglial activation and Aβ load. Thus, NFκB/C3/C3aR signaling has been shown to mediate the Aβ response by involving astrocytes and microglia [46, 47] (Fig. 1B). Besides, transforming growth factor-beta (TGF-β)-mediated modulation in microglia has been reported [48]. Impaired TGF-β signaling in the AD brain is shown [47, 49]. Also, the extracellular ATP release from N-methyl-D-aspartate (NMDA) induced stimulated neurons and astrocytes activates microglia by binding to purinergic receptors [50, 51]. And, there are many more factors by which astrocytes regulate microglia function, but the underlying mechanism is poorly understood.
Furthermore, there are reports which have shown that microglia could alter the astrocyte status in CNS. The activated microglia induce A1 astrocyte, a subtype of reactive astrocyte, by secreting cytokines IL-1α, TNF, and C1q. A1 astrocytes are neurotoxic (Fig. 1B). The presence of A1 astrocyte in human AD post-mortem tissue is reported [52]. The research studies have highlighted the beneficial effect of neuropeptide in reducing neuroinflammation in AD. Recent study has shown that glucagon-like peptide-1, a gut-derived incretin agonist has reduced Aβ-induced microglia activation. In turn, executes the formation of reactive astrocytes concomitantly leading to inhibition of neuroinflammation in AD model [53]. Thus, evidence implies that astrocytes and microglia regulation are interdependent. The impairment in existing intercommunication may lead to neurotoxicity.
Role of cellular mechanisms in the regulation of microglial-mediated neuroinflammation
Studies have focussed on the biosynthetic mechanisms, derived metabolites, and mediators regulating microglia in AD pathogenesis. The sphingosine kinase1 (SphK1), an ATP-dependent lipid kinase, regulates microglia. SphK1 causes the catalytic reaction of acetyl COA and sphingosine to generate N-acetyl sphingosine (N-AS). N-AS acetylates cyclo-oxygenase 2(COX 2) and increases pro-resolving mediators (SPM). The treatment of NAS in APP/PS1 mice has shown to increase SPM, which resolves neuroinflammation by upregulation of microglia genes linked with phagocytosis in comparison to untreated APP/PS1 mice. In addition, Aβ-treated microglia showed less N-AS while treatment of N-AS has increased the production of SPM, consequently resolving neuroinflammation [54]. Studies have shown a key role of COX/prostaglandin E 2(PGE2) in the pathogenesis of AD. Genetic deletion of the PGE2 EP2 receptor on microglia has shown beneficial effects by enhancing phagocytosis of Aβ, resolving neuroinflammation, maintaining trophic factors, and signalling. Moreover, improvement in cognitive behavior and synaptic proteins in the mice model of AD [55, 56]. Neuro-inflammation has been associated with disturbed metabolic processes leading to over-production of reactive oxygen species. NADPH oxidase is a multi-subunit enzyme complex that drives the production of reactive oxygen species, and its role in microglia-mediated neurotoxicity is shown [57, 58]. Microglia exhibits classical M1 and alternative M2 phenotypes in pathological conditions. M1 phenotype facilitates the production of inflammatory cytokines leading to a neurotoxic effect, while M2 produces anti-inflammatory cytokines. The study has shown that deletion of NADPH oxidase causes microglia to switch from M1 to M2 phenotype, thus protecting neurons against neuroinflammation [59]. The alteration of the microglia state from M2 to M2 has shown a beneficial effect in Alzheimer’s model. Research studies have found induction of the M2 microglia state ameliorates Aβ-induced toxicity and improves cognition in Alzheimer’s disease model [60]. The advent of single-cell RNA sequencing/ScRNA and single nucleus sequencing/snRNA has enabled researchers to identify the heterogeneity of microglia subpopulations under normal and pathological conditions. ScRNA/snRNA datasets have paved the way to develop a therapeutic strategy against DAM and resolve neuroinflammation [61]. Researchers are focussing on the implication of microglia deletion in a pathological context. Studies have found that upon microglia deletion the niche is repopulated by microglia that come from differentiated microglia progenitor cells, residual microglia, or infiltrating peripheral cells [62,63,64]. In the context of AD, the beneficial effect of microglia depletion has shown by the administration of a colony-stimulating factor 1 receptor (CSF1 R) inhibitor in the mice model of AD. CSF1R is an essential factor for maintaining microglia health. The inhibition of microglial CSF1R has improved cognition [65], rescued dendritic formation, resolved neuroinflammation [66], and reduced amyloid-beta plaque formation [67]. Notably, depletion of microglia followed by repopulation of newborn microglia has opened the way to explore therapeutic approaches in a pathological context. Taken together, microglia modulation is a tantalizing attempt to modulate neuroinflammation-mediated neurotoxicity.
Calcium and P2XY signaling in regulating microglia-mediated inflammation
Calcium homeostasis plays a crucial role in microglia activation. Emerging studies have found a role of calcium homeostasis modulators (Calhms) in regulating calcium homeostasis in neurological disorders. Calhms are the voltage-gated calcium ion channel that allows the permeability of ions and ATP in a voltage-dependent manner. It gets activated upon the abrupt extracellular concentration of calcium and plays a significant role in controlling neuronal excitability, taste signalling, and pathologies of depression and AD. Genetic study has identified that CALHM1 P86L polymorphism is associated with incidence of AD. The role of calhm1 in regulating calcium homeostasis, Aβ production, and neuronal cell viability against Aβ-induced neurotoxicity has been shown [68]. The increased level of calhm2 has been noticed in AD pathogenesis. Moreover, the dissection of the mechanistic pathway reveals that calhm2 regulates calcium homeostasis, and inflammation in the microglia mice model of AD [69]. Calhm2 regulates the functional interaction of purinergic receptor P2XY (a cation channel expressed highly on microglia) with NLRP3. Inhibition of P2XY has reduced inflammation and increased the phagocytic potential of microglia [70], which in turn provides a beneficial effect on AD pathogenesis [71, 72]. In the study, the knockout of calhm2 decreases P2XY leading to a reduction in its interaction with NLRP3, consequently reducing neuroinflammation [69]. Research lines have identified the protein structure of calhm2 [73, 74], which has paved the way for understanding and developing therapeutic strategies against neurological diseases [Table 1].
