
Abstract
Background
Despite rigorous confirmation with reliable assays, some individuals showing the neuromyelitis optica spectrum disorder (NMOSD) phenotype remain negative for both aquaporin-4 (AQP4) and myelin oligodendrocyte glycoprotein (MOG) antibodies.
Objective
We aimed to investigate whether double seronegative NMOSD (DN-NMOSD) and NMOSD with AQP4 antibody (AQP4–NMOSD) share the same pathophysiological basis, astrocytopathy, by measurement of cerebrospinal fluid (CSF) glial fibrillary acidic protein (GFAP) levels as a marker of astrocyte damage.
Methods
Seventeen participants who (1) satisfied the 2015 diagnostic criteria for NMOSD, and (2) tested negative for AQP4 and MOG antibodies confirmed with repeated cell-based assays, and (3) had available CSF samples obtained at the point of clinical attacks, were enrolled from 4 medical centers (South Korea, Germany, Thailand, and Denmark). Thirty age-matched participants with AQP4–NMOSD, 17 participants with MOG antibody associated disease (MOGAD), and 15 participants with other neurological disorders (OND) were included as controls. The concentration of CSF GFAP was measured using enzyme-linked immunosorbent assay.
Results
CSF GFAP levels in the DN-NMOSD group were significantly lower than those in the AQP4–NMOSD group (median: 0.49 versus 102.9 ng/mL; p < 0.001), but similar to those in the OND (0.25 ng/mL) and MOGAD (0.39 ng/mL) control groups. The majority (90% (27/30)) of participants in the AQP4–NMOSD group showed significantly higher CSF GFAP levels than the highest level measured in the OND group, while no participant in the DN-NMOSD and MOGAD groups did.
Conclusions
These results suggest that DN-NMOSD has a different underlying pathogenesis other than astrocytopathy, distinct from AQP4–NMOSD.
Similar content being viewed by others


Introduction
Since the discovery of aquaporin-4 (AQP4) antibodies, neuromyelitis optica spectrum disorder (NMOSD) is defined as a neuro-inflammatory disease of the central nervous system (CNS), separate from multiple sclerosis [1]. The majority (up to 90%) of individuals with NMOSD defined by the 2015 revised diagnostic criteria show seropositivity for AQP4 antibody [2, 3]. However, the criteria allow the diagnosis of NMOSD in individuals without AQP4 antibodies [4,5,6]. A subset of individuals with a NMOSD phenotype, but without AQP4 antibodies, test positive for the myelin oligodendrocyte glycoprotein (MOG) antibody, which primarily targets myelin antigens [7]. Although some clinical features overlap with AQP4 antibody positive NMOSD (AQP4–NMOSD), MOG antibody associated disease (MOGAD) has been recently defined as a distinctive disease entity, with elucidation of plausible different pathophysiology [8]. Despite repeated rigorous antibody measurements with reliable assays, some individuals with NMOSD phenotype remain persistently negative for both AQP4 and MOG antibodies [9,10,11,12]. Such double seronegative NMOSD (DN-NMOSD) poses diagnostic and therapeutic challenges in clinical practice and the classification of DN-NMOSD within the neuro-inflammatory diseases of the CNS remains unknown.
AQP4–NMOSD is a primary astrocytopathy mediated by antibodies selectively targeting the water channel protein AQP4, which is abundantly located on astrocytic foot processes in the CNS [1]. Glial fibrillary acidic protein (GFAP), a major constituent of the astrocyte cytoskeleton, can reflect astrocyte injury in AQP4–NMOSD when measured in body fluids, such as cerebrospinal fluid (CSF) [13,14,15]. This study aimed to investigate whether DN-NMOSD and AQP4–NMOSD share the same pathophysiological basis, astrocytopathy, by comparing the CSF GFAP levels at clinical exacerbation.
