
Abstract
Background
Infants born preterm due to chorioamnionitis are frequently affected by a fetal inflammatory response syndrome (FIRS) and then by subsequent postnatal infections. FIRS and postnatal systemic inflammatory events independently contribute to poor neurocognitive outcomes of preterm infants. Developmental integrity of the hippocampus is crucial for intact neurocognitive outcomes in preterms and hippocampally dependent behaviors are particularly vulnerable to preterm systemic inflammation. How FIRS modulates the hippocampal immune response to acute postnatal inflammatory events is not well understood.
Methods
Prenatal LPS exposed (FIRS) and control neonatal rats received i.p. LPS or saline at postnatal day (P) 5. On P7, immune response was evaluated in the hippocampus of four treatment groups by measuring gene expression of inflammatory mediators and cytosolic and nuclear NFκB pathway proteins. Microglial activation was determined by CD11b+ and Iba1+ immunohistochemistry (IHC) and inflammatory gene expression of isolated microglia. Astrocyte reactivity was measured using Gfap+ IHC.
Results
Postnatal LPS resulted in a robust hippocampal inflammatory response. In contrast, FIRS induced by prenatal LPS attenuated the response to postnatal LPS exposure, evidenced by decreased gene expression of inflammatory mediators, decreased nuclear NFκB p65 protein, and fewer activated CD11b+ and Iba1+ microglia. Isolated microglia demonstrated inflammatory gene upregulation to postnatal LPS without evidence of immune tolerance by prenatal LPS.
Conclusion
Prenatal LPS exposure induced immune tolerance to subsequent postnatal LPS exposure in the hippocampus. Microglia demonstrate a robust inflammatory response to postnatal LPS, but only a partial immune tolerance response.
Similar content being viewed by others


Background
Preterm infants are frequently exposed to both prenatal and postnatal inflammatory events, generally due to infectious agents. Chorioamnionitis is often the first event and is the cause of 50% of preterm births < 28 weeks gestation [1,2,3,4,5]. Many of these infants develop fetal inflammatory response syndrome (FIRS) [5], a severe, innate immune reaction to the mother’s infection that results in major consequences to the preterm infant in spite of the brain not being directly infected. These include higher mortality, greater neonatal morbidity, and higher rates of long-term neurocognitive disability than preterm infants without FIRS [4,5,6,7,8,9,10,11].
Preterm infants also commonly experience postnatal systemic inflammatory illnesses such as early-onset sepsis (EOS), late-onset sepsis (LOS), or necrotizing enterocolitis (NEC). The incidence of EOS ranges between 2 and 4% [3, 12,13,14,15,16], LOS between 16 and 31% [12, 14,15,16,17], and NEC between 5 and 10% [12,13,14,15,16]. Similar to infants with FIRS, long-term neurodevelopmental outcomes for infants that experience postnatal inflammatory events without direct infection of the brain are worse than for infants that do not [15, 18].
Intact neurocognitive outcomes, especially long-term learning and memory, are dependent on developmental integrity of the hippocampus, which begins rapid development at 28 weeks post-conceptional age [19, 20]. The preterm hippocampus is particularly vulnerable to injury from adverse conditions including inflammation [21,22,23]. Preterm infants exposed to chorioamnionitis have smaller volume hippocampi compared to preterms without chorioamnionitis exposure [23]. A similar reduction of hippocampal volume occurs in preclinical models of chorioamnionitis [24]. The functional consequence of smaller hippocampal volumes in former preterm infants is poor working memory at 2 years of age [25]. Thus, it is likely that systemic inflammation is a key contributor to poor preterm infant outcomes that are associated with hippocampal development; however, the mechanism by which inflammation affects the hippocampus is not well understood.
To address this question, we previously studied the inflammatory response of the hippocampus to a prenatal inflammatory event in a rat model of FIRS and reported persistent postnatal altered expression of pro-inflammatory genes and microglial activation associated with gray matter dysregulation [26]. In the current study, we assessed how a postnatal inflammatory challenge in the setting of FIRS would affect the hippocampus with the hypothesis that a prenatal inflammatory event would modulate the response to the postnatal event.
Immune training (sensitization) and tolerance (desensitization) [27, 28] describe conditioning such that either exaggerated or suppressed responses occur, respectively, to a second exposure [27]. This phenomenon is well established in adult models, but understudied across the prenatal-postnatal spectrum such that a prenatal exposure may affect inflammatory responses to a secondary postnatal exposure. One recent study demonstrated that prenatal exposure to an inflammatory challenge with the viral mimic poly I:C early in gestation results in decreased expression of TNFα, IL-6, and IFNγ and increased expression of IL-1β when an LPS challenge is given at adulthood [29], suggesting that immune conditioning effects are long-lived. Whether immune training or tolerance responses occur acutely in the postnatal period following late gestation prenatal inflammation, such as FIRS, has not been studied.
While the combination of prenatal and postnatal inflammation is common in preterm infants, data on outcomes in infants experiencing both events are limited. In this study, we challenged FIRS-exposed neonatal rats with postnatal LPS to better understand how prenatal inflammation affects the hippocampal tissue and glial cell immune response to a postnatal inflammatory challenge within the first week of life.
Methods
Animals
Animals were studied with the approval of the Institutional Animal Care and Use Committee at the University of Minnesota. Timed-pregnant Sprague-Dawley rat dams were purchased (Charles River, Wilmington, MA) and housed for 5 days before interventions and 1 week before birth of pups. Rats were housed in a temperature and humidity-controlled animal care facility with 12 h:12 h light:dark cycle and allowed food and water ad libitum.
Model of fetal and postnatal inflammation
On gestational day 20 (term = 22 days) dams underwent laparotomy to facilitate intra-amniotic (i.a.) injections to model FIRS as previously reported [26]. Dams were premedicated with long-acting buprenorphine (1.2 mg/kg) 4 h prior to surgery. Under general anesthesia with isoflurane (3% for induction and 1.5% for maintenance) via facemask, a laparotomy was performed. Each dam was randomly assigned to receive doses of 1 μg of LPS (Escherichia coli O155:B5, Sigma, St. Louis, MO) dissolved in 50 μL normal saline (NS) to model FIRS or an equivalent volume of NS to each amniotic sac. After uterine horns were exposed, every amniotic sac was injected with the assigned treatment. Abdominal incisions were closed in two layers with nylon sutures, staples, and tissue glue. Incisions were treated along their lengths with bupivacaine (2 mg/kg s.c.). Delivery was performed operatively on gestational day 22 as reported [26]; dams were under general anesthesia with isoflurane. Following delivery, dams were killed using an overdose of sodium pentobarbital (100 mg/kg, i.p.). Each pup was removed from the amniotic sac and resuscitated by clamping the umbilical cord, drying with gauze, and placing on a warming pad. Mouths of pups were gently opened repeatedly to stimulate breathing until skin color became pink. Pups were then placed in the nest of a foster dam. Weights were obtained at postnatal days (P) 5 and 7. To model the effect of postnatal inflammatory episodes, pups were randomly administered either LPS i.p. (0.1 μg/kg) or an equivalent volume of saline on P5 and then returned to the nest [30, 31]. Four experimental groups were created: prenatal saline-postnatal saline (SS), prenatal LPS-postnatal saline (LS), prenatal saline-postnatal LPS (SL), and prenatal LPS-postnatal LPS (LL).
