
Abstract
Background
Neuromyelitis optica spectrum disorders (NMOSD) are central nervous system (CNS) autoimmune inflammatory demyelinating diseases characterized by recurrent episodes of acute optic neuritis and transverse myelitis. Aquaporin-4 immunoglobulin G (AQP4-IgG) autoantibodies, which target the water channel aquaporin-4 (AQP4) on astrocytic membrane, are pathogenic in NMOSD. Glutamate excitotoxicity, which is triggered by internalization of AQP4-glutamate transporter complex after AQP4-IgG binding to astrocytes, is involved in early NMOSD pathophysiologies. We studied the effects of memantine, a N-methyl-D-aspartate (NMDA) receptor antagonist, on motor impairments and spinal cord pathologies in mice which received human AQP4-IgG.
Methods
Purified IgG from AQP4-IgG-seropositive NMOSD patients were passively transferred to adult C57BL/6 mice with disrupted blood-brain barrier. Memantine was administered by oral gavage. Motor impairments of the mice were assessed by beam walking test. Spinal cords of the mice were assessed by immunofluorescence and ELISA.
Results
Oral administration of memantine ameliorated the motor impairments induced by AQP4-IgG, no matter the treatment was initiated before (preventive) or after (therapeutic) disease flare. Memantine profoundly reduced AQP4 and astrocyte loss, and attenuated demyelination and axonal loss in the spinal cord of mice which had received AQP4-IgG. The protective effects of memantine were associated with inhibition of apoptosis and suppression of neuroinflammation, with decrease in microglia activation and neutrophil infiltration and reduction of increase in levels of proinflammatory cytokines including interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α). In addition, memantine elevated growth factors including brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), and vascular endothelial growth factor (VEGF) in the spinal cord.
Conclusions
Our findings support that glutamate excitotoxicity and neuroinflammation play important roles in complement-independent pathophysiology during early development of NMOSD lesions, and highlight the potential of oral memantine as a therapeutic agent in NMOSD acute attacks.
Similar content being viewed by others

Introduction
Neuromyelitis optica spectrum disorders (NMOSD) are inflammatory demyelinating disorders of the central nervous system (CNS) clinically characterized by recurrent attacks of acute optic neuritis, transverse myelitis, and less frequently encephalitis. Patients with severe NMOSD can develop blindness, paraplegia, and even mortality [1]. The majority of NMOSD patients are seropositive for aquaporin-4 immunoglobulin G autoantibodies (AQP4-IgG), which target the water channel aquaporin-4 (AQP4) [2, 3] that is highly expressed on the membrane of astrocytic foot processes [4]. NMOSD seropositive for AQP4-IgG are considered as an autoimmune astrocytopathy [5, 6].
Diverse lesion pathologies have been observed in the CNS of NMOSD patients, suggesting that multiple pathophysiologies are involved in acute attacks of NMOSD including both complement-dependent and -independent mechanisms [7]. Notably, the initiator of complement cascade, C1q, is absent in the quiescent CNS. Complement-independent events likely contribute to the early development of NMOSD lesions. These events include AQP4 loss from internalization and degradation of the antigen-autoantibody complex [8], glutamate excitotoxicity from reduction of the glutamate transporter excitatory amino acid transporter 2 (EAAT2) [9, 10], neuroinflammation due to release of proinflammatory cytokines and chemokines from astrocytes [11], and antibody-dependent cell-mediated cytotoxicity (ADCC) which involves microglia/macrophages and granulocytes [12].
Our recent study showed that mice which received IgG from AQP4-IgG-seropositive NMOSD patients developed complement-independent spinal cord pathologies, including AQP4 and glial fibrillary acidic protein (GFAP) loss, EAAT2 decrease, microglia activation, neutrophil infiltration, demyelination, and axonal loss, which were associated with motor impairments [13]. These findings support that AQP4-IgG binding to astrocytes triggers glutamate excitotoxicity secondary to EAAT2 decrease, neuroinflammation, and astrocyte cytotoxicity through ADCC by activated microglia and infiltrated granulocytes. Targeting glutamate excitotoxicty caused by AQP4-IgG binding to astrocytes may be a potential therapeutic intervention in acute attacks of NMOSD.
Memantine (1-amino-3,5-dimethyladamantane hydrochloride) is a non-competitive N-methyl-D-aspartate (NMDA) receptor antagonist. It is an approved drug for the treatment of dementia in Alzheimer’s disease and Parkinson’s disease [14, 15]. Memantine has been shown to be neuroprotective in different animal models of CNS damages. In an experimental autoimmune encephalomyelitis model with optic neuritis, memantine reduced optic nerve demyelination and protected axons and retinal ganglion cells [16]. In experimental glaucomatous neurodegeneration, memantine prevented astrocytic dysfunction of the optic nerves by enhancing mitochondrial fission, increasing mitochondrial volume density and length, and reducing auto-phagosome formation [17]. Following subarachnoid hemorrhage, memantine suppressed apoptotic cascade via reducing neuronal nitric oxide synthase expression, peroxynitrite formation, and subsequent oxidative stress [18]. In this study, we investigated if memantine exerts neuroprotective effects in mice which received passive transfer of IgG from AQP4-IgG-seropositive NMOSD patients.
Materials and methods
Patient samples and IgG purification
Sera/plasma were obtained from 13 NMOSD patients who were AQP4-IgG-seropositive as detected by cell-based indirect immunofluorescence assay [19] and 3 healthy subjects. IgG from sera or plasma was isolated using HiTrap Protein G Sepharose columns (GE Healthcare Bio-sciences, USA). Samples were further purified with Slide-A-Lyzer Dialysis Cassettes (Thermo Scientific, USA) and concentrated with Amicon Ultra-15 centrifugal filters (Merck Millipore, Germany). Protein concentration was measured by Bradford assay (Bio-Rad, USA). Pooled IgG isolated from AQP4-IgG-seropositive NMOSD patients was termed IgG(AQP4+). Pooled IgG isolated from healthy subjects was termed IgG(con).