Microglia expressed gene variants associated with AD
Genetic studies have elucidated different immunomodulatory gene variants associated with AD pathogenesis. Mutation in APP and γ-secretase components presenilin 1 and 2 causes rare familial AD [75]. Gene variants expressed on microglia have shown an association with the development of LOAD. The most studied and established risk factor is allele E4 of the apolipoprotein E gene. And, gene variants of TREM2, phosphoinositide phospholipase Cγ2 (PLCG2), CD33, TRPM2 are associated with LOAD [76, 77].
The role of APOE in microglial regulation
Human genetics studies have elucidated the majority of genes expressed on microglia and associated with AD risk [9]. Transcriptomics studies have suggested microglia acquire alteration in gene expression profile during the transition from homeostatic phenotype to molecular phenotype called DAM phenotype [78,79,80]. Genome-wide association studies (GWAS), cellular and animal model studies have shown a strong association of apolipoprotein E with the high risk of AD. Strittmatter and Roses were the first to suggest the role of APOE in AD [81], were further supported by other studies. The APOE plays physiological functions within the brain, but a significant role is to transport cholesterol, high-density lipoprotein particles. In humans, three polymorphic forms of APOE are present such as APOE2, APOE3, and APOE4. APOE4 is strongly associated with a high risk of AD. Numerous studies emphasize that the APOE4 modulates microglia function and contributes to neurotoxicity. A human cellular model study has shown the isogenic conversion of human iPSC-derived microglia from APOE3/E3 AD patients to APOE4/E4. Transitioned APOE4 microglia was found transcriptionally similar to APOE4 of the human AD brain. The study has configured that in human AD DAM phenotype of microglia might be induced by APOE4 [82]. APOE and its receptor have crucial role in AD. Studies have suggested that APOE4 is less effective in clearing Aβ aggregates because it has less tendency to bind Aβ peptides in comparison to APOE3 [83]. Noticeably, the APOE4 regulates the aggregation and clearance of Aβ aggregates and contributes to Aβ plaque accumulation [84]. The APOE4-mediated increase in the levels of pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL12p40) more efficiently than APOE3 has shown [85]. The human clinical study data obtained from two Chinese populations corroborate the above finding where APOE4 carriers had an increased level of pro-inflammatory (TNF-α, IL-6, and IL-1β) cytokines than the carriers of APOE2 and APOE3 [86]. Thus, APOE4 plays a crucial role in AD pathology and could be a target for therapeutic intervention.
The role of TREM2 in microglia regulation
GWAS studies have identified some rare gene variants expressed on microglia which contribute to AD pathology. In 2013, two independent studies from distinct populations deciphered the whole-exome and whole-genome sequencing and showed that R47H options of TREM2 had an increased risk of developing AD [87, 88]. TREM2 is a microglia cell surface receptor that provides several physiological functions to microglia. TREM2 stimulation (triggered by tyrosine phosphorylation) promotes microglial chemotaxis, phagocytosis, proliferation, and survival [89,90,91,92]. TREM2 has reported to regulate microbial function by maintaining the cellular energetics and biosynthetic metabolism [93]. The common extracellular ligands of TREM2 are anionic lipid, APOE, Aβ, and apoptotic cells [94, 95]. Impairment in TREM2–ligand interactions have been noticed in TREM2 variants, which can be a causation factor promoting the increased risk of AD [94, 96]. Aβ aggregates bind efficiently to lipoproteins, which in interaction with TREM2 microglia triggers downstream activation of target genes that eventually mediate Aβ clearance. However, TREM2-deficient microglia have reduced uptake of Aβ-lipoprotein complexes [94, 97]. Also, the TREM2 deficiency have shown higher Aβ plaque accumulation and dysfunctional microglia in mouse model of AD [95, 98]. The supraphysiological expression of TREM2 has shown improvement in microglia function, and amelioration in neuropathological and behavioral deficit in AD mouse models [99]. Also, a study has shown that TREM2 facilitates microglia-mediated phagocytosis of APOE bounded apoptotic neurons [100]. Thus, TREM2 could be an important regulatory factor of microglia, as its impairment may lead to Aβ pathology.
The role of TRPM2 channel in microglia regulation
Recently, the research line has focused on a receptor TRPM2 which is majorly expressed on microglia and has a role in microglia regulation. The TRPM2 channel is a ligand-gated calcium-permeable cation channel, co-activated by intracellular ADP ribose (ADPR) and calcium and has shown high sensitivity toward oxidative stress or ROS [101,102,103,104]. TRPM2 channel has critical physiological functions such as body temperature regulation, immune response, and apoptosis [105,106,107,108]. In 2004, Kraft et al. were the first to examine the expression of the TRPM2 channel in rodent’s microglial cells. The research has revealed novel calcium influx/signaling pathways mediated by the TRPM2 channel upon activation by H2O2 and ADPR [109]. And it was supported by the study, which has shown oxidative stress-induced calcium influx through the TRPM2 channel in human cultured microglia [110]. Moreover, the genetic deletion of TRPM2 showed to reverse the Aβ-induced perturbations such as microglial activation, neuronal toxicity, and memory impairment in APP/PS1 mice [111]. Continuingly, the research line has revealed the molecular mechanism of Aβ-induced activation of TRMP2 in cultured mouse microglia. Aβ-induced ROS (produced in protein kinase C/NADPH oxidase-dependent manner) activates poly (ADPR) polymerase‐1 (PARP-1), which acts on NAD to generate ADPR. Subsequently, the ADPR induces the activation of the TRPM2 channel. Also, TRPM2 channel-mediated activation of PYK2/MEK/ERK signaling acts as a fuel for PARP-1 and TRPM2 signaling. This leads to the sustained activation of TRPM2 channel with cytotoxic affects [112]. Thus, based on considerable evidence, TRPM2 can be regarded as a new player in neuroinflammation and could be used as a therapeutic intervention against AD.