Tissue and cell preparation
Tissue was collected from equal numbers of male and female pups at P7 after overdose of sodium pentobarbital (100 mg/kg, i.p.). For pups used for qPCR and protein analysis, the brain was removed and both hippocampi were quickly dissected on ice, flash-frozen in liquid nitrogen, and stored at – 80 °C until analysis. Pups used for immunohistochemistry underwent transcardial perfusion-fixation before removal of brain as previously described [26]. Microglia were isolated from whole brain tissue using the Gentle MACS Octo Dissociator with Heaters and Neural Tissue Dissociation Kit (Miltenyi Biotech, Auburn, CA). Whole brains were digested according to the manufacturer’s protocol for animal age ≤ P7 to obtain single cell preparations. Debris removal and red blood cell lysis steps were performed according to Adult Brain Dissociation Kit protocol (Miltenyi Biotech) following completion of Neural Tissue Dissociation protocol. Microglia were obtained by positive selection using CD11b/c beads and magnetic separating column based on manufacturer’s protocol (Miltenyi Biotech).
Quantitative RT-PCR
qPCR experiments were performed as previously described (n = 6–8) [26]. Total RNA was isolated from hippocampi or isolated microglia using an RNA extraction kit (Thermo Fischer Scientific, Waltham, MA and Qiagen Inc., Germantown, MD, respectively) and cDNA was obtained using 500 ng of RNA in a high capacity RNA to cDNA kit (Applied Biosystems, Foster City, CA). The qPCR experiments were performed using 4 μL of diluted cDNA and 0.5 μL × 20 Taqman primer/probe (Applied Biosystems, Supplementary Table 1). Each sample was assayed in duplicate and normalized against ribosomal protein S18 (hippocampi) or RPP30 (isolated microglia). Reference genes were selected following analysis of 8 genes in RefFinder [32].
Immunohistochemistry
CD11b and GFAP immunohistochemistry was performed to identify microglia and astrocytes respectively as previously described (n = 5–7) [26]. Sections were incubated with mouse monoclonal anti-rat CD11b/c (ab1211, 1:1000; Abcam) or rabbit polyclonal GFAP (ab7260, 1:500, Abcam) followed by anti-mouse and anti-rabbit biotinylated secondary antibody and avidin-horseradish peroxidase conjugate solution (Vector Laboratories, Burlingame, CA). The protein/antibody complex was visualized using DAB (Vector Laboratories). For co-localization experiments, hippocampal slides were incubated overnight at 4 °C with goat Iba-1 (011-27991,1:500, Wako) and mouse MHCII (Ab23880,1:50, Abcam). They were subsequently incubated at room temperature for 2 h with Alexa Fluor 488 donkey anti-goat IgG (H+L) (A11055, 1:200, Invitrogen) and Alexa Fluor 555 donkey anti-mouse IgG (A31570, 1:200, Invitrogen). The slides were cover-slipped using Vectashield PLUS (Vector Laboratories).
To determine area and activated microglial cell counts, four × 20 hippocampal images (CA1 and CA3 bilaterally) were obtained from a single section. Microglia and astrocyte areal coverage ratios in the hippocampus were measured as has been reported [26]. Using set intensity thresholds, three measurements of non-stained pixels were made of each image and then averaged. The area of CD11b or GFAP positive cells was calculated by subtracting non-stained pixels from total pixel area (Photoshop, Adobe, San Jose, CA). The mean was calculated among four images for each animal and then each treatment group mean was established. Areal coverage ratio was measured by dividing CD11b or GFAP positive area by total area of the image. The number of activated hippocampal microglia was determined by counting CD11b+ and Iba1+ cells with thickened bodies and blunted processes at equivalent sites for each animal [33]. The mean was calculated for each animal and then each treatment group mean was established. Co-localization cell counts were obtained from six hippocampal images (CA1, CA2, and CA3 bilaterally). The total number of double positive cells was determined for each animal and the mean was calculated for each treatment group. The scorer was blinded to animal ID and group assignments for area calculation and cell counting.
Western analysis
Cytoplasmic and nuclear protein from individual frozen hippocampi were extracted using the EpiQuik Nuclear Extraction Kit (EpiGentek, Farmingdale, NY) per manufacturer’s protocol. The protein was quantified with a Bradford assay (Bioworld, St. Louis Park, MN) after which reducing agent (Thermo Scientific, Waltham, MA) and LDS (Thermo) was added to the samples to prepare the protein for gel separation. Twenty micrograms of protein per sample was separated on NuPage 4–12% Bis-Tris Gels (Thermo) and then transferred to a nitrocellulose membrane (Millipore, Burlington, MA). The membranes were blocked in Rockland (Pottstown, PA) Blocking Buffer for Fluorescent Western Blotting for 1 h at room temperature and incubated overnight at 4 oC with primary antibodies. After incubation with the secondary antibodies for 45 min at room temperature, the membranes were imaged and analyzed with Odyssey infrared scanning (LiCor Bioscience, Lincoln, NE). Target proteins were standardized to β-actin or α-tubulin for cytoplasmic protein and histone deacetylase 1 (HDAC1) for nuclear protein. Primary antibodies were purchased from Cell Signaling Technologies (Danvers, MA) against: p65 (8242S, 1:1000), p50 (13586S, 1:1000), IκBα (4812S, 1:500), IκBβ (94101S, 1:1000), β-actin (5356S, 1:50,000), HDAC1 (5356S,1:50,000), and α-Tubulin (3873S, 1:50,000). Secondary antibodies: Alexa Fluor 680 conjugated anti-mouse IgG (1:12,500, Jackson Laboratory, Bar Harbor, ME), Alexa Fluor 790 conjugated anti-rabbit IgG (1:12,500, Jackson Laboratory).
Statistical analysis
Data are reported as mean ± SEM. All comparisons were specified a priori. One-way ANOVA was used to test multigroup comparisons with Tukey’s multiple comparison tests (GraphPad Prism v6, La Jolla, CA). Statistical significance was accepted at p < 0.05.