Methods
This international collaborative study enrolled participants from 4 medical centers (South Korea, Germany, Thailand, and Denmark). Participants who (1) satisfied the 2015 diagnostic criteria for NMOSD, [2] (2) were seronegative for both AQP4 and MOG antibodies following rigorous confirmation with reliable assays, and (3) had CSF samples available that were obtained at the point of clinical attack, were included. Seventeen CSF samples were obtained from individuals with DN-NMOSD (11 from South Korea, 2 from Germany, 1 from Thailand, and 3 from Denmark). Thirty age-matched participants with AQP4–NMOSD and 17 participants with MOGAD were included as controls. The MOGAD group included 10 patients who satisfied and 7 who did not satisfy the 2015 criteria for NMOSD. In addition, 15 age-matched controls with other neurological disorders (OND: 4 primary headache, 3 idiopathic sixth cranial neuropathies, 1 compressive myelopathy, 1 sub-acute combined degeneration, 1 hemi-facial spasm, 1 diabetic polyneuropathy, 1 brachial plexopathy, 1 non-specific white matter disease, 1 adrenoleukodystrophy, and 1 suspected motor neuron disease), not expected to present with underlying astrocytopathy, were included. All CSF samples were collected within 1 month of clinical exacerbation and most of the samples were obtained before the initiation of high dose steroid therapy (94% (16/17) in DN-NMOSD, 83% (25/30) in AQP4–NMOSD, and 71% (12/17) in MOGAD).
The serostatus of AQP4 antibodies was rigorously confirmed using two different methods: an in-house live cell-based assay (CBA) conducted at the National Cancer Center (NCC, South Korea) [16], and a commercial CBA (Euroimmun, Luebeck, Germany). The serostatus of anti-MOG antibodies was determined using the in-house CBA at the NCC with live transfected cells with a full-length human MOG [17]. Additional file 1: Table S1 shows titers of AQP4 and MOG antibodies at the time of CSF sampling. Repeated examinations were performed during the course of the disease, at minimum of two different timepoints including at least one acute exacerbation in participants with DN-NMOSD. The seronegative status of the AQP4 and MOG antibodies in the sera, at the same timepoint with the CSF sample collection, 6 participants from Germany, Thailand, and Denmark was double-checked with CBA at each center and NCC. Antibody tests in CSF were also performed to eliminate the possibility of AQP4 and MOG antibodies detectable only in the CSF [18,19,20]. CSF GFAP concentrations were evaluated in duplicate by an independent investigator who was blinded to the diagnosis using an enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s protocol (BioVendor, Minneapolis, MN, USA).
Statistical analysis
GFAP levels across groups were compared by the Kruskal–Wallis test and post-hoc analysis was conducted using Dunn’s multiple comparisons test.
Results
Table 1 shows the demographics of the participants. The mean age at sampling of the participants with DN-NMOSD, AQP4–NMOSD, MOGAD, and OND was 32.3 ± 9.6, 35.2 ± 11.1, 31.2 ± 7.3 and 36.8 ± 7.4 years, respectively. Table 2 demonstrates the clinical and para-clinical features of the participants with DN-NMOSD.
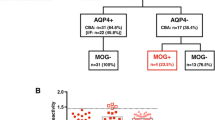
As shown in Fig. 1, the concentration of CSF GFAP in the DN-NMOSD group was significantly lower than that in the AQP4–NMOSD group (median: 0.49 (range 0.25–1.36) versus 102.9 (range 0.62–90,242.6) ng/mL; p < 0.001), but similar to that in the OND (0.25 (0.25–1.48) ng/mL) and MOGAD (0.39 (0.25–1.35) ng/mL) control groups.

Cerebrospinal fluid (CSF) glial fibrillary acidic protein (GFAP) levels (DN-NMOSD: double seronegative neuromyelitis optica spectrum disorder, AQP4–NMOSD: anti-aquaporin-4 antibody positive neuromyelitis optica spectrum disorder, MOGAD: anti-myelin oligodendrocyte glycoprotein antibody associated diseases, ONDs: other neurological disorders; dot line: the highest measured GFAP level in ONDs, ***p < 0.0001)
None of the DN-NMOSD and MOGAD groups showed higher CSF GFAP levels than the highest measured CSF GFAP level in participants with ONDs (primary headache, 1.48 ng/mL). In contrast, the CSF GFAP levels of most participants (27/30, 90%) in the AQP4–NMOSD group were higher than the highest measured CSF GFAP level in the OND group, and the remaining 3 participants with AQP4–NMOSD experienced relatively mild clinical symptoms (2 with optic neuritis and 1 with sensory-only partial myelitis).
Discussion
The CSF GFAP levels in the rigorously confirmed DN-NMOSD group were significantly lower than those in the AQP4–NMOSD group, but did not differ from those in the OND or MOGAD group. Consistent with previous results, [12,13,14,15] CSF GFAP levels in participants with AQP4–NMOSD were significantly higher than those in participants with ONDs or MOGAD. These findings suggest a discriminative underlying pathophysiology in DN-NMOSD other than astrocytopathy, distinguishing it from AQP4–NMOSD.