Genetic variants linked with astrocytes
Few genes expressed on astrocytes have an immunomodulatory effect and undergo altered expression in AD [113]. The APOE gene variant APOE4 has shown an impact on Aβ species degradation and clearance. Here we discuss the role of astrocytes APOE gene variants in AD pathogenesis.
The role of APOE in astrocyte regulation
Inconsistent with microglia, astrocytes APOE gene variants too associate with neuroprotective or neurotoxic effects in AD by regulating Aβ dynamics. The study has shown that the APOE-deficient mouse astrocytes were inefficient in clearing Aβ plaque compared to wild-type mouse astrocytes in vitro [114]. The cell culture, animal model, and human study have deciphered a strong association of astrocytes APOE4 with AD pathogenesis. But the underlying mechanism is poorly understood [115,116,117,118]. The APOE4 astrocytes showed to inhibit Aβ clearance and promote Aβ accumulation. It affects initial amyloid plaque development [115]. In vivo study has highlighted a mechanistic view of APOE4 astrocytes-mediated impairment in Aβ clearance. APOE4 astrocytes induce the acidification of endosomal pH by downregulating Na + /H + exchanger isoform NHE6. It is a critical leak pathway for endosomal protons. It has led to the intracellular sequestration of low-density lipoprotein receptor-related protein (LRP1), leading to the loss of Aβ clearance. Epigenetic modifiers have restored NHE6 expression, normalized APOE4-specific defects in endosome pH, LRP1 trafficking, and Aβ clearance. The study has proposed endosomal pH as a target for the correction of amyloid disorders [119]. Besides, several studies have shown a higher contribution of APOE4 in tau pathogenesis and neuroinflammation relative to other APOE isoforms. Research study has shown APOE4 exerts neurotoxicity by influencing tau pathology and neuroinflammation. However, the absence of APOE was protective [120]. Taken together, APOE performs a complex set of interrelated functions in astrocytes, and impaired APOE expression affects neuronal survival.
Connecting link between neuroinflammation and autophagy
Autophagy is a catabolic process that maintains intracellular homeostasis by clearing misfolded protein and damaged organelles. Autophagy impairment has been associated with several neurodegenerative diseases like AD. The autophagy process involves a series of crucial steps responsible for delivering damaged organelles or protein aggregates to lysosomal degradation. The accumulated autophagic vacuoles (AVs) within dystrophic neurites indicate incomplete autophagy in AD [121] Studies have shown connecting link between the accumulation of Aβ aggregates and dysfunctional autophagy [122,123,124,125]. Emerging evidence has found the existing relation between neuroinflammation and autophagy. In vitro study has shown that the IL-1beta cytokine induces autophagy indicated by the accumulation of autophagosomes loaded with p62 and LC3 in triculture and pure culture of microglial [126]. Another study has found that IL-1 beta reduces inflammation and enhances microglial phagocytosis of Aβ, providing a beneficial effect in the 3xTgAD model [127]. Autophagy plays a role in facilitating the secretion of IL-1beta [128]. The anti-inflammatory role of autophagy has come into consideration due to its role in scavenging mitochondrial ROS and suppressing NLRP3 inflammasome activation [129, 130]. Moreover, defect in autophagy protein has been associated with microglia-mediated neuroinflammation. The study has shown the defect in microglial beclin 1, an essential protein for induction of autophagy, mediates neuroinflammation [131]. Researchers are interested in exploring the therapeutic use of autophagy enhancers against neurodegenerative diseases by employing animal models. The translation of preclinical outcomes could be different when implicated for clinical use because of species variation. The scientific community is utilizing human-induced pluripotent stem cells (hi-PSCs) that have paved the way to mirror the effect of therapeutic strategies in vivo. The majority of cases of AD are sporadic, few are familial, and modeling sporadic AD in rodents does not exist. The hi-PSCs neuronal model for sporadic AD has paved the way for exploring the therapeutic strategies encompassing autophagy modulators [132]. Studies have suggested diverse ways of autophagy enhancement that could provide potential benefits concerning neurodegenerative diseases. The autophagy inducer rapamycin, an inhibitor of mTOR, has shown a beneficial effect in several cellular and animal models of AD [133,134,135].
Rapamycin along with other compounds has shown enhancement in autophagy. The combinatorial effect of autophagy modulators has been suggested to provide better output in AD pathogenesis [136]. It can be hypothesized that autophagy enhancers could reduce neuroinflammation, in turn, protects neurons against the toxicity of protein aggregates in neurodegenerative conditions like AD.
Therapeutic approaches to combat neuroinflammation in AD
Considerably, neuroinflammation occurs before the appearance of histopathological or pathological features in AD. Therefore, existing as well as ongoing therapeutic interventions rely on the asymptomatic and prodromal stages of AD. Herbal medicines have shown beneficial effects in AD pathogenesis, probably by dampening neuroinflammation.