Results
Neonatal outcomes
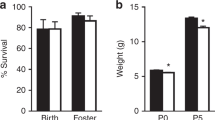
Survival was measured among the four treatment groups on P7 following i.p. LPS or saline administered on P5 (Fig. 1a). We previously showed no difference in survival between neonatal animals exposed to prenatal LPS or saline [26]. Pups receiving postnatal LPS had increased mortality compared to both groups receiving postnatal saline. Prenatal treatment did not affect survival. There was minimal mortality after P7 for all groups. Growth at P5 and P7 was affected for animals that received prenatal or postnatal LPS (Fig. 1b). We previously showed that birthweights were lower for animals that received prenatal LPS [26]. Among the four treatment groups, both prenatal LPS treated groups (LS, LL) had poorer weight gain at P5 (prior to postnatal injection) than both prenatal saline groups. At P7, LPS treatment administered prenatally, postnatally or at both time points caused a decrease in weight compared to controls, but were not different among each other.

Effect of FIRS and postnatal inflammation on survival and growth. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create 4 treatment groups. a The percentage of pups from each litter surviving at 7 days of life following fostering is depicted (n = 20–22 litters). There was significantly lower survival in both groups that received postnatal LPS (SL and LL); prenatal treatment did not change survival outcomes (*p < 0.05 vs SS and LS). b Weights of pups that received i.a. LPS (LS and LL) were decreased on postnatal (P) day 5 prior to postnatal i.p. LPS injection (*p < 0.05 vs SS and SL; n = 12–30). At P7, all groups that received LPS prenatally or postnatally had lower weights than the controls (#p < 0.05 vs SS; n = 6–17). Data presented as mean ± SEM
Hippocampal inflammatory mediators
To determine how pre- and postnatal LPS challenge altered the hippocampal immune response at P7, we measured transcription of several inflammatory genes (Fig. 2). Expressions of IL1B (gene for IL-1β), NOS2 (gene for iNOS), CD86, CXCL10, IL6, and CCL2 were all upregulated 1.5–2-fold in the SL group compared to controls. In contrast, CXCL10, IL6, and CCL2 were all downregulated by ~ 50% in the LL group compared to the SL group. Aif1, the gene for Iba1, demonstrated a similar pattern, but did not reach significance. NOS2 expression in the LL group was not significantly different from the SL group in the four-group comparison, but also not significantly different from the control group. CD86 was not different between the two postnatal LPS groups (SL, LL). In contrast, IL1B was further upregulated in the LL group by 1.3-fold compared to the SL group. TNF, the gene for cytokine TNFα, responded differently from other inflammatory markers to pre- and postnatal LPS treatment with a 50% downregulation in the SL group compared to the control group. However, similar to several other markers, the LL group was also downregulated by 50% compared to the SL group.

Effect of FIRS and postnatal inflammation on hippocampal gene expression of pro-inflammatory mediators at P7. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create four treatment groups. Bars above the graphs indicate significant differences between groups (p < 0.05). Data presented as mean ± SEM. Genes are normalized to ribosomal protein S18
Hippocampal anti-inflammatory markers are shown in Supplementary Fig. 1a. Significant hippocampal gene expression differences across groups were limited to IL10, a cytokine responsible for suppressing macrophage function [34]. IL10 was downregulated by 50% in the LS group and 30% in the LL group compared to controls, differences that are consistent with increased pro-inflammatory mediators. There were no differences in expression of Arg1 (the gene for arginase), MRC1 (the gene for CD206) or STAT6. Overall, prenatal LPS alters the hippocampal inflammatory gene response to postnatal LPS.
Hippocampal NFκB regulation
We measured the effect of pre- and postnatal LPS on the NFκB pathway as a further measure of immune response in the hippocampus (Fig. 3). NFκB is a transcription factor activated by several upstream signaling pathways to regulate transcription of pro-inflammatory cytokines. The NFκB family consists of five proteins from which dimers are formed; the most abundant dimer is composed of p65 and p50 [35]. Dimers translocate from the cytoplasm to the nucleus to initiate transcription of target genes. We found that gene expression of NFkb1, the gene for the p50 subunit, in the LL group was downregulated by 30% compared to control and SL treatment groups (Fig. 3a). There were not significant differences of RelA expression, the gene for the p65 subunit, in the four-group comparison. We measured p65 and p50 protein by western blot in both the nucleus and cytosol as an indication of functional activity (Fig. 3b, c). The p65 subunit protein was decreased by 65% in the nuclear fraction of the LL group compared to the SL group (Fig. 3b). Again, attenuation of the response to postnatal LPS was demonstrated when a prenatal LPS dose was administered. The p65 nuclear findings were accompanied by a 70% decrease of cytosolic p65 in the LS group compared to controls. There was a non-significant trend decrease in the SL group compared to controls. We did not find differences of the p50 subunit protein in the nucleus or cytosol (Fig. 3c).

Effect of FIRS and postnatal inflammation on NFκB pathway gene expression and protein in the hippocampus at P7. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create four treatment groups. a Relative gene expression of indicated NFκB pathway genes in the hippocampus at P7 normalized to ribosomal protein S18 (n = 5–7/group). Protein product of each gene is in parentheses. b Western blot analyses of nuclear and cytosolic protein fractions of NFκB p65 in the hippocampus at P7 (n = 3–6/group). c Western blot analyses of nuclear and cytosolic protein fractions of NFκB p50 in the hippocampus at P7 (n = 4–6/group). d Western blot analyses of cytosolic fractions of IκBα and IκBβ proteins in the hippocampus at P7 (n = 3–5/group). In all graphs, bars above the graphs indicate significant differences between groups (p < 0.05). Data presented as mean ± SEM
NFκB is prevented from translocating to the nucleus by its inhibitor IκB [36]. When phosphorylated by upstream effectors, IκB is degraded by ubiquitin, and NFκB is able to translocate [36]. As a further characterization of the NFκB pathway, we measured gene and protein expression of IκBα and IκBβ, two IκB proteins with similar properties but some functional differences (Fig. 3a, d) [37]. We found that gene expression of Nfkbia, the gene for IκBα, was downregulated by 30% in the LL group compared to the SL group (Fig. 3a). The protein expression of cytosolic IκBα was increased by 1.5-fold in the SL group compared to the control group (Fig. 3d). There was a non-significant trend downregulation of IκBα in the LL group compared to the SL group. There were no significant differences in IκBβ gene or protein expression among the groups.