It is important to confirm the truly negative status of AQP4 and MOG antibodies to facilitate a fair comparison between DN-NMOSD and AQP4–NMOSD. Therefore, repeated tests using reliable CBAs were performed at least two different timepoints, particularly in the clinical exacerbation, to detect the positive conversion of AQP4 and MOG antibodies during subsequent clinical relapse [21]. At least two different assays with high sensitivity and specificity were used [16, 17], in addition to CSF assays, because antibodies in some cases may be only detectable in CSF but not in serum [18,19,20]. A previous study reported that CSF GFAP levels in 2 of 3 participants with DN-NMOSD were elevated above those in non-neurological controls (highest level: 2.3 ng/mL) and increased over the median CSF GFAP level (5.40 ng/mL) of AQP4–NMOSD [12]; whereas no elevation in CSF GFAP levels was observed in 17 participants with DN-NMOSD relative to those in controls with ONDs (highest level: 1.48 ng/mL) in current study. The reason for this discrepancy is yet to be uncovered. Given the possible heterogeneity of DN-NMOSD, there would be a subset of patients with astrocytopathy due to yet unidentified autoantibodies or other causes in 3 DN-NMOSD patients of previous study, but not in 17 patients of current study. Alternatively, it might simply result from the difference in the confirmation of double negativity against AQP4 and MOG antibodies. The median CSF GFAP level of controls with ONDs (0.25 ng/mL) in the current study was comparable to that of previous studies (median 0.5 ng/mL in non-neurological controls [12] and mean 0.6–0.7 ng/mL in controls with ONDs [13, 14]).
Conclusions
The current evidence of the pathophysiological distinction between DN-NMOSD and AQP4–NMOSD raises an issue whether these two potentially different diseases should be within the same umbrella of NMOSD. Two recent clinical trials that applied the 2015 NMOSD criteria revealed that the efficacy of therapeutic agents for AQP4-negative NMOSD was unclear compared to that for AQP4-positive NMOSD [22, 23]. To establish adequate therapeutic strategies on the basis of appropriate pathophysiology, further investigations to identify other potential immunological target(s) specific to DN-NMOSD are warranted.
Availability of data and materials
Anonymized data not published within this article will be made available upon request from any appropriately qualified investigator.
Abbreviations
- AQP4:
-
Aquaporin 4
- NMOSD:
-
Neuromyelitis optica spectrum disorder
- CNS:
-
Central nervous system
- MOG:
-
Myelin oligodendrocyte glycoprotein
- AQP4–NMOSD:
-
AQP4 antibody positive NMOSD
- MOGAD:
-
MOG antibody associated disease
- DN-NMOSD:
-
Double seronegative NMOSD
- GFAP:
-
Glial fibrillary acidic protein
- CSF:
-
Cerebrospinal fluid
- OND:
-
Other neurological disorder
- CBA:
-
Cell based assay
References
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7.
Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–89.
Hyun JW, Jeong IH, Joung A, et al. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology. 2016;86:1772–9.
Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14.
Marignier R, Bernard V, Pascale G, et al. Aquaporin-4 antibody-negative neuromyelitis optica: distinct assay sensitivity-dependent entity. Neurology. 2013;80:2194–200.
Jiao Y, Fryer JP, Lennon VA, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology. 2013;81:1197–204.
Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017;264:2088–94.
Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. 2019;15:89–102.
Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474–81.
Hoftberger R, Sepulveda M, Armangue T, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler. 2015;21:866–74.
Sepulveda M, Armangue T, Sola-Valls N, et al. Neuromyelitis optica spectrum disorders: comparison according to the phenotype and serostatus. Neurol Neuroimmunol Neuroinflamm. 2016;3:e225.
Kleerekooper I, Herbert MK, Kuiperij HB, et al. CSF levels of glutamine synthetase and GFAP to explore astrocytic damage in seronegative NMOSD. J Neurol Neurosurg Psychiatry. 2020;91:605–11.
Misu T, Takano R, Fujihara K, Takahashi T, Sato S, Itoyama Y. Marked increase in cerebrospinal fluid glial fibrillar acidic protein in neuromyelitis optica: an astrocytic damage marker. J Neurol Neurosurg Psychiatry. 2009;80:575–7.