Curcumin and its metabolite tetrahydrocurcumin a derived from the spice turmeric, has shown a neuroprotective effect in cell culture and animal model studies of AD [137,138,139,140]. Curcumin-mediated beneficial effects such as modulation in inflammatory responses, reduction in Aβ plaque accumulation, protection against synaptic toxicity, and improvement in cognitive function enforce the use of curcumin in clinical trials for treating AD. A review of Voulgaropoulou et al., 2019 has summarized preclinical and clinical data encompassing the curcumin effect on cognitive impairment in AD and normal aging. Curcumin has a cognition enhancer property shown in AD and non-pathological aging [141]. Moreover, quercetin flavonoid derived from mulberry fruit Morus alba is rich in phenolics, vitamin c, linoleic acid, etc., and possesses anti-inflammatory and anti-oxidant properties [142]. Quercetin inhibits the NF-kappa B pathway and upregulates the anti-oxidant defense system NRf2/HO-1 axis [143].
Resveratrol, a polyphenol, commonly present in grapevine and other fruits, has shown beneficial effects in AD. Studies suggest that resveratrol has anti-oxidant, anti-inflammatory, and neuroprotective properties. The anti-oxidant and anti-inflammatory effect of resveratrol is due to the activation of Silent Information Regulator-1 [144]. Treatment of resveratrol in AD patients has shown amelioration in inflammatory responses, a decrease in Aβ levels, and improved cognitive function [145]. Furthermore, terpenoids such as Ginkgo biloba have shown neuroprotection in neurodegenerative conditions like AD. Ginkgo biloba has anti-inflammatory, anti-oxidant, and anti-apoptotic effects in the neurological diseases model [146]. It increases SIRT-1 expression, inhibits NF-kB, upregulates heme oxygenase-1 (HO-1), anti-apoptotic protein expression, and down-regulates pro-apoptotic protein expression [147]. It ameliorates mitochondrial dysfunction and protects SH-SY5Y cells against the toxic incidence of ROS [148]. In Aβ25–35-induced cultured hippocampal neurons, Ginkgo biloba has upregulated brain-derived neurotrophic factors and inhibited apoptosis [149]. Studies have also shed light on the compound Sinomenine, an alkaloid, extracted from the Chinese medicinal plant, Sinomenium acutum. In China, Sinomenine is a recommended medicine for the patient against rheumatoid arthritis. It has shown neuroprotection against Aβ-induced glial-mediated toxicity to neurons [150, 151]. The mechanism of Sinomenine is not clearly understood. The study by Qian and colleagues has shown that Sinomenine inhibits NADPH oxidase and protects neurons against the toxic effect of lipopolysaccharide (LPS) and the 1-methyl-4 phenylpyridinium (MPP +)- in a model of PD [152]. The dissection of the molecular mechanism of Sinomenine will be interesting and could uncover the therapeutic targets for AD. Many more natural compounds have shown potential efficacy toward AD, but have not included because this article has other orientations too.
Besides natural compounds, several synthetic drugs are in clinical research of AD and have modulated inflammatory responses. Drugs targeting cell surface receptors (TLRs, RAGE) or transducers NF-κB/effector (TNF-α) of the innate immune system are in the research.
GC021109 binds a purinergic receptor (P2Y6) expressed on microglia. It stimulates microglial phagocytosis and inhibits the release of inflammatory cytokines. GC021109 is in the early phase of a clinical trial [153]. Secondly, Azeliragon, an antagonist of RAGE, blocks the interaction of Aβ − RAGE and has shown neuroprotection in AD transgenic mouse models [154]. Pioglitazone is an agonist of peroxisome-proliferator-activated receptor gamma (PPARγ), which regulates astrocyte/neuron metabolic coupling, dendritic formation, and synapses. Pioglitazone has shown beneficial effects against several pathological features of AD including disturbed bioenergetic, inflammation, oxidative stress, microglial defects, etc. It improves learning and memory, synaptic activity, and amyloid and tau pathology [155]. Despite promising preclinical data, the drug failed in the clinical trial due to a lack of efficacy.
Besides, epidemiological studies have shown that the use of nonsteroidal anti-inflammatory drugs (NSAIDs) reduces the risk of developing AD by modulating the inflammatory process [156, 157]. But most of the tested NSAIDs failed in AD clinical trials majorly due to a lack of efficacy in improving AD symptoms [158]. Researchers have now focused on a new approach to combat AD pathology. The combination therapy of ibuprofen and cromolyn (ALZT-OP1) is on a clinical trial to combat the early stages of AD [159, 160]. Few drugs regulating cytokine production and activity are under investigation. Neflamapimod, an inhibitor of the intracellular enzyme p38 MAPK α, has shown promising results in cellular and animal studies of AD. Neflamapimod showed a reduction in Aβ load, ameliorate synaptic function, and reverses memory deficits [159,160,161]. The drug has passed the phase 2a clinical trial with a positive outcome and forwarded for phase 2b clinical trial. Conclusively, preclinical data of anti-inflammatory medications are reliable. But for clinical use, they should be accessible to CNS and could target mediators of neuroinflammation [Table 2].