Microglial activation
Microglia are the tissue-specific macrophages of the brain. They respond to pathogen-associated molecular patterns (PAMPs) through TLR4 and IFNγ receptors in a characteristic cell response known as microglial activation to produce cytokines, present antigen, and induce adaptive immunity [38, 39]. Histologically, activation is measured by increased density and altered morphology [26, 33, 40]. We measured microglial activation in the hippocampus, as a response to pre- and postnatal inflammation, using CD11b and Iba1 immunohistochemistry at P7 (Fig. 4). The number of CD11b+ (Fig. 4a) and Iba1+ (Fig. 4b) cells with a thickened cell body and blunted processes were counted. There was a 5- and 3-fold increase in the number of activated microglia measured by CD11b+ or Iba1+ cells, respectively, in the SL group compared to the SS group. In contrast, the LL group had 70% fewer activated microglia (CD11b+ and Iba1+) than the SL group. The total number of CD11b+ cells did not differ among groups. To account for complex morphology, we also calculated the cross-sectional areal coverage of CD11b+ cells as a ratio of total image area (Supplementary Fig. 2). Areal measurement yielded similar findings as the cell counts. There was a 1.4-fold increase in microglial area in the SL group compared to the SS group. Prenatal LPS prior to postnatal LPS prevented this activation. Prenatal LPS appears to prevent the microglial activation response that follows postnatal LPS alone.

Postnatal LPS increases activated microglia in the hippocampus, but the effect is attenuated by FIRS. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create four treatment groups. On P7, microglial activation was quantified in each treatment group by counting activated microglia and total microglia. a Activated microglia count (top bar graph) and total microglia count (bottom bar graph) of CD11b+ cells. Activated microglia were identified by thickened cell bodies and blunted processes. Examples of surveillance (non-activated) and activated microglia are shown below pictures. b Activated microglia count of Iba1+ cells identified by thickened cell bodies and blunted process. Bars above the graphs indicate significant differences between groups (p < 0.05; n = 5–7/group). Data presented as mean ± SEM. Pictures representative of each group are shown at × 20. Scale bar represents 100 μm
Astrocyte activation
We measured cross-sectional area coverage ratio of Gfap+ astrocytes in the hippocampus at P7 to determine changes in astrocyte reactivity following pre- and postnatal inflammatory challenge (Fig. 5a). We found no differences among treatment groups in amount of Gfap. We also measured the expression of multiple genes associated with astrocyte activation (Fig. 5b) [41]. Lcn2 expression was increased by 5.5 fold in the SL group compared to the control group. There was a trend decrease in the LL group compared to the SL group in a pattern similar to our measurements of microglial activation. Steap4 expression demonstrated a trend increase in the SL group compared to controls. There were no expression differences in Vim or Gfap.

Effect of FIRS and postnatal inflammation on astrocytes in the hippocampus at P7. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create four treatment groups. a On P7, reactive astrocytosis was quantified in each treatment group by calculating areal coverage ratio of Gfap+ cells in the hippocampus. Scale bar represent 100 μm. Pictures representative of each group are shown at × 20. b Expression of reactive astrocyte genes in the hippocampus at P7. Bars above the graphs indicate significant differences between groups (p < 0.05). Data presented as mean ± SEM
Microglia-specific inflammation
Microglial activation in response to pathogens is commonly described as M1 activation and is characterized by increased transcription factor activation (e.g., NFκB and STAT1), increased expression of surface markers (e.g., CD86, MHCII), increased iNOS to produce NO for pathogen killing, and increased production of cytokines (e.g., IL-1β, TNFα, IL-6) and chemokines (e.g., CCL2 and CXCL10) [38, 42]. Based on hippocampal expression patterns of these genes and morphologic changes signifying microglial activation, we sought to determine whether microglia expressed similar immune tolerance and training gene expression and thus the major contributor to these responses in the hippocampus (Fig. 6). In the NFκB pathway, there was a 1.3-fold upregulation of RelA and 1.5-fold upregulation of Nfkbib in the SL group compared to controls. Nfkb1 demonstrated a similar trend upregulation in the SL group. Nos2, CD86, and Aif1 were upregulated in the SL group compared to SS and LS groups. There was a trend increase of IL1B in the SL group compared to the SS group. TNF was downregulated by postnatal LPS in both SL and LL groups. Differential expression was not measured between SL and LL groups, indicating prenatal LPS did not affect response to postnatal LPS of these genes in microglia. Other pro-inflammatory cytokines and chemokines did not demonstrate significant differences.

Effect of FIRS and postnatal inflammation on gene expression of M1 activation markers in isolated microglia at P7. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create four treatment groups. Bars above the graphs indicate significant differences between groups (p < 0.05). Data presented as mean ± SEM. Genes are normalized to the ribonuclease RPP30
M2 activation is the microglial response to repair tissue and dampen inflammation and is characterized by anti-inflammatory markers such as IL-10, Arg1, and CD206 [38, 39]. Among the anti-inflammatory markers measured in isolated microglia, only IL10 displayed a difference (Supplementary Fig. 1b). The SL group was downregulated in comparison to the LS group, but not different compared to SS or LL groups.
Co-localization of MHCII+ cells with Iba1+ microglia was quantified to further measure activated microglia in the hippocampus and correlate with upregulation of M1 activation markers (Fig. 7). Iba1+MHCII+ cells increased by 2.5 fold in the SL group compared to the control group indicating activation caused by postnatal LPS. Prenatal LPS did not cause differences between the SL and LL groups although LL was also not different from the controls.

Co-localization of M1 marker MHCII with Iba1+ microglia in the hippocampus at P7. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create four treatment groups. Total double positive Iba1 (green) and MHCII (red) cells were quantified in each treatment group (blue = DAPI). Bars above the graph indicate significant differences between groups (p < 0.05; n = 4–6/group). Data presented as mean ± SEM. Pictures representative of each group are shown at × 20. Scale bar represents 100 μm
Discussion
FIRS and postnatal inflammation are independently linked to long-term neurodevelopmental deficits for preterm infants [11, 15]. We sought to better understand how inflammatory exposures occurring both pre- and postnatally affect immune tolerance and training in the hippocampus. The hippocampus was chosen because of its role in learning and memory, its rapid development in the late fetal/early neonatal time period, and the prominence of learning and memory deficits in preterm infants. We found that pro-inflammatory mediator gene expression, microglial activation, and astrocyte activation gene expression increased following postnatal inflammation. We also found signals of hippocampal immune tolerance; there was an attenuation of many of these effects when prenatal inflammatory challenge was administered prior to postnatal challenge. Although histologic microglial activation caused by postnatal LPS was attenuated by prenatal LPS, gene expression in isolated microglia did not demonstrate a similar immune tolerance.