Mice
Female wild-type C57BL/6 mice of age 6–8 weeks were used. Mice were housed in the animal facilities at the Laboratory Animal Unit of The University of Hong Kong. They were maintained in groups of five per cage under a 12 h dark/light cycle and provided with free access of water and chow.
Disruption of BBB and passive transfer of IgG from NMOSD patients
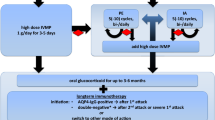
Animal procedures are summarized in Fig. 1a. To disrupt the BBB, mice were anaesthetized with intraperitoneal (i.p.) injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). They received subcutaneous injections of complete Freund’s adjuvant (CFA, BD Biosciences, USA) containing heat-killed H37Ra Mycobacterium tuberculosis (Difco, USA) at 4 sites (50 μg in 50 μl CFA each site) on the hind flank on 7 days before IgG transfer. In addition, mice received i.p. injections of pertussis toxin (PTx, 200 ng in 0.2 ml PBS, List Biological Laboratories, USA) on 7 and 3 days prior to IgG transfer.

Oral administration of memantine ameliorates human AQP4-IgG-induced motor impairments in mice. a Animal groups and experimental procedures. b, c Time required (b) and number of paw slips (c) during walking across a 1.2 × 80 cm (width × length) beam in beam walking test on IgG(AQP4+) mice treated with preventive memantine or vehicle at 1, 4, and 8 days post-injection (dpi) or with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. d, e Time required (d) and number of paw slips (e) during walking across a 0.6 × 80 cm (width × length) beam in beam walking test on mice in different groups. n = 8 per group. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001
Drug treatments
Memantine (Sigma-Aldrich, USA) was prepared by adding the compound to corn oil (Sigma-Aldrich, USA). The mixture was sonicated at room temperature for 20 min. It was freshly made immediately before each administration. Following BBB disruption, mice were randomly assigned to four sets of experiments. For preventive treatment, IgG(AQP4+) mice in set 1 received i.p. injection of 4 mg purified IgG from AQP4-IgG-seropositive NMOSD patients and oral gavage of vehicle or 60 mg/kg body weight memantine on the day of IgG passive transfer (day 0). Animals were sacrificed at 1 day post-injection (dpi). In set 2, beginning from day 0, IgG(AQP4+) mice received daily i.p. injection of purified IgG from AQP4-IgG-seropositive NMOSD patients and oral gavage of vehicle or memantine till 3 dpi. Animals were sacrificed at 4 dpi. In set 3, beginning from day 0, IgG(AQP4+) mice received daily i.p. injection of purified IgG from AQP4-IgG-seropositive NMOSD patients and oral gavage of vehicle or memantine till 7 dpi. Animals were sacrificed at 8 dpi. For therapeutic treatment in set 4, IgG(AQP4+) mice received daily i.p. injection of purified IgG from AQP4-IgG-seropositive NMOSD patients beginning from day 0 till 7 dpi. Oral gavage of vehicle or memantine was begun on the 4 dpi and continued once daily immediately after IgG injection till 7 dpi. Animals were sacrificed at 8 dpi. IgG(con) mice which received vehicle were used as sham controls in all sets of experiments. The dose of memantine was adopted from a previous study [16].
Detection of motor impairments
Motor impairments were detected using beam walking test, in which examines the animal’s ability to keep upright and walk across an elevated narrow beam to a platform [20]. The apparatus consisted of 80-cm-long beams with width of 1.2 or 0.6 cm, resting 50 cm above a table top on two stands. Prior to CFA and PTx treatment, mice were trained for 2 days, with 3 trials a day, with walking across each of the 1.2- and 0.6-cm-wide beams. One day after completion of IgG transfer and drug treatment, the time for the mice to cross each beam and the number of paw slips during the process were recorded. A slip is defined as one or both feet coming off the top of the beam [20]. The investigators were blinded to the experimental groups during beam walking test.
Tissue preparation and immunofluorescence
After beam walking test, mice were sacrificed by pentobarbital overdose and received intracardiac perfusion with ice-cold PBS and paraformaldehyde. Cervical spinal cords were harvested, and sectioned at 10-μm thickness with a cryostat (Thermo Fisher Scientific, USA). Immunofluorescence was performed with standard procedure. The following primary antibodies were used: (1) rabbit anti-human IgG (1:600, Dako, USA), (2) rabbit anti-AQP4 (1:200, Sigma-Aldrich, USA), (3) mouse anti-glial fibrillary acidic protein (GFAP, 1:200, Santa Cruz Biotechnology, USA), (4) goat anti-myelin basic protein (MBP, 1:200, Dako, USA), (5) rabbit anti-neurofilament heavy polypeptide (NF-H, 1:400, Sigma-Aldrich, USA), (6) rabbit anti-NR2B (1:200, Abcam, UK), (7) rabbit anti-ionized calcium-binding adapter molecule 1 (Iba-1, 1:200, Wako, Japan), (8) rat anti-lymphocyte antigen 6 complex locus G6D (Ly6G, 1:400, Abcam, UK), (9) rat anti-CD4 (1:100, Santa Cruz Biotechnology, USA), and (10) rat anti-CD8 (1:100, Santa Cruz Biotechnology, USA). Sections were then incubated with the appropriate Alexa-Fluor-conjugated secondary antibodies (Thermo Fisher Scientific, USA) at room temperature for 1 h. They were counterstained with DAPI and mounted with anti-fade reagent (Thermo Fisher Scientific, USA).
Image processing and quantification
Measurement of immunofluorescent intensity was performed on eight rostral-to-caudal alternate cross sections of the ventrolateral white matter of the cervical spinal cord. All signals were captured with the same microscope (Nikon Eclipse Ni, Japan) and digitized with SPOT software 5.0 (Diagnostic Instruments, USA) in identical settings. Signal intensity was quantified using ImageJ software (Wayne Rasband, NIH, USA).