Conclusion and future perspectives
Neuroinflammation is a central mechanism in the pathogenesis of AD. It consists of a complex network of interrelated processes involving roles played by microglia and astrocytes. Unbalanced activation of astrocytes and microglia instigates abnormal inflammatory responses leading to neuronal death. Also, a perturbation in existing cross-talk between microglia, astrocytes, and neurons causes neurotoxicity and cognitive deterioration. Moreover, genetic variants associated with astrocytes and microglia contribute to AD progression. Targeting genetic variants could be a promising therapeutic intervention but challenging too. Drugs are in the clinical research line, aiming to reduce AD progression or revert AD pathology by targeting neuroinflammation. Natural compounds have shown a minimum level of side effects with a promising outcome. Therefore, combination therapy involving natural compounds with other interventions could lead to a better therapeutic approach toward AD. Besides, ongoing recent studies target microglia and astrocyte activation state, trying to revert them into the homeostatic phenotype, which may lower the consequences of inflammatory responses. The existing link between inflammation and autophagy has attracted the attention of researchers towards improving autophagy defects. The available literature suggests that enhancing autophagy could prove to be a promising therapeutic strategy against the toxicity of protein aggregates in neurodegenerative conditions. Improving autophagy defects may ameliorate neuroinflammation and could be a therapeutic intervention toward AD progression.
Availability of data and materials
The author declares that the relevant data are included in the article.
Abbreviations
- CNS:
-
Central nervous system
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid beta
- NFTs:
-
Neurofibrillary tangles
- APP:
-
Amyloid precursor protein
- NO:
-
Nitric oxide
- ROS:
-
Reactive oxygen species
- TLRs:
-
Toll-like receptors
- RAGE:
-
Receptors for advanced glycoxidation end-product
- SphK1:
-
Sphingosine Kinase 1
- N-AS:
-
N-Acetyl sphingosine
- SPM:
-
Pro resolving factor
- PGE2:
-
Prostaglandin E2
- CSF1R:
-
Colony stimulating factor 1-receptor
- Calhms:
-
Calcium homeostasis modulators
- LOAD:
-
Late onset of AD
- TREM2:
-
Triggering receptor expressed on myeloid cells
- TRPM2:
-
Transient receptor potential melastatin 2
- DAM:
-
Death-associated microglia
- PRRs:
-
Pattern recognition receptors
- NOD:
-
Nucleotide binding oligomerization domain
- NLRs:
-
NOD-like receptors
- RNS:
-
Reactive nitrogen species
- iNOS:
-
Inducible nitric oxide synthase
- NSCs:
-
Neural stem cells
- GFAP:
-
Glial fibrillar acidic protein
- CN:
-
Calcium/calcineurin
- NFAT:
-
Nuclear factor of activated T cells
- NF-κB:
-
Nuclear factor Kappa B
- HO-1:
-
Heme-oxygenase-1
- C3aR:
-
C3a receptor
- TGF-β:
-
Transforming growth factor
- NMDA:
-
N-Methyl-D-aspartate
- GWAS:
-
Genome-wide association studies
- ADPR:
-
ADP ribose
- PARP-1:
-
Ploy (ADPR) polymerase-1
- LRP1:
-
Lipoprotein receptor-related protein
- NSAIDs:
-
Nonsteroidal anti-inflammatory drugs
References
Murphy MP, LeVine H III. Alzheimer’s disease and the amyloid-β peptide. J Alzheimers Dis. 2010;19(1):311–23.
Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68(2):270–81.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421.
Kitazawa M, Yamasaki TR, LaFerla FM. Microglia as a Potential Bridge between the Amyloid β-Peptide and Tau. Ann N Y Acad Sci. 2004;1035(1):85–103.
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29(38):11982–92.
Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schüffer W, Fassbender K. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem. 2007;20(6):947–56.
Du Yan S, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382(6593):685–91.
Paudel YN, Angelopoulou E, Piperi C, Othman I, Aamir K, Shaikh M. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s disease (AD): from risk factors to therapeutic targeting. Cells. 2020;9(2):383.
Chun H, Marriott I, Lee CJ, Cho H. Elucidating the interactive roles of glia in Alzheimer’s disease using established and newly developed experimental models. Front Neurol. 2018;9:797.
Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217(2):459–72.
Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63(3):287–303.
Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45.
Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, Yamashita T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16(5):543–51.
Araujo DM, Cotman CW. Basic FGF in astroglial, microglial, and neuronal cultures: characterization of binding sites and modulation of release by lymphokines and trophic factors. J Neurosci. 1992;12(5):1668–78.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8.
Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. 2017;79:619–43.
Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi KI. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2008;5(1):1.
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi KI. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain. 2006;129(11):3006–19.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–8.
Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y 12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9(12):1512–9.
Hidetoshi TS, Makoto T, Inoue K. P2Y receptors in microglia and neuroinflammation. Wiley Interdisciplinary Reviews: Membrane Transport and Signaling. 2012;1(4):493–501.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20.
Weitz TM, Town T. Microglia in Alzheimer’s disease: it’s all about context. Int J Alzheimer’s Dis. 2012;2012:1.
Wisniewski HM, Wegiel J, Wang KC, Kujawa M, Lach B. Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can J Neurol Sci. 1989;16(S4):535–42.
Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 1992;84(2):117–27.
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med. 2003;9(4):453–7.
Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39(1):1–6.
Shaftel SS, Kyrkanides S, Olschowka JA, Jen-nie HM, Johnson RE, O’Banion MK. Sustained hippocampal IL-1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Investig. 2007;117(6):1595–604.
Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, Zubair AC, Dickson D, Golde TE, Das P. Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24(2):548–59.
Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–84.
Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32(12):638–47.
Farhy-Tselnicker I, Allen NJ. Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev. 2018;13(1):1–2.
Kato S, Gondo T, Hoshii Y, Takahashi M, Yamada M, Ishihara T. Confocal observation of senile plaques in Alzheimer’s disease: senile plaque morphology and relationship between senile plaques and astrocytes. Pathol Int. 1998;48(5):332–40.
Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APPsw mice. Neurobiol Aging. 2001;22(1):49–61.
Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, Brown J, Wilhelmsson U, Restivo JL, Cirrito JR. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013;27(1):187–98.
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, Diwan A. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34(29):9607–20.