Whether repeated exposure to inflammatory events causes immune training or tolerance appears to be dependent on timing. Adult mice receiving repeated dosing of i.p. LPS over four consecutive days have decreasing concentrations of plasma and brain cytokines after each dose, suggesting immune tolerance [28]. LPS tolerance is long-lived in the brain after systemic injection to adults and neonatal mice [43, 44]. LPS dosing to adult mice repeated after 270 days results in attenuated inflammatory response, including IL-1β and IL-6 in the hippocampus [43]. Similarly, systemic LPS to 10-day-old mice results in downregulated expression of IL1B and TNF in the hypothalamus after adult LPS challenge [44]. In contrast, prenatal LPS followed by LPS injection at adult ages resulted in exaggerated (immune training) expression of IL1B, TNF, and IL6 in the hippocampus [45]. Our study is unique in that we studied how prenatal LPS dosing affects hippocampal response to postnatal LPS in the acute neonatal period. Despite prenatal exposure, the results are consistent with other studies in showing that LPS induces immune tolerance in the brain.
Mechanisms that underlie the attenuated hippocampal responses caused by prenatal inflammation are not known. Because NFκB has a central role in inflammation, we explored the possibility that p50 could be involved because the p50/p50 homodimer is a repressor of inflammatory response [46]. The p50 subunit has no transcriptional activity itself and is typically a regulator of activity when paired with other subunits [47]. In contrast, the p65/p50 heterodimer is a transcription factor that promotes pro-inflammatory responses [35]. In our model, decreased nuclear p65 protein following pre- and postnatal LPS without a parallel change in p50 is consistent with an inhibitory phenotype and resulted in the decreased expression of several downstream inflammatory markers. Changes in hippocampal inflammation appear to be dependent on p65 rather than p50 and responsible mechanisms likely occur upstream from IκB and NFκB phosphorylation.
Whether hippocampal immune tolerance is beneficial or harmful to neonates is not known. Neonatal inflammation is a well-established mediator of brain injury resulting in poorer behavioral outcomes in humans and preclinical models [6,7,8,9,10, 24, 48, 49]. Preclinical studies show that attenuation of the neuroinflammatory response, by inducing tolerance or inhibiting inflammatory cell activity, is associated with improved developmental outcomes [43, 50,51,52]. In contrast, harmful effects may result from multiple inflammatory exposures. For example, exaggerated IL-1β response following multiple doses of LPS is associated with adverse learning and memory behavioral consequences [53, 54]. In our model, expression of IL1B demonstrated an immune training response that was consistent with these studies despite other signals of hippocampal immune tolerance. Regulation of IL-1β in the hippocampus is important for proper development of learning and memory [53]. Absent or excessive IL-1β results in impairments [55, 56]. Alterations of IL-1β, such as that seen in our model, may lead to long-term learning impairment.
Microglial activation is also associated with poor neurodevelopmental outcomes [50, 51]. In rodent models, microglial activation is associated with long-term learning impairments, motor deficits, and anxiety, results that mirror several domains of poor neurodevelopmental outcomes of infants born preterm [57,58,59]. Under normal developmental conditions, microglia have critical homeostatic functions that include synaptogenesis, axonal elongation, promotion of myelination, growth factor production, and they are the primary source for IL-1β during normal hippocampal-dependent learning [45, 53, 60, 61]. These functions are likely disrupted by microglial activation.
We hypothesized that immune tolerance signals in hippocampal tissue gene and protein expression were a reflection of microglia based on CD11b and Iba1 histologic findings and therefore isolated microglia to explore that possibility. Gene expression in microglia in the SL group was similar to expression in the hippocampus for several M1 activation markers suggesting that microglia are an important driver of the acute hippocampal inflammatory response. There was similar upregulation of Nos2, CD86, IL1B, and Aif1, and similar downregulation of TNF. We also measured upregulation of RelA and Nfkbib in the NFκB pathway. However, there were two notable differences between hippocampal tissue and microglia suggesting that microglia are not the only cells contributing to inflammatory response. First, unlike in the hippocampus, we did not measure significant attenuation of expression in any marker in microglia when prenatal challenge was administered prior to a postnatal challenge. Second, whereas IL6, CXCL10, and CCL2 demonstrated robust upregulation in the SL group and attenuation in the LL group in the hippocampus, these genes were unaffected in microglia by pre- or postnatal LPS.
The upregulation of M1 markers in SL microglia is consistent with the significant increase in histologic-activated microglia that we measured by CD11b and Iba1. In contrast, there was significant attenuation of histologic activated microglia in the LL group, but without a similar attenuation of M1 marker expression in microglia. This suggests different phenotypes of microglia between SL and LL groups. Single cell gene studies have shown numerous phenotypes of microglia [62]. Several factors could influence this difference in phenotype. We considered that upregulation of M2 marker expression in microglia in the LL group could inhibit morphologic changes caused by postnatal inflammation. However, there were no significant differences between SL and LL groups in the M2 genes that we measured. Another possibility is that exogenous signaling of IL-6, CXCL10, and CCL2 influence microglial activation in this model. They are highly upregulated in the SL hippocampus, attenuated in the LL hippocampus, both in a pattern similar to CD11b and Iba1 results. Neurons are one possible exogenous source. Preclinical models show that neurons under adverse conditions produce CCL2 [63, 64] and CXCL10 [65, 66] that increase microglial activation. IL-6 is also produced by neurons but less studied [67].
Astrocytes are another potential source of IL6, CXCL10, and CCL2 expression. Similar to microglia, astrocytes undergo a transformation to reactive astrocytosis in response to injury and disease. Reactive astrocytes have a gene expression pattern distinct from non-reactive astrocytes [41], produce pro-inflammatory cytokines by an NFκB-mediated mechanism [52, 68, 69], and have immune tolerance responses [68, 70]. To determine whether astrocytes may contribute to hippocampal inflammatory mediator results and demonstrate immune tolerance in our pre- and postnatal LPS model, we measured Gfap+ astrocytes in the hippocampus. In contrast to microglia, we found no histological differences in Gfap+ astrocytes among treatment groups. However, upregulation of Lcn2 and Steap4, in addition to CXCL10, genes associated with reactive astrocytosis [41], suggest a change in astrocyte reactivity following postnatal inflammatory challenge. Lcn2 and CXCL10 also demonstrated immune tolerance in the LL group.