Histochemistry
Luxol fast blue (LFB) staining was performed according to the manufacturer’s instructions (Abcam, UK). Briefly, spinal cord sections were rehydrated and incubated in 0.1% Luxol fast blue solution at 40 oC overnight. Excess stain was rinsed with 95% ethanol. Slides were washed in distilled water, immersed in 0.05% lithium carbonate solution for 30s, and then dehydrated in serial ethanols and mounted in Permount mounting medium (Sigma-Aldrich, USA). In addition, selected sections were stained with hematoxylin and eosin (H&E) using standard procedure.
Detection of apoptotic cells
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining was performed using the In-situ Cell Death Detection Kit (Roche, Germany) according to the manufacturer’s instructions. Sections were mounted by counterstained with DAPI and mounted with anti-fade reagent (Thermo Fisher Scientific, USA). To quantify apoptotic cells, cells positive for TUNEL with condensed nuclei were counted on eight rostral-to-caudal alternate cross sections of the ventrolateral white matter of the cervical spinal cord.
ELISA for cytokines and growth factors
Following transcardiac perfusion with ice-cold PBS, spinal cords of mice were harvested and homogenized using a lysis buffer containing protease inhibitor cocktail and phosphatase inhibitor. Levels of proinflammatory cytokines including interleukin-1β (IL-1β; RayBiotech, USA), interleukin-6 (IL-6; RayBiotech, USA) and tumor necrosis factor-α (TNF-α; RayBiotech, USA), and growth factors including brain-derived neurotrophic factor (BDNF; MyBioSource, USA), glial cell line-derived neurotrophic factor (GDNF; MyBioSource, USA) and vascular endothelial growth factor (VEGF; MyBioSource, USA) in the homogenates were determined using mouse ELISA kits according to the manufacturer’s instructions.
Statistical analysis
Differences between groups were compared by one-way analysis of variance (ANOVA) followed by Tukey-Kramer post hoc test. Data are shown as mean ± SEM. Levels of significance are indicated with *p < 0.05, **p < 0.01, and ***p < 0.001. Calculation was performed using IBM SPSS Statistics Version 24 software.
Results
Memantine improved AQP4-IgG-induced motor impairments
Passive transfer of human AQP4-IgG to mice with disrupted BBB causes motor impairments [13]. To assess whether memantine improved motor performance in this animal model, beam walking test was performed in IgG(AQP4+) mice which had received either preventive or therapeutic treatment of memantine. We found that 60 mg/kg/day of memantine is effective in improving clinical severity, and therefore this dose was used in subsequent experiments (Additional file 1 a–d). H&E staining revealed no infiltration of inflammatory cells in hind limb muscles of mice in different groups; hence, no AQP4-IgG-induced myositis in hind limbs of mice contributing to motor impairments (Additional file 1e).
On a 1.2 × 80 cm (width × length) beam, no significant differences on motor performance were observed between sham, vehicle-treated, and memantine-treated mice at 1 dpi. Onset of motor impairments was found at 4 dpi, when vehicle-treated IgG(AQP4+) mice required significantly longer time to cross the beam and displayed significantly more paw slips during walking on the beam than sham mice. Preventive treatment with memantine at 60 mg/kg/day, when it was begun on the same day of IgG(AQP4+) passive transfer, completely prevented the development of motor impairments in terms of time to cross the beam and number of paw slips during walking on the beam. Therapeutic treatment with memantine at 60 mg/kg/day, begun at 4 days after the first IgG(AQP4+) passive transfer, also prevented the development of motor impairments. The efficacy of therapeutic treatment was comparable to that of preventive treatment (Fig. 1b, c, Additional files 7, 8, 9, 10). Similar findings were observed in walking test using a narrower 0.6 × 80 cm (width × length) beam that allowed detection of more subtle motor impairments. Onset of motor impairments was observed at 4 dpi. Both preventive and therapeutic treatment with memantine significantly reduced the time to cross the beam and the number of paw slips during walking on the beam (Fig. 1d, e, Additional files 11, 12, 13, 14). These findings indicated that memantine markedly ameliorated motor impairments triggered by human AQP4-IgG in mice.
Additional files 7. Representative video clip showing beam walking test of mouse received IgG(con) and vehicle, IgG(AQP4+) and vehicle, IgG(AQP4+) and preventive memantine, and IgG(AQP4+) and therapeutic memantine on the 1.2 x 80 cm beam
Additional file 8. Representative video clip showing beam walking test of mouse received IgG(con) and vehicle, IgG(AQP4+) and vehicle, IgG(AQP4+) and preventive memantine, and IgG(AQP4+) and therapeutic memantine on the 1.2 x 80 cm beam
Additional file 9. Representative video clip showing beam walking test of mouse received IgG(con) and vehicle, IgG(AQP4+) and vehicle, IgG(AQP4+) and preventive memantine, and IgG(AQP4+) and therapeutic memantine on the 1.2 x 80 cm beam
Additional file 10. Representative video clip showing beam walking test of mouse received IgG(con) and vehicle, IgG(AQP4+) and vehicle, IgG(AQP4+) and preventive memantine, and IgG(AQP4+) and therapeutic memantine on the 1.2 x 80 cm beam
Additional file 11. Representative video clip showing beam walking test of mouse received IgG(con) and vehicle, IgG(AQP4+) and vehicle, IgG(AQP4+) and preventive memantine, and IgG(AQP4+) and therapeutic memantine on the 0.6 x 80 cm beam
Additional file 12. Representative video clip showing beam walking test of mouse received IgG(con) and vehicle, IgG(AQP4+) and vehicle, IgG(AQP4+) and preventive memantine, and IgG(AQP4+) and therapeutic memantine on the 0.6 x 80 cm beam
Additional file 13. Representative video clip showing beam walking test of mouse received IgG(con) and vehicle, IgG(AQP4+) and vehicle, IgG(AQP4+) and preventive memantine, and IgG(AQP4+) and therapeutic memantine on the 0.6 x 80 cm beam
Additional file 14. Representative video clip showing beam walking test of mouse received IgG(con) and vehicle, IgG(AQP4+) and vehicle, IgG(AQP4+) and preventive memantine, and IgG(AQP4+) and therapeutic memantine on the 0.6 x 80 cm beam.