Takano R, Misu T, Takahashi T, et al. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology. 2010;75:208–16.
Kaneko K, Sato DK, Nakashima I, et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J Neurol Neurosurg Psychiatry. 2016;87:1257–9.
Kim Y, Kim G, Kong BS, et al. Large-scale in-house cell-based assay for evaluating the serostatus in patients with neuromyelitis optica spectrum disorder based on new diagnostic criteria. J Clin Neurol. 2017;13:175–80.
Kim Y, Hyun JW, Woodhall MR, et al. Refining cell-based assay to detect MOG-IgG in patients with central nervous system inflammatory diseases. Mult Scler Relat Disord. 2020;9(40):101939.
McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76:1108–10.
Mariotto S, Gajofatto A, Batzu L, et al. Relevance of antibodies to myelin oligodendrocyte glycoprotein in CSF of seronegative cases. Neurology. 2019;93:e1–6.
Akaishi T, Takahashi T, Misu T, et al. Difference in the source of anti-AQP4-IgG and anti-MOG-IgG antibodies in CSF in patients with neuromyelitis optica spectrum disorder. Neurology. 2021. https://doi.org/10.1212/WNL.0000000000012175.
Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665–71.
Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:2114–24.
Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a doubleblind randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394:1352–63.
Hyun JW, Lee HL, Park J, et al. Brighter spotty lesions on spinal MRI help differentiate AQP4 antibody-positive NMOSD from MOGAD. Mult Scler. 2021;6:13524585211060326.
Acknowledgements
The authors would like to thank the participants in current study.
Funding
This study was supported by the National Cancer Center (2011510-1) and National Research Foundation of Korea (NRF-2020R1F1A1072174 & NRF-2018R1A5A2023127).
Author information
Authors and Affiliations
Contributions
J-WH and HJK had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. J-WH and HJK contributed to the study concept and design. All the authors contributed to the acquisition, analysis, and interpretation of the data. J-WH and HJK performed the statistical analysis. All the authors contributed to drafting of the manuscript and critically revised it for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Institutional Review Board of each center approved the study (no. NCC 2014-0146, 157/2552, EA 1/041/14, S-20130137), and written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
Siritho, Asgari, Kim Y, Kim KH report no financial disclosures. Olesen’s current affiliation is H Lundbeck A/S. Hyun has received grants from the National Cancer Center and National Research Foundation of Korea. Kim SH has lectured, consulted, and received honoraria from Bayer Schering Pharma, Biogen, Genzyme, Merck Serono, and UCB and received a grant from the National Research Foundation of Korea. Kim HJ received a grant from the National Research Foundation of Korea and research support from Aprilbio and Eisai; received consultancy/speaker fees from Alexion, Aprilbio, Biogen, Celltrion, Daewoong, Eisai, GC Pharma, HanAll BioPharma, MDimune, Merck Serono, Novartis, Roche, Sanofi Genzyme, Teva-Handok, UCB, and Viela Bio; is a co-editor for the Multiple Sclerosis Journal and an associated editor for the Journal of Clinical Neurology. Paul served on the scientific advisory boards of Novartis and MedImmune; received travel funding and/or speaker honoraria from Bayer, Novartis, Biogen, Teva, Sanofi-Aventis/Genzyme, Merck Serono, Alexion, Chugai, MedImmune, and Shire; is an associate editor of Neurology: Neuroimmunology & Neuroinflammation; is an academic editor of PLoS ONE; consulted for Sanofi Genzyme, Biogen, MedImmune, Shire, and Alexion; received research support from Bayer, Novartis, Biogen, Teva, Sanofi-Aventis/Geynzme, Alexion, and Merck Serono; and received research support from the German Research Council, Werth Stiftung of the City of Cologne, German Ministry of Education and Research, Arthur Arnstein Stiftung Berlin, EU FP7 Framework Program, Arthur Arnstein Foundation Berlin, Guthy-Jackson Charitable Foundation, and NMSS.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Supplementary table 1.
The titers of AQP4 and MOG antibodies.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hyun, JW., Kim, Y., Kim, K.H. et al. CSF GFAP levels in double seronegative neuromyelitis optica spectrum disorder: no evidence of astrocyte damage. J Neuroinflammation 19, 86 (2022). https://doi.org/10.1186/s12974-022-02450-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-022-02450-w