Reactive astrocyte gene expression is likely a direct effect of activated microglia. Liddelow et al. demonstrated that activated microglia are required to induce transformation of cultured A1 reactive astrocytes (nomenclature similar to M1 microglia) from non-reactive astrocytes after 7 days [41]. Our study is consistent with this process because (1) outcomes measured 2 days after postnatal LPS administration was a timeframe adequate to measure a robust histologic microglial activation but only a partial astrocyte gene response; (2) the responses of Lcn2 and CXCL10 to pre- and postnatal LPS mirror hippocampal responses.
Non-resident immune cells including infiltrating macrophages and neutrophils could also contribute to the hippocampal immune response in this model, but their impact is likely much less than microglia and astrocytes. A study by Hutton et al. used a preterm fetal ovine model to identify infiltrating macrophages by morphology 72 h following inflammatory challenge [71]. They identified 60–100 macrophages per mm2 in white matter areas. In contrast, they identified 1–3 macrophages per mm2 in the hippocampus. We did not specifically identify infiltrating cells in our study, but found a far greater density of activated microglia in the hippocampus that in the similar model. Infiltrating cells may be a more significant mediator of inflammation in white matter regions than gray matter regions such as the hippocampus.
Conclusions
Postnatal LPS administered to neonatal rats at P5 to model sepsis resulted in a pro-inflammatory immune response in the hippocampus. Evidence of immune tolerance resulted if prenatal LPS to model FIRS was administered first. Microglia demonstrate a robust inflammatory response to postnatal LPS, but partial immune tolerance response. Hippocampal immune tolerance likely results due to the responses of multiple cell types.
Availability of data and materials
All data generated or analyzed during this study are included in the published article or supplementary files.
Abbreviations
- Aif1:
-
Allograft inflammatory factor 1
- Arg:
-
Arginase
- BDNF:
-
Brain-derived neurotrophic factor
- CCL2:
-
C-C motif chemokine ligand 2
- CXCL10:
-
C-X-C motif chemokine ligand 10
- EOS:
-
Early-onset sepsis
- FIRS:
-
Fetal inflammatory response syndrome
- Gfap:
-
Glial fibrillary acidic protein
- HDAC1:
-
Histone deacetylase 1
- iNOS:
-
Inducible nitric oxide synthase
- i.a.:
-
Intra-amniotic
- i.p.:
-
Intraperitoneal
- Lcn2:
-
Lipocalin 2
- LL:
-
Prenatal LPS-postnatal LPS group
- LOS:
-
Late-onset sepsis
- LS:
-
Prenatal LPS-postnatal saline group
- NEC:
-
Necrotizing enterocolitis
- NS:
-
Normal saline
- PAMP:
-
Pathogen-associated molecular patterns
- P(x):
-
Postnatal day x
- SL:
-
Prenatal saline-postnatal LPS group
- Steap4:
-
Steap 4 metalloreductase
- SS:
-
Prenatal saline-postnatal saline group
- Vim:
-
Vimentin
References
Gravett MG, Rubens CE, Nunes TM, GAPPS Review Group. Global report on preterm birth and stillbirth (2 of 7): discovery science. BMC Pregnancy Childbirth. 2010;10 Suppl 1:S2.
Hamilton BE, Martin JA, Osterman MJ, Curtin SC, Matthews TJ. Births: Final Data for 2014. Natl Vital Stat Rep. 2015;64:1–64.
Stoll BJ, Hansen N. Infections in VLBW infants: studies from the NICHD Neonatal Research Network. Semin Perinatol. 2003;27:293–301.
Gotsch F, Romero R, Kusanovic JP, Mazaki-Tovi S, Pineles BL, Erez O, Espinoza J, Hassan SS. The fetal inflammatory response syndrome. Clin Obstet Gynecol. 2007;50:652–83.
Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM. The fetal inflammatory response syndrome. Am J Obstet Gynecol. 1998;179:194–202.
Salas AA, Faye-Petersen OM, Sims B, Peralta-Carcelen M, Reilly SD, McGwin G Jr, Carlo WA, Ambalavanan N. Histological characteristics of the fetal inflammatory response associated with neurodevelopmental impairment and death in extremely preterm infants. J Pediatr. 2013;163:652–7 e1-2.
Tsiartas P, Kacerovsky M, Musilova I, Hornychova H, Cobo T, Sävman K, Jacobsson B. The association between histological chorioamnionitis, funisitis and neonatal outcome in women with preterm prelabor rupture of membranes. J Matern Fetal Neonatal Med. 2013;26:1332–6.
Rovira N, Alarcon A, Iriondo M, Ibañez M, Poo P, Cusi V, Agut T, Pertierra A, Krauel X. Impact of histological chorioamnionitis, funisitis and clinical chorioamnionitis on neurodevelopmental outcome of preterm infants. Early Hum Dev. 2011;87:253–7.
Hendson L, Russell L, Robertson CM, Liang Y, Chen Y, Abdalla A, Lacaze-Masmonteil T. Neonatal and neurodevelopmental outcomes of very low birth weight infants with histologic chorioamnionitis. J Pediatr. 2011;158:397–402.
Suppiej A, Franzoi M, Vedovato S, Marucco A, Chiarelli S, Zanardo V. Neurodevelopmental outcome in preterm histological chorioamnionitis. Early Hum Dev. 2009;85:187–9.
Pappas A, Kendrick DE, Shankaran S, Stoll BJ, Bell EF, Laptook AR, Walsh MC, Das A, Hale EC, Newman NS, Higgins RD, Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Chorioamnionitis and early childhood outcomes among extremely low-gestational-age neonates. JAMA Pediatr. 2014;168:137–47.
Stensvold HJ, Klingenberg C, Stoen R, Moster D, Braekke K, Guthe HJ, Astrup H, Rettedal S, Gronn M, Ronnestad AE. Norwegian Neonatal Network. Neonatal morbidity and 1-year survival of extremely preterm infants. Pediatrics. 2017;139:e20161821.
Travers CP, Carlo WA, McDonald SA, Das A, Bell EF, Ambalavanan N, Jobe AH, Goldberg RN, D'Angio CT, Stoll BJ, Shankaran S, Laptook AR, Schmidt B, Walsh MC, Sánchez PJ, Ball MB, Hale EC, Newman NS, Higgins RD, Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Mortality and pulmonary outcomes of extremely preterm infants exposed to antenatal corticosteroids. Am J Obstet Gynecol. 2018;218:130 e1-130.e13.
Chee YY, Wong MS, Wong RM, Wong KY. Neonatal outcomes of preterm or very-low-birth-weight infants over a decade from Queen Mary Hospital, Hong Kong: comparison with the Vermont Oxford Network. Hong Kong Med J. 2017;23:381–6.