Memantine decreased AQP4-IgG-induced AQP4 and GFAP loss
Our previous study has shown that passive transfer of human AQP4-IgG to mice with disrupted BBB does not cause complement activation in the spinal cord [13], consistent with the findings from others [21]. To evaluate the effects of memantine on AQP4-IgG-induced astrocytopathy, immunofluorescence analyses were performed. With pretreatment of CFA and PTx to disrupt the BBB, we found human IgG infiltration in the spinal cord parenchyma in all groups of mice (Additional file 2a, b). Moreover, we observed profound infiltration of human IgG in the area postrema, where there is a lack of BBB, as well as in the optic nerves (Additional file 2c, d). Double immunofluorescence staining revealed colocalization of AQP4 and GFAP in the spinal cord white matter of the mice. At 1 dpi, no differences on AQP4 and GFAP immunofluorescences in the spinal cords were observed between sham, vehicle-treated and memantine-treated IgG(AQP4+) mice. Loss of AQP4 and GFAP immunofluorescences began at 4 dpi in vehicle-treated IgG(AQP4+) mice. Both preventive and therapeutic treatment with memantine decreased the loss of AQP4 and GFAP immunofluorescences at 4 and 8 dpi compared to vehicle-treated IgG(AQP4+) mice (Fig. 2a). Evaluation of immunofluorescence intensity confirmed that the increases in AQP4 and GFAP levels in memantine-treated mice at 4 and 8 dpi compared to vehicle-treated IgG(AQP4+) mice were statistically significant (Fig. 2b, c). Similar findings were observed in the area postrema of the brain stem and the optic nerve (Additional file 5).

Memantine decreases AQP4 and GFAP loss in IgG(AQP4+) mice. Pictures are representative photomicrographs of cross sections showing the white matter in ventrolateral region of cervical spinal cord. a Double immunofluorescence staining of AQP4 and GFAP (astrocyte marker) in IgG(AQP4+) mice treated with preventive memantine or vehicle at 1, 4, and 8 dpi or with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. Sections were counterstained with DAPI. Scale bar = 100 μm. b Relative intensity of AQP4 immunofluorescence in the spinal cord. c Relative intensity of GFAP immunofluorescence in the spinal cord. n = 5 per group. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; *p < 0.05; ***p < 0.001
Memantine ameliorated demyelination and axonal loss
We next examined whether memantine preserved myelin integrity and prevented axonal loss secondary to AQP4-IgG binding to astrocytes. MBP and NF-H immunofluorescence revealed, beginning from 4 dpi, prominent and patchy loss of myelin and axons in the spinal cord white matter of vehicle-treated IgG(AQP4+) mice compared to sham. Preventive and therapeutic treatment with memantine greatly increased MBP and NF-H signals at 4 and 8 dpi compared to vehicle-treated IgG(AQP4+) mice (Fig. 3a). Evaluation of immunofluorescence intensity confirmed that the increases in MBP level in memantine-treated mice at 4 and 8 dpi were statistically significant compared to vehicle-treated IgG(AQP4+) mice (Fig. 3b). Counting of NF-H-positive spots confirmed that at 4 and 8 dpi memantine-treated mice had more axons in the spinal cord than vehicle-treated IgG(AQP4+) mice (Fig. 3c). Luxol fast blue staining further showed that memantine preserved myelin integrity in the spinal cord of IgG(AQP4+) mice (Additional file 3). Similar findings were observed in the area postrema of the brain stem and the optic nerve (Additional file 6). These results indicated a protective effect of memantine against human AQP4-IgG-triggered demyelination and axonal loss in the mouse CNS.

Memantine reduces demyelination and axonal loss in IgG(AQP4+) mice. a Immunofluorescence staining of MBP (myelin marker) and NF-H (axon marker) in the spinal cords of IgG(AQP4+) mice treated with preventive memantine or vehicle at 1, 4, and 8 dpi; or with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. Sections were counterstained with DAPI. Scale bar = 100 μm. b Relative intensity of MBP immunofluorescence in the spinal cord. c Quantitative analysis of the number of NF-H spots in the spinal cord normalized to sham. n = 5 per group. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001
Memantine reduced NR2B expression and prevented apoptosis of astrocytes
Memantine has been shown to downregulate the NMDA receptor subunit NR2B, which is predominantly localized to extrasynaptic NMDA receptors [22,23,24]. To examine whether memantine decreased NR2B expression in our model, immunofluorescence analysis was performed. At 1 dpi, the level of NR2B expression in the spinal cord white matter of vehicle-treated IgG(AQP4+) mice were higher than that in sham mice. Memantine treatment profoundly reduced the NR2B expression in the spinal cord of IgG(AQP4+) mice to sham level (Fig. 4a). Evaluation of immunofluorescence intensity confirmed that the reduction in NR2B level in memantine-treated mice compared to vehicle-treated IgG(AQP4+) mice was statistically significant (Fig. 4b).

Memantine reduces NR2B expression and inhibits apoptosis in IgG(AQP4+) mice. a Immunofluorescence staining of NR2B in the spinal cords of IgG(AQP4+) mice treated with memantine or vehicle at 1 dpi. IgG(con) mice treated with vehicle were used as a sham control. Sections were counterstained with DAPI. Scale bar = 100 μm. b Relative intensity of NR2B immunofluorescence in the spinal cord. n = 5 per group. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; ***p < 0.001. c TUNEL staining of apoptotic cells in the spinal cords of IgG(AQP4+) mice treated with memantine or vehicle at 1 dpi. IgG(con) mice treated with vehicle were used as a sham control. Sections were counterstained with DAPI. Scale bar = 100 μm. d Quantitative analysis of the number of TUNEL-positive cells in the spinal cord. n = 5 per group. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; ***p < 0.001
To investigate whether memantine prevented cell loss by apoptosis, we detected DNA fragmentation using the TUNEL assay. At 1 dpi, apoptotic cells in the spinal cord white matter of vehicle-treated IgG(AQP4+) mice were more than that in sham mice. Memantine treatment markedly reduced apoptotic cells (Fig. 4c). Quantitative analysis revealed that the number of TUNEL-positive cells was significantly reduced in the memantine-treated mice, compared to the vehicle-treated IgG(AQP4+) mice (Fig. 4d). At 4 and 8 dpi, no apoptotic cells were found in the spinal cord of mice in all groups (data not shown). These results suggested that astrocytic apoptosis is an early event upon AQP4-IgG binding to astrocytes, and memantine treatment could prevent apoptotic cell death of astrocytes induced by human AQP4-IgG in mouse spinal cord.