Gomez-Arboledas A, Davila JC, Sanchez-Mejias E, Navarro V, Nuñez-Diaz C, Sanchez-Varo R, Sanchez-Mico MV, Trujillo-Estrada L, Fernandez-Valenzuela JJ, Vizuete M, Comella JX. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia. 2018;66(3):637–53.
Liu C, Cui G, Zhu M, Kang X, Guo H. Neuroinflammation in Alzheimer’s disease: chemokines produced by astrocytes and chemokine receptors. Int J Clin Exp Pathol. 2014;7(12):8342.
Perez-Nievas BG, Serrano-Pozo A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front Aging Neurosci. 2018;10:114.
Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation. 2011;8(1):1–7.
Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of A β-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2(6): e167.
Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32(46):16129–40.
Griffin WS. Inflammation and neurodegenerative diseases. Am J Clin Nutr. 2006;83(2):470S-S474.
Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187(12):6539–49.
Von Bernhardi R, Eugenín J. Microglial reactivity to β-amyloid is modulated by astrocytes and proinflammatory factors. Brain Res. 2004;1025(1–2):186–93.
Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 2015;85(1):101–15.
Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci. 2016;36(2):577–89.
Eyüpoglu IY, Bechmann I, Nitsch R. Modification of microglia function protects from lesion-induced neuronal alterations and promotes sprouting in the hippocampus. FASEB J. 2003;17(9):1110–1.
Lee HG, Ueda M, Zhu X, Perry G, Smith MA. Ectopic expression of phospho-Smad2 in Alzheimer’s disease: Uncoupling of the transforming growth factor-β pathway? J Neurosci Res. 2006;84(8):1856–61.
Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB, MacVicar BA. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci. 2014;34(32):10511–27.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–8.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Park JS, Kam TI, Lee S, Park H, Oh Y, Kwon SH, Lee S. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol Commun. 2021;9(1):1–15.
Lee JY, Han SH, Park MH, Song IS, Choi MK, Yu E, Bae JS. N-AS-triggered SPMs are direct regulators of microglia in a model of Alzheimer’s disease. Nat Commun. 2020;11(1):1–19.
Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 as a New Target to Increase Amyloid β Phagocytosis and Decrease Amyloid β-Induced Damage to Neurons. Brain Pathol. 2005;15(2):134–8.
Johansson JU, Woodling NS, Wang Q, Panchal M, Liang X, Trueba-Saiz A, Andreasson KI. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J Clin Investig. 2015;125(1):350–64.
Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279(2):1415–21.
Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS. Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem. 2002;83(4):973–83.
Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem. 2012;120(2):292–301.
Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11(1):1–15.
Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, Haass C. Emerging microglia biology defines novel therapeutic approaches for Alzheimer’s disease. Neuron. 2020;108(5):801–21.
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–97.
Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, Peng B. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. 2018;21(4):530–40.
Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, Jucker M. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci. 2012;109(44):18150–5.
Dagher NN, Najafi AR, Kayala KMN, Elmore MR, White TE, Medeiros R, Green KN. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015;12(1):1–14.
Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, Green KN. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016;139(4):1265–81.
Sosna J, Philipp S, Albay R, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, Glabe CG. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener. 2018;13(1):1–11.
Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Marambaud P. A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer’s disease risk. Cell. 2008;133(7):1149–61.
Cheng J, Dong Y, Ma J, Pan R, Liao Y, Kong X, Yuan Z. Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer’s disease pathology. Sci Adv. 2021;7(35):e3600.
Ni J, Wang P, Zhang J, Chen W, Gu L. Silencing of the P2X7 receptor enhances amyloid-β phagocytosis by microglia. Biochem Biophys Res Commun. 2013;434(2):363–9.
Chen X, Hu J, Jiang L, Xu S, Zheng B, Wang C, Wang Q. Brilliant Blue G improves cognition in an animal model of Alzheimer’s disease and inhibits amyloid-β-induced loss of filopodia and dendrite spines in hippocampal neurons. Neuroscience. 2014;279:94–101.
Martin E, Amar M, Dalle C, Youssef I, Boucher C, Le Duigou C, Delarasse C. New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol Psychiatry. 2019;24(1):108–25.
Choi W, Clemente N, Sun W, Du J, Lü W. The structures and gating mechanism of human calcium homeostasis modulator 2. Nature. 2019;576(7785):163–7.
Syrjanen JL, Michalski K, Chou TH, Grant T, Rao S, Simorowski N, Furukawa H. Structure and assembly of calcium homeostasis modulator proteins. Nat Struct Mol Biol. 2020;27(2):150–9.
Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9(10):768–78.
Fernandez CG, Hamby ME, McReynolds ML, Ray WJ. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front Aging Neurosci. 2019;11:14.
Misra A, Chakrabarti SS, Gambhir IS. New genetic players in late-onset Alzheimer’s disease: Findings of genome-wide association studies. Indian J Med Res. 2018;148(2):135.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–90.
Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Investig. 2017;127(9):3240–9.
Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, Duong DM, Pennington MW, Lah JJ, Seyfried NT, Levey AI. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):1–25.
Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19(1):53–77.
Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98(6):1141–54.
Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10(5):333–44.
Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81(4):740–54.
Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30(9):1350–60.
Fan YY, Cai QL, Gao ZY, Lin X, Huang Q, Tang W, Liu JH. APOE ε4 allele elevates the expressions of inflammatory factors and promotes Alzheimer’s disease progression: a comparative study based on Han and She populations in the Wenzhou area. Brain Res Bull. 2017;132:39–43.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–27.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–16.
Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trümbach D, Wurst W, Brunner B. TREM 2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017;18(7):1186–98.
Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6(243):243.
Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Investig. 2015;125(5):2161–70.