Adams-Chapman I, Heyne RJ, DeMauro SB, Duncan AF, Hintz SR, Pappas A, Vohr BR, McDonald SA, Das A, Newman JE, Higgins RD, Follow-up study of the Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Neurodevelopmental impairment among extremely preterm infants in the Neonatal Research Network. Pediatrics. 2018;141:e20173091.
Boghossian NS, Geraci M, Edwards EM, Horbar JD. Sex differences in mortality and morbidity of infants born at less than 30 weeks' gestation. Pediatrics. 2018;142:e20182352.
Kim JK, Chang YS, Sung S, Ahn SY, Park WS. Trends in the incidence and associated factors of late-onset sepsis associated with improved survival in extremely preterm infants born at 23-26 weeks' gestation: a retrospective study. BMC Pediatr. 2018;18:172.
Shah DK, Doyle LW, Anderson PJ, Bear M, Daley AJ, Hunt RW, Inder TE. Adverse neurodevelopment in preterm infants with postnatal sepsis or necrotizing enterocolitis is mediated by white matter abnormalities on magnetic resonance imaging at term. J Pediatr. 2008;153:170–5.
Riggins T, Miller NC, Bauer PJ, Georgieff MK, Nelson CA. Consequences of low neonatal iron status due to maternal diabetes mellitus on explicit memory performance in childhood. Dev Neuropsychol. 2009;34(6):762–79.
Fretham SJ, Carlson ES, Wobken J, Tran PV, Petryk A, Georgieff MK. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus. 2012;22:1691–702.
Strahle JM, Triplett RL, Alexopoulos D, Smyser TA, Rogers CE, Limbrick DD Jr, Smyser CD. Impaired hippocampal development and outcomes in very preterm infants with perinatal brain injury. Neuroimage Clin. 2019;22:101787.
Lodygensky GA, Seghier ML, Warfield SK, Tolsa CB, Sizonenko S, Lazeyras F, Hüppi PS. Intrauterine growth restriction affects the preterm infant's hippocampus. Pediatr Res. 2008;63:438–43.
Hatfield T, Wing DA, Buss C, Head K, Muftuler LT, Poggi DE. Magnetic resonance imaging demonstrates long-term changes in brain structure in children born preterm and exposed to chorioamnionitis. Am J Obstet Gynecol. 2011;205:384 e1-8.
Dada T, Rosenzweig JM, Al Shammary M, Firdaus W, Al Rebh S, Borbiev T, Tekes A, Zhang J, Alqahtani E, Mori S, Pletnikov MV, Johnston MV, Burd I. Mouse model of intrauterine inflammation: sex-specific differences in long-term neurologic and immune sequelae. Brain Behav Immun. 2014;38:142–50.
Beauchamp MH, Thompson DK, Howard K, Doyle LW, Egan GF, Inder TE, Anderson PJ. Preterm infant hippocampal volumes correlate with later working memory deficits. Brain. 2008;131(Pt 11):2986–94.
Gisslen T, Singh G, Georgieff MK. Fetal inflammation is associated with persistent systemic and hippocampal inflammation and dysregulation of hippocampal glutamatergic homeostasis. Pediatr Res. 2019;85:703–10.
Neher JJ, Cunningham C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019;40:358–74.
Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A, Blank T, Staszewski O, Datta M, Centeno TP, Capece V, Islam MR, Kerimoglu C, Staufenbiel M, Schultze JL, Beyer M, Prinz M, Jucker M, Fischer A, Neher JJ. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–8.
Clark SM, Notarangelo FM, Li X, Chen S, Schwarcz R, Tonelli LH. Maternal immune activation in rats blunts brain cytokine and kynurenine pathway responses to a second immune challenge in early adulthood. Prog Neuropsychopharmacol Biol Psychiatry. 2019;89:286–94.
Le Rouzic V, Wiedinger K, Zhou H. Attenuated mRNA expression of inflammatory mediators in neonatal rat lung following lipopolysaccharide treatment. J Inflamm Res. 2012;5:99–109.
Correa F, Ljunggren E, Patil J, Wang X, Hagberg H, Mallard C, Sandberg M. Time-dependent effects of systemic lipopolysaccharide injection on regulators of antioxidant defence Nrf2 and PGC-1α in the neonatal rat brain. Neuroimmunomodulation. 2013;20(4):185–93.
Xie F, Xiao P, Chen D, Xu L, Zhang B. miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol. 2012;80:75–84
Kannan S, Saadani-Makki F, Muzik O, Chakraborty P, Mangner TJ, Janisse J, Romero R, Chugani DC. Microglial activation in perinatal rabbit brain induced by intrauterine inflammation: detection with 11C-(R)-PK11195 and small-animal PET. J Nucl Med. 2007;48:946–54.
Murphy K, Weaver C. Janeway’s Immunobiology. 9th ed. New York: Garland Science, Taylor and Frances Group; 2017.
Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034.
Hayden MS, Ghosh S. Shared Principles in NF-kappaB Signaling. Cell. 2008;132:344–62.
Tran K, Merika M, Thanos D. Distinct functional properties of IkappaB alpha and IkappaB beta. Mol Cell Biol. 1997;17:5386–99.
Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 Polarization and Metabolic States. Br J Pharmacol. 2016;173:649–65.
Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39:3–18.
Schmidt AF, Kannan PS, Chougnet CA, Danzer SC, Miller LA, Jobe AH, Kallapur SG. Intra-amniotic LPS causes acute neuroinflammation in preterm rhesus macaques. J Neuroinflammation. 2016;13:238.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–7.
Subramaniam SR, Federoff HJ. Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front Aging Neurosci. 2017;9:176.
Zhou XY, Gao R, Hu J, Gao DP, Liao YL, Yang JJ. Trained innate immunity by repeated low-dose lipopolysaccharide injections displays long-term neuroprotective effects. Mediators Inflamm. 2020;2020:8191079.
Iwasa T, Matsuzaki T, Kinouchi R, Fujisawa S, Murakami M, Kiyokawa M, Kuwahara A, Yasui T, Irahara M. Neonatal LPS injection alters the body weight regulation systems of rats under non-stress and immune stress conditions. Int J Dev Neurosci. 2010;28:119–24.
Schaafsma W, Basterra LB, Jacobs S, Brouwer N, Meerlo P, Schaafsma A, Boddeke E, Eggen B. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol Dis. 2017;106:291–300.
Taetzsch T, Benusa S, Shannon Levesque S, Mumaw CL, Block ML. Loss of NF-κB p50 function synergistically augments microglial priming in the middle-aged brain. J Neuroinflammation. 2019;16:60.
Cartwright T, Perkins ND, Wilson CL. NFKB1: A Suppressor of Inflammation, Ageing and Cancer. FEBS J. 2016;283:1812–22.