Memantine attenuated microglia activation and neutrophil infiltration
To examine whether memantine influenced neuroinflammation triggered by human AQP4-IgG, immunofluorescence analyses on microglia activation and neutrophil infiltration were performed. At 1 dpi, no differences on microglia activation, as evaluated by Iba-1 immunofluorescence, and neutrophil infiltration, as evaluated by Ly6G immunofluorescence, were observed between the spinal cords of sham, vehicle-treated, and memantine-treated IgG(AQP4+) mice. Profound increases in Iba-1 and Ly6G immunofluorescences were observed since 4 dpi in vehicle-treated IgG(AQP4+) mice. At 4 and 8 dpi, both preventive and therapeutic treatment with memantine decreased Iba-1 and Ly6G immunofluorescences compared to vehicle-treated IgG(AQP4+) mice (Fig. 5a). Evaluation of immunofluorescence intensity confirmed that the decreases in Iba-1 and Ly6G levels in memantine-treated mice at 4 and 8 dpi compared to vehicle-treated IgG(AQP4+) mice were statistically significant (Fig. 5b, c). H&E staining revealed tissue damage and inflammatory cell infiltration in the spinal cord parenchyma of vehicle-treated IgG(AQP4+) mice, while these were markedly reduced with memantine treatments (Additional file 3). These findings indicated that memantine suppressed AQP4-IgG-induced microglia activation and neutrophil infiltration in the spinal cord. To further examine if T cells infiltrated into the spinal cord in our model, immunofluorescence staining of CD4 (T helper cells) and CD8 (T cytotoxic cells) were performed. No T cell infiltration was observed in the spinal cords of sham, vehicle-treated, and memantine-treated IgG(AQP4+) mice (Additional file 4). These findings suggested that the beneficial effects of memantine did not involve modulation of T cell infiltration into the spinal cord.

Memantine suppresses microglia activation and neutrophil infiltration. a Immunofluorescence staining of Iba-1 (microglia marker) and Ly6G (neutrophil marker) in the spinal cords of IgG(AQP4+) mice treated with preventive memantine or vehicle at 1, 4, and 8 dpi or with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. Sections were counterstained with DAPI. Scale bar = 100 μm. b Relative intensity of Iba-1 immunofluorescence in the spinal cord. c Relative intensity of Ly6G immunofluorescence in the spinal cord. n = 5 per group. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; **p < 0.01; ***p < 0.001
Memantine reduced concentrations of proinflammatory cytokines in the spinal cord
Activated microglia releases proinflammatory cytokines such as IL-1β, IL-6, and TNF-α [25]. AQP4-IgG can induce astrocytes to release a broad spectrum of proinflammatory mediators including IL-1β and IL-6 [11]. To investigate whether memantine treatment reduced neuroinflammation, ELISA of IL-1β, IL-6 and TNF-α were performed in spinal cord homogenates of mice in different groups. Results revealed that concentrations of IL-1β and IL-6 in the spinal cords of vehicle-treated IgG(AQP4+) mice were markedly higher than that of sham mice at all the time points examined. Both preventive and therapeutic treatment with memantine significantly reduced IL-1β and IL-6 concentrations compared to vehicle-treated IgG(AQP4+) mice (Fig. 6a, b). The concentration of TNF-α in the spinal cords of vehicle-treated IgG(AQP4+) mice was markedly higher than that of sham mice at 1 and 4 dpi, and then it decreased to that similar to sham. Preventive treatment with memantine significantly reduced TNF-α concentration at 1 and 4 dpi compared to vehicle-treated IgG(AQP4+) mice (Fig. 6c).

Reduction in proinflammatory cytokines in memantine-treated IgG(AQP4+) mice. a–c ELISA analyses of IL-1β (a), IL-6 (b), and TNF-α (c) in the spinal cords of IgG(AQP4+) mice treated with preventive memantine or vehicle at 1, 4, and 8 dpi or with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. n = 3 per group. Each ELISA was performed in duplicates. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001
Memantine elevated concentrations of growth factors in the spinal cord
To further characterize the protective effects of memantine, we performed ELISA to evaluate the concentrations of BDNF, GDNF, and VEGF in spinal cord homogenates of mice in different groups. Results revealed that BDNF concentration in the spinal cords of vehicle-treated IgG(AQP4+) mice was markedly lower than that of sham mice at all the time points examined. Both preventive and therapeutic treatment with memantine significantly increased BDNF concentration compared to vehicle-treated IgG(AQP4+) mice (Fig. 7a). GDNF concentration in the spinal cord of vehicle-treated IgG(AQP4+) mice did not differ significantly compared to sham mice at all the time points examined. Both preventive and therapeutic treatment with memantine significantly increased GDNF concentration compared to sham- and vehicle-treated IgG(AQP4+) mice at 4 and 8 dpi (Fig. 7b). Similarly, VEGF concentration in the spinal cord of vehicle-treated IgG(AQP4+) mice did not differ significantly compared to sham mice. The memantine treatments significantly increased VEGF concentration compared to sham and vehicle-treated IgG(AQP4+) mice at all the time points examined (Fig. 7c).