Zheng H, Jia L, Liu CC, Rong Z, Zhong L, Yang L, Chen XF, Fryer JD, Wang X, Zhang YW, Xu H. TREM2 promotes microglial survival by activating Wnt/β-catenin pathway. J Neurosci. 2017;37(7):1772–84.
Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell. 2017;170(4):649–63.
Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91(2):328–40.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–71.
Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, Wang Y, Tanzi RE, Colonna M. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017;13(4):381–7.
Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213(5):667–75.
Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J Neurosci. 2017;37(3):637–47.
Lee CD, Daggett A, Gu X, Jiang LL, Langfelder P, Li X, Wang N, Zhao Y, Park CS, Cooper Y, Ferando I. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron. 2018;97(5):1032–48.
Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, Li X, Rademakers R, Kang SS, Xu H, Younkin S. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem. 2015;290(43):26043–50.
Wang L, Fu TM, Zhou Y, Xia S, Greka A, Wu H. Structures and gating mechanism of human TRPM2. Science. 2018;362:6421.
Huang Y, Winkler PA, Sun W, Lü W, Du J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature. 2018;562(7725):145–9.
Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9(1):163–73.
Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, Masker K, Miller BA. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem. 2003;278(18):16222–9.
Tan CH, McNaughton PA. The TRPM2 ion channel is required for sensitivity to warmth. Nature. 2016;536(7617):460–3.
Song K, Wang H, Kamm GB, Pohle J, de Castro RF, Heppenstall P, Wende H, Siemens J. The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science. 2016;353(6306):1393–8.
Knowles H, Heizer JW, Li Y, Chapman K, Ogden CA, Andreasen K, Shapland E, Kucera G, Mogan J, Humann J, Lenz LL. Transient Receptor Potential Melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc Natl Acad Sci. 2011;108(28):11578–83.
Hecquet CM, Zhang M, Mittal M, Vogel SM, Di A, Gao X, Bonini MG, Malik AB. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ Res. 2014;114(3):469–79.
Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286(1):C129–37.
Lee M, Cho T, Jantaratnotai N, Wang YT, McGeer E, McGeer PL. Depletion of GSH in glial cells induces neurotoxicity: relevance to aging and degenerative neurological diseases. FASEB J. 2010;24(7):2533–45.
Ostapchenko VG, Chen M, Guzman MS, Xie YF, Lavine N, Fan J, Beraldo FH, Martyn AC, Belrose JC, Mori Y, MacDonald JF. The transient receptor potential melastatin 2 (TRPM2) channel contributes to β-amyloid oligomer-related neurotoxicity and memory impairment. J Neurosci. 2015;35(45):15157–69.
Alawieyah SMS, Sim JA, Neubrand VE, Jiang LH. A critical role of TRPM2 channel in Aβ42-induced microglial activation and generation of tumor necrosis factor-α. Glia. 2018;66(3):562–75.
Sekar S, McDonald J, Cuyugan L, Aldrich J, Kurdoglu A, Adkins J, Serrano G, Beach TG, Craig DW, Valla J, Reiman EM. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol Aging. 2015;36(2):583–91.
Koistinaho M, Lin S, Wu XI, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10(7):719–26.
Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, Bu G. ApoE4 accelerates early seeding of amyloid pathology. Neuron. 2017;96(5):1024–32.
Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci. 2013;110(19):E1807–16.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774.
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Science translational medicine. 2011;3(89):89ra57
Prasad H, Rao R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc Natl Acad Sci. 2018;115(28):E6640–9.
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–7.
Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120(23):4081–91.
Uddin M, Stachowiak A, Mamun AA, Tzvetkov NT, Takeda S, Atanasov AG, Stankiewicz AM. Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Frontiers in Aging Neuroscience. 2018;10:4.
Ling D, Salvaterra PM. A central role for autophagy in Alzheimer type neurodegeneration. Autophagy. 2009;5(5):738–40.
Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE. 2009;4(1): e432.
Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Aβ-induced cytotoxicity. Autophagy. 2009;5(4):502–10.
François A, Terro F, Janet T, Bilan AR, Paccalin M, Page G. Involvement of interleukin-1β in the autophagic process of microglia: Relevance to Alzheimer’s disease. J Neuroinflammation. 2013;10(1):1–22.
Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187(12):6539–49.
Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30(23):4701–11.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–5.
Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Kanneganti TD. Receptor interacting protein kinase 2–mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14(5):480–8.
Houtman J, Freitag K, Gimber N, Schmoranzer J, Heppner FL, Jendrach M. Beclin1‐driven autophagy modulates the inflammatory response of microglia via NLRP 3. The EMBO J. 2019 5;38(4):e99430.
Jiménez-Moreno N, Stathakos P, Caldwell MA, Lane JD. Induced pluripotent stem cell neuronal models for the study of autophagy pathways in human neurodegenerative disease. Cells. 2017;6(3):24.
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and tau effects on cognitive impairments. J Biol Chem. 2010;285(17):13107–20.
Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE. 2011;6:9.
Liu Y, Su Y, Wang J, Sun S, Wang T, Qiao X, Liang Z. Rapamycin decreases tau phosphorylation at Ser214 through regulation of cAMP-dependent kinase. Neurochem Int. 2013;62(4):458–67.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113–22.
Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21(21):8370–7.
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280(7):5892–901.
Mishra S, Mishra M, Seth P, Sharma SK. Tetrahydrocurcumin confers protection against amyloid β-induced toxicity. NeuroReport. 2011;22(1):23–7.
Hoppe JB, Haag M, Whalley BJ, Salbego CG, Cimarosti H. Curcumin protects organotypic hippocampal slice cultures from Aβ1–42-induced synaptic toxicity. Toxicol In Vitro. 2013;27(8):2325–30.