Novak CM, Ozen M, McLane M, Alqutub S, Lee JY, Lei J, Burd I. Progesterone improves perinatal neuromotor outcomes in a mouse model of intrauterine inflammation via immunomodulation of the placenta. Am J Reprod Immunol. 2018;79:e12842.
Smith CJ, Kingsbury MA, Dziabis JE, Hanamsagar R, Malacon KE, Tran JN, Norris HA, Gulino M, Bordt EA, Bilbo SD. Neonatal immune challenge induces female-specific changes in social behavior and somatostatin cell number. Brain Behav Immun. 2020;90:332–45.
Hsieh CT, Lee YJ, Dai X, Ojeda NB, Lee HJ, Tien LT, Fan LW. Systemic lipopolysaccharide-induced pain sensitivity and spinal inflammation were reduced by minocycline in neonatal rats. Int J Mol Sci. 2018;19:2947.
Niño DF, Zhou Q, Yamaguchi Y, Martin LY, Wang S, Fulton WB, Jia H, Lu P, Prindle T Jr, Zhang F, Crawford J, Hou Z, Mori S, Chen LL, Guajardo A, Fatemi A, Pletnikov M, Kannan RM, Kannan S, Sodhi CP, Hackam DJ. Cognitive Impairments Induced by Necrotizing Enterocolitis Can Be Prevented by Inhibiting Microglial Activation in Mouse Brain. Sci Transl Med. 2018;10:eaan0237.
Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia. 2016;64:300–16.
Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal Infection-Induced Memory Impairment After Lipopolysaccharide in Adulthood Is Prevented via caspase-1 Inhibition. J Neurosci. 2005;25:8000–9.
Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and Memory: Modulation by Early-Life Infection. J Neurosci. 2011;31:15511–21.
Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Memory for Context Is Impaired by a Post Context Exposure Injection of interleukin-1 Beta Into Dorsal Hippocampus. Behav Brain Res. 2002;134:291–8.
Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, Levy-Lahad E, Yirmiya R. A Dual Role for interleukin-1 in Hippocampal-Dependent Memory Processes. Psychoneuroendocrinology. 2007;32:1106–15.
Girard S, Kadhim H, Beaudet N, Sarret P, Sébire G. Developmental motor deficits induced by combined fetal exposure to lipopolysaccharide and early neonatal hypoxia/ischemia: a novel animal model for cerebral palsy in very premature infants. Neuroscience. 2009;158:673–82.
Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology. 2005;48:903–17.
Golan H, Lev V, Mazar Y, Hallak M, Huleihel M. Alterations in behavior in adult offspring mice following maternal inflammation during pregnancy. Dev Psychobiol. 2006;48(2):162–8.
Harry GJ. Microglia during development and aging. Pharmacol Ther. 2013;139:313–26.
Tran PV, Fretham SJ, Carlson ES, Georgieff MK. Long-term reduction of hippocampal brain-derived neurotrophic factor activity after fetal-neonatal iron deficiency in adult rats. Pediatr Res. 2009;65:493–8.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. A Unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017;169:1276–90 e17.
Zhang L, Tan J, Jiang X, Qian W, Yang T, Sun X, Chen Z, Zhu Q. Neuron-derived CCL2 contributes to microglia activation and neurological decline in hepatic encephalopathy. Biol Res. 2017;50:26.
Chun S, Kwon YB. The CCL2 elevation in primary afferent fibers produces zymosan-induced hyperalgesia through microglia-mediated neuronal activation in the spinal dorsal horn. Brain Res Bull. 2019;149:53–9.
Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HW, Nitsch R, Kettenmann H. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24:8500–9.
Listwak SJ, Rathore P, Herkenham M. Minimal NF-κB activity in neurons. Neuroscience. 2013;250:282–99.
Jüttler E, Tarabin V, Schwaninger M. Interleukin-6 (IL-6): a possible neuromodulator induced by neuronal activity. Neuroscientist. 2002;8:268–75.
Chistyakov DV, Astakhova AA, Azbukina NV, Goriainov SV, Chistyakov VV, Sergeeva MG. Cellular Model of Endotoxin Tolerance in Astrocytes: Role of Interleukin 10 and Oxylipins. Cells. 2019;8:1553.
Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–60.
Beurel E, Jope RS. Glycogen synthase kinase-3 regulates inflammatory tolerance in astrocytes. Neuroscience. 2010;169:1063–70.
Hutton LC, Castillo-Melendez M, Smythe GA, Walker DW. Microglial activation, macrophage infiltration, and evidence of cell death in the fetal brain after uteroplacental administration of lipopolysaccharide in sheep in late gestation. Am J Obstet Gynecol. 2008;198:117–e1-11.
Acknowledgements
We would like to thank Haley Campbell for her contributions to these experiments.
Funding
This project was supported by grants from The National Institutes of Health (K12 HD068322, K08 HD099246 (TG), K08 GM11305 (BJS); Bethesda, MD), and the Viking Children’s Fund, Department of Pediatrics, University of Minnesota, Minneapolis, MN.
Author information
Authors and Affiliations
Contributions
GS, MG, and TG formulated the concept and designed the study. GS and TG collected and analyzed data. All authors interpreted the results, contributed to manuscript writing, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were conducted with the approval of the Institutional Animal Care and Use Committee at the University of Minnesota under protocol ID 1804-35798A.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure 1
. Effect of FIRS and postnatal inflammation on gene expression of anti-inflammatory mediators in the hippocampus and isolated microglial at P7. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create four treatment groups. (a) Relative expression measured in hippocampus. (b) Relative expression measured in isolated microglia. Bars above the graphs indicate significant differences between groups (p<0.05). Data presented as mean ± SEM. Genes are normalized to S18 (hippocampus) or RPP30 (microglia).
Additional file 2: Supplementary Figure 2
. Postnatal LPS increases activated microglia in the hippocampus, but the effect is attenuated by FIRS. Rat pups were exposed to prenatal i.a. LPS (L) or saline (S) (listed first) and then postnatal i.p. LPS or saline at P5 (listed second) to create four treatment groups. On P7, microglial activation was quantified in each treatment group by calculating areal coverage ratio of CD11b+ cells in the hippocampus. Bars above the graph indicate significant differences between groups (p<0.05). Data presented as mean ± SEM.
Additional file 3: Supplementary Table 1
. List of Taqman Probes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Singh, G., Segura, B.J., Georgieff, M.K. et al. Fetal inflammation induces acute immune tolerance in the neonatal rat hippocampus. J Neuroinflammation 18, 69 (2021). https://doi.org/10.1186/s12974-021-02119-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-021-02119-w