Elevation of growth factors in memantine-treated IgG(AQP4+) mice. a–c ELISA analyses of BDNF (a), GDNF (b), and VEGF (c) in the spinal cords of IgG(AQP4+) mice treated with preventive memantine or vehicle at 1, 4, and 8 dpi or with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. n = 3 per group. Each ELISA was performed in duplicates. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001
Discussion
We recently reported that passive transfer of human AQP4-IgG to mice with disrupted BBB led to complement-independent NMOSD-like spinal cord pathologies of AQP4 and astrocyte loss, EAAT2 loss, microglia activation, neutrophil infiltration, demyelination, and axonal loss, which were associated with motor impairments [13]. Here, we showed that memantine, when administered either simultaneously with or 4 days after passive transfer of human AQP4-IgG, ameliorated motor impairments in mice associated with marked alleviation of NMOSD-like pathologies, including decrease of AQP4 and GFAP loss, and reduction of demyelination and axonal loss. The mechanisms underlying the preventive and therapeutic effects of memantine involved prevention of apoptotic cell death, suppression of microglia activation and neutrophil infiltration, reduction of proinflammatory cytokines, and elevation of growth factors. These results indicate important pathological roles of glutamate excitotoxicity and neuroinflammation in NMOSD acute attacks, and highlight the potential of memantine as a therapeutic agent in NMOSD acute attacks.
We found memantine markedly prevented and ameliorated astrocyte loss, demyelination and axonal loss in IgG(AQP4+) mice. The primary mechanism of memantine’s beneficial effects may be attributed to the reduction of glutamate excitotoxicity against astrocytes, oligodendrocytes and neurons. Interestingly, memantine could not completely reverse the AQP4 loss (up to 70-80% as shown in Figure 2b), suggesting that the upstream immunological activity, including AQP4 loss due to internalization of autoantigen-autoantibody complexes, persisted after memantine treatment. Our previous study found that AQP4-IgG caused reduction of glutamate transporter EAAT2, suggesting a decrease in glutamate uptake [13]. Memantine has been shown to downregulate NR2B in different models of CNS insults [22,23,24]. In this study, we found that AQP4-IgG induced upregulation of NR2B expression, which was reversed by memantine treatment. The protective effects of memantine may be attributed to its inhibitory action on extrasynaptic glutamate receptors, which may stimulate cell death pathways in glia and neurons. Moreover, astrocyte is an important component of the BBB. Prevention of glutamate excitotoxicity-induced astrocyte loss by memantine may help to preserve BBB integrity.
Memantine is approved for the treatment of Alzheimer’s disease. It is an open-channel blocker of NMDA receptor with faster blocking/unblocking kinetics and stronger voltage dependency than other competitive and non-competitive NMDA receptor antagonists [26]. These properties account for the better safety and tolerability of memantine compared to other NMDA receptor blockers, such as MK-801 and phencyclidine, which may cause negative psychotropic effects [26, 27]. Memantine is an effective blocker of NMDA receptor only during pathological conditions, without affecting normal physiological functions. We found that both preventive and therapeutic treatment with memantine improved motor performance in mice which had received human AQP4-IgG. To our knowledge, this is the first study to show a clinical benefit of memantine in a NMOSD model.
Following AQP4-IgG binding to astrocytic AQP4, complement-independent pathophysiologies have been suggested to be important early processes in the development of NMOSD lesions [10, 28]. AQP4-IgG binding causes internalization of AQP4 together with the glutamate transporter EAAT2 from astrocytic membrane. This reduces the uptake of extracellular glutamate by astrocytes and disrupts glutamate homeostasis, leading to glutamate excitotoxicity against NMDA receptor-expressing glial cells and neurons [9, 29,30,31,32]. A recent study demonstrated that co-endocytosis of AQP4 and EAAT2 on astrocytic membrane upon AQP4-IgG binding required astrocytic Fcγ receptor, supporting a role of glutamate excitotoxicity in the early pathophysiologies of NMOSD acute attacks [10]. AQP4-IgG from NMOSD patients caused astrocyte injury with secondary damage to oligodendrocytes mediated by glutamate excitotoxicity, which may contribute to the demyelination observed in our model [32]. Overstimulation of NMDA receptors by glutamate triggers excessive Ca2+ influx, mitochondrial dysfunction, oxidative stress, and apoptotic cell death [33]. We found that memantine prevented astrocyte loss, demyelination, and axonal loss in mice which had received human AQP4-IgG. We also detected significantly more apoptotic cells in the spinal cord white matter of vehicle-treated mice than that of memantine-treated mice early (1 day) after passive transfer of AQP4-IgG. Our previous study found that human AQP4-IgG did not cause significant loss of oligodendrocytes and neurons in this mouse model [13]. The apoptotic cells in the white matter observed in the current study were likely astrocytes. These findings suggested that memantine protected astrocytes from glutamate excitotoxicity and apoptosis that were triggered by AQP4-IgG binding. Indeed, loss of EAAT2 was observed in the spinal cord lesion of NMOSD patients, supporting a role of glutamate exicitotoxicity in NMOSD pathophysiologies in human [5].
AQP4-IgG binding stimulates astrocytes to release proinflammatory cytokines, chemokines, and other inflammatory mediators that can activate microglia, thus promote further production of inflammatory mediators in a vicious cycle [11]. This aggravates neuroinflammation which involves microglia activation and parenchymal infiltration of macrophages, granulocytes, and natural killer cells from the peripheral circulation. These activated immune cells with Fc receptors can lead to astrocyte cytotoxicity via ADCC [12, 34, 35]. The protective effect of memantine on astrocytes likely attenuated the release of proinflammatory cytokines and chemokines from astrocytes triggered by AQP4-IgG binding, hence reducing microglia activation. In the EAE model of multiple sclerosis, memantine reduced neurological symptoms and decreased the expression of proinflammatory cytokines in the brain [36]. In the mouse cuprizone model of demyelination, memantine inhibits the production of proinflammatory cytokines by astrocytes via modulation of NMDA receptor [37]. Moreover, the anti-inflammatory property of memantine was shown to be mediated through inhibition of microglia activation [38]. Consistent with these studies, we found that memantine suppressed human AQP4-IgG-triggered microglia activation and neutrophil infiltration, and reduced proinflammatory IL-1β, IL-6, and TNF-α levels in mouse spinal cord. These findings suggested that memantine could exert anti-inflammatory effects through cellular mechanisms secondary to blocking NMDA receptors.