Voulgaropoulou SD, van Amelsvoort TA, Prickaerts J, Vingerhoets C. The effect of curcumin on cognition in Alzheimer’s disease and healthy aging: A systematic review of pre-clinical and clinical studies. Brain Res. 2019;1725: 146476.
Khan H, Ullah H, Aschner M, Cheang WS, Akkol EK. Neuroprotective effects of quercetin in Alzheimer’s disease. Biomolecules. 2019;10(1):59.
Davies DA, Adlimoghaddam A, Albensi BC. Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells. 2021;10(8):1884.
Gomes BA, Silva JP, Romeiro CF, Dos Santos SM, Rodrigues CA, Gonçalves PR, Sakai JT, Mendes PF, Varela EL, Monteiro MC. Neuroprotective mechanisms of resveratrol in Alzheimer’s disease: role of SIRT1. Oxidative medicine and cellular longevity. 2018 ;2018.
Moussa C, Hebron M, Huang X, Ahn J, Rissman RA, Aisen PS, Turner RS. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J Neuroinflammation. 2017;14(1):1.
Fang W, Deng Y, Li Y, Shang E, Fang F, Lv P, Mao L. Blood brain barrier permeability and therapeutic time window of Ginkgolide B in ischemia–reperfusion injury. Eur J Pharm Sci. 2010;39(1–3):8–14.
Mohammad Nabavi S, Habtemariam S, Daglia M, Braidy N, Rosa Loizzo M, Tundis R, Fazel Nabavi S. Neuroprotective effects of ginkgolide B against ischemic stroke: A review of current literature. Curr Top Med Chem. 2015;15(21):2222–32.
Li L, Zhang QG, Lai LY, Wen XJ, Zheng T, Cheung CW, Xu SY. Neuroprotective effect of ginkgolide B on bupivacaine-induced apoptosis in SH-SY5Y cells. Oxidative medicine and cellular longevity; 2013;5:89.
Xiao Q, Wang C, Li J, Hou Q, Li J, Ma J, Wang Z. Ginkgolide B protects hippocampal neurons from apoptosis induced by beta-amyloid 25–35 partly via up-regulation of brain-derived neurotrophic factor. Eur J Pharmacol. 2010;647(1–3):48–54.
Shukla SM, Sharma SK. Sinomenine inhibits microglial activation by Aβ and confers neuroprotection. J Neuroinflammation. 2011;8(1):1–1.
Singh D, Agrawal A, Singal CM, Pandey HS, Seth P, Sharma SK. Sinomenine inhibits amyloid beta-induced astrocyte activation and protects neurons against indirect toxicity. Mol Brain. 2020;13(1):1.
Qian L, Xu Z, Zhang W, Wilson B, Hong JS, Flood PM. Sinomenine, a natural dextrorotatory morphinan analog, is anti-inflammatory and neuroprotective through inhibition of microglial NADPH oxidase. J Neuroinflammation. 2007;4(1):1–14.
Fu WY, Wang X, Ip NY. Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: mechanisms, drug candidates, and new opportunities. ACS Chem Neurosci. 2018;10(2):872–9.
Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, Thiyagarajan M. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J Clin Investig. 2012;122(4):1377–92.
Reassessment of pioglitazone for Alzheimer’s disease. Front Neurosci. P. 702.
Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48(3):626–32.
McGeer PL, Rogers J, McGeer EG. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9(s3):271–6.
Miguel-Álvarez M, Santos-Lozano A, Sanchis-Gomar F, Fiuza-Luces C, Pareja-Galeano H, Garatachea N, Lucia A. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: a systematic review and meta-analysis of treatment effect. Drugs Aging. 2015;32(2):139–47.
Wyss-Coray T, Mucke L. Ibuprofen, inflammation and Alzheimer disease. Nat Med. 2000;6(9):973–4.
Zhang C, Griciuc A, Hudry E, Wan Y, Quinti L, Ward J, Tanzi RE. Cromolyn reduces levels of the Alzheimer’s disease-associated amyloid β-protein by promoting microglial phagocytosis. Sci Rep. 2018;8(1):1–9.
Duffy JP, Harrington EM, Salituro FG, Cochran JE, Green J, Gao H, Bemis GW, Evindar G, Galullo VP, Ford PJ, Germann UA. The discovery of VX-745: A novel and selective p38α kinase inhibitor. ACS Med Chem Lett. 2011;2(10):758–63.
Alam JJ. Selective brain-targeted antagonism of p38 MAPKα reduces hippocampal IL-1β levels and improves Morris water maze performance in aged rats. J Alzheimers Dis. 2015;48(1):219–27.
Alam J, Blackburn K, Patrick D. Neflamapimod: clinical phase 2b-ready oral small molecule inhibitor of p38alpha to reverse synaptic dysfunction in early Alzheimer’s disease. J Prev Alzheimers Dis. 2017;4:273–8.
Acknowledgements
I thank Ritu Borah and Deepti Dama for their helpful suggestion.
Funding
The author is funded by ICMR-Research Associate fellowship (5/3/8/58/ITR-F/2020); the funding body has no involvement in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
DS conceived the idea, analyzed the data, and drafted the manuscript. The author read and approved the final manuscript.
Author’s information
The author is a PhD in Cellular and Molecular biology from CSIR-Indian Institute of Toxicology Research Lucknow, India specializing in inflammation biology associated with neurodegeneration. Currently, the author is a Research Associate in the Department of Cellular and Molecular Neurosciences, National Brain Research Centre, Manesar, Haryana, India. She is investigating challenges in the remediation of neurotoxicity in Alzheimer’s disease.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Singh, D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J Neuroinflammation 19, 206 (2022). https://doi.org/10.1186/s12974-022-02565-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-022-02565-0