Neurotrophic factors play a critical role in the maintenance and survival of glial cells and neurons in the adult CNS. Astrocytes have been found to be a major source of neurotrophic factors [39]. Memantine markedly increased the levels of BDNF and its receptor trkB across the brain [40]. In dopaminergic neuron-glia cocultures, the neuroprotective effects of memantine was mediated by stimulating GDNF release from astrocytes [38]. In a streptozotocin-induced astrocytotoxicity model for Alzheimer’s disease, memantine ameliorated BDNF and GDNF decline in astrocytes along with phosphorylation of IRS-1, Akt, and GSK-3α/β [41]. On the other hand, astrocytes were shown to produce VEGF upon brain injury, ischemia, and neuroinflammation for regulating vascular remodeling and angiogenesis during the repair process [42, 43]. Memantine improved stroke outcomes via increasing BDNF, GDNF, and VEGF levels, reducing reactive astrogliosis and enhancing vascular density [24, 44]. In a rat model of chronic cerebral hypoperfusion, memantine enhanced neovascularization with increases in BDNF and VEGF expressions [45]. The precise mechanism of how memantine enhances the production of growth factors is not clear. Interestingly, in addition to its antagonistic effect on NMDA receptors, memantine is a highly lipophilic agent that can readily enter cells and act on intracellular processes [27]. We found that memantine prevented astrocyte loss and simultaneously increased BDNF, GDNF, and VEGF levels in the spinal cord of mice which had received human AQP4-IgG. These results suggested that the elevated growth factor levels likely associated with the protective effect of memantine on astrocytes.
A limitation of our study is that complement-dependent pathophysiologies were absent in our model. In addition, the dose of memantine used in this study was higher than that in human patients with Alzheimer’s disease (maintenance dose: 20 mg orally per day). The serum half-life of memantine in rodents is shorter than that in human [46]. Memantine will likely exert neuroprotective effects on patients with NMOSD at commonly used doses such as that for Alzheimer’s disease. Another limitation of this study is that we did not examine if memantine blocks NMDA-R in our model.
Conclusions
In summary, we found that memantine markedly ameliorated complement-independent motor impairments and spinal cord pathologies induced by human AQP4-IgG in mice. These findings suggested important roles of glutamate excitotoxicity and neuroinflammation in early pathophysiologies of NMOSD attacks. Memantine is a well-tolerated oral drug. Our results support perspectives for clinical studies on the use of memantine as preventive and therapeutic treatments of NMOSD acute attacks.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- ANOVA:
-
Analysis of variance
- AQP4:
-
Aquaporin-4
- AQP4-IgG:
-
Aquaporin-4 immunoglobulin G
- BDNF:
-
Brain-derived neurotrophic factor
- CFA:
-
Complete Freund’s adjuvant
- CNS:
-
Central nervous system
- EAAT2:
-
Excitatory amino acid transporter 2
- GDNF:
-
Glial cell line-derived neurotrophic factor
- GFAP:
-
Glial fibrillary acidic protein
- H&E:
-
Hematoxylin and eosin
- Iba-1:
-
Ionized calcium-binding adapter molecule 1
- IL-1β:
-
Interleukin-1β
- IL-6:
-
Interleukin-6
- LFB:
-
Luxol fast blue
- Ly6G:
-
Lymphocyte antigen 6 complex locus G6D
- MBP:
-
Myelin basic protein
- NF-H:
-
Neurofilament heavy polypeptide
- NMDA:
-
N-methyl-D-aspartate
- NMOSD:
-
Neuromyelitis optica spectrum disorders
- PTx:
-
Pertussis toxin
- TNF-α:
-
Tumor necrosis factor-α
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick-end labeling
- VEGF:
-
Vascular endothelial growth factor
References
Pittock SJ, Lucchinetti CF. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: A decade later. Ann N Y Acad Sci. 2016;1366:20–39.
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet. 2004;364:2106–12.
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7.
Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. 2014;10:493–506.
Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: Lessons learned from neuromyelitis optica. Brain Pathol. 2014;24:83–97.
Popescu BF, Lucchinetti CF. Immunopathology: Autoimmune glial diseases and differentiation from multiple sclerosis. 1st ed. Handb. Clin. Neurol. Elsevier B.V. 2016.
Misu T, Höftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, Nakashima I, Konno H, Bradl M, Garzuly F, Itoyama Y, Aoki M, Lassmann H. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013;125:815–27.
Hinson SR, Pittock SJ, Lucchinetti CF. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221–31.
Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL, Howe CL, Pittock SJ, Lennon VA. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med. 2008;205:2473–81.
Hinson SR, CliftI C, Luo N, Kryzer TJ, Lennon VA. Autoantibody-induced internalization of CNS AQP4 water channel and EAAT2 glutamate transporter requires astrocytic Fc receptor. Proc Natl Acad Sci. 2017;114:5491–6.
Howe CL, Kaptzan T, Magaña SM, Ayers-Ringler JR, Lafrance-Corey RG, Lucchinetti CF. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia. 2014;62:692–708.
Ratelade J, Asavapanumas N, Ritchie AM, Wemlinger S, Bennett JL, Verkman AS. Involvement of antibody-dependent cell-mediated cytotoxicity in inflammatory demyelination in a mouse model of neuromyelitis optica. Acta Neuropathol. 2013;126:699–709.
Yick LW, Ma OKF, Ng RCL, Kwan JSC, Chan KH. Aquaporin-4 autoantibodies from neuromyelitis optica spectrum disorder patients induce complement-independent immunopathologies in mice. Front Immunol. 2018;9:1438.
Aarsland D, Ballard C, Walker Z, Bostrom F, Alves G, Kossakowski K, Leroi I, Pozo-Rodriguez F, Minthon L, Londos E. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: A double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8:613–8.
Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348:1333–41.
Suhs KW, Fairless R, Williams SK, Heine K, Cavalie A, Diem R. N-methyl-D-aspartate receptor blockade is neuroprotective in experimental autoimmune optic neuritis. J Neuropathol Exp Neurol. 2014;73:507–18.
Ju WK, Kim KY, Noh YH, Hoshijima M, Lukas TJ, Ellisman MH, Weinreb RN, Perkins GA. Increased mitochondrial fission and volume density by blocking glutamate excitotoxicity protect glaucomatous optic nerve head astrocytes. Glia. 2015;63:736–53.
Huang CY, Wang LC, Wang HK, Pan CH, Cheng YY, Shan YS, Chio CC, Tsai KJ. Memantine alleviates brain injury and neurobehavioral deficits after experimental subarachnoid hemorrhage. Mol Neurobiol. 2015;51:1038–52.
Chan KH, Kwan JSC, Ho PWL, Ho JWM, Chu ACY, Ramsden DB. Aquaporin-4 autoantibodies in neuromyelitis optica spectrum disorders: comparison between tissue-based and cell-based indirect immunofluorescence assays. J Neuroinflammation. 2010;7:50.
Luong TN, Carlisle HJ, Southwell A, Patterson PH. Assessment of motor balance and coordination in mice using the balance beam. J Vis Exp. 2011;49:2376.
Ratelade J, Verkman AS. Inhibitor(s) of the classical complement pathway in mouse serum limit the utility of mice as experimental models of neuromyelitis optica. Mol Immunol. 2014;62:104–13.
Rai S, Kamat PK, Nath C, Shukla R. A study on neuroinflammation and NMDA receptor function in STZ (ICV) induced memory impaired rats. J Neuroimmunol. 2013;254:1–9.
Jang CH, Lee S, Park IY, Song A, Moon C, Cho GW. Memantine attenuates salicylate-induced tinnitus possibly by reducing NR2B expression in auditory cortex of rat. Exp Neurobiol. 2019;28:495–503.
Wang YC, Sanchez-Mendoza EH, Doeppner TR, Hermann DM. Post-Acute delivery of memantine promotes post-ischemic neurological recovery, peri-infarct tissue remodeling, and contralesional brain plasticity. J Cereb Blood Flow Metab. 2017;37:980–93.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–68.
Parsons C, Rammes G, Danysz W. Pharmacodynamics of memantine: an update. Curr Neuropharmacol. 2008;6:55–78.
Lipton SA. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx. 2004;1:101–10.
Hinson SR, Lennon VA, Pittock SJ. Autoimmune AQP4 channelopathies and neuromyelitis optica spectrum disorders. Handb Clin Neurol 1st ed. Elsevier B.V. 2016;377–403.
Skowrońska K, Obara-Michlewska M, Czarnecka A, Dąbrowska K, Zielińska M, Albrecht J. Persistent overexposure to N-Methyl-d-Aspartate (NMDA) calcium-dependently downregulates glutamine synthetase, aquaporin 4, and Kir4.1 channel in mouse cortical astrocytes. Neurotox Res. 2018;35:271–80.
Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells. 2019;8:184.
Filous AR, Silver J. Targeting astrocytes in CNS injury and disease: A translational research approach. Prog Neurobiol. 2016;144:173–87.
Marignier R, Nicolle A, Watrin C, Touret M, Cavagna S, Varrin-Doyer M, Cavillon G, Rogemond V, Confavreux C, Honnorat J, Giraudon P. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain. 2010;133:2578–91.
Olloquequi J, Cornejo-Córdova E, Verdaguer E, Soriano FX, Binvignat O, Auladell C, Camins A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J Psychopharmacol. 2018;32:265–75.
Marignier R, Ruiz A, Cavagna S, Nicole A, Watrin C, Touret M, Parrot S, Malleret G, Peyron C, Benetollo C, Auvergnon N, Vukusic S, Giraudon P. Neuromyelitis optica study model based on chronic infusion of autoantibodies in rat cerebrospinal fluid. J Neuroinflammation. 2016;13:111.
Felix CM, Levin MH, Verkman AS. Complement-independent retinal pathology produced by intravitreal injection of neuromyelitis optica immunoglobulin G. J Neuroinflammation. 2016;13:275.
Sulkowski G, Dabrowska-Bouta B, Chalimoniuk M, Struzyńska L. Effects of antagonists of glutamate receptors on pro-inflammatory cytokines in the brain cortex of rats subjected to experimental autoimmune encephalomyelitis. J Neuroimmunol. 2013;261:67–76.
Sühs KW, Gudi V, Eckermann N, Fairless R, Pul R, Skripuletz T, Stangel M. Cytokine regulation by modulation of the NMDA receptor on astrocytes. Neurosci Lett. 2016;629:227–33.
Wu HM, Tzeng NS, Qian L, Wei SJ, Hu X, Chen SH, Rawls SM, Flood P, Hong JS, Lu RB. Novel neuroprotective mechanisms of memantine: Increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology. 2009;34:2344–57.
Singh S, Swarnkar S, Goswami P, Nath C. Astrocytes and microglia: Responses to neuropathological conditions. Int J Neurosci. 2011;121:589–97.
Marvanová M, Lakso M, Pirhonen J, Nawa H, Wong G, Castrén E. The neuroprotective agent memantine induces brain-derived neurotrophic factor and trkB receptor expression in rat brain. Mol Cell Neurosci. 2001;18:247–58.
Rajasekar N, Nath C, Hanif K, Shukla R. Inhibitory effect of memantine on streptozotocin-induced insulin receptor dysfunction, neuroinflammation, amyloidogenesis, and neurotrophic factor decline in astrocytes. Mol Neurobiol. 2016;53:6730–44.
Koyama Y, Maebara Y, Hayashi M, Nagae R, Tokuyama S, Michinaga S. Endothelins reciprocally regulate VEGF-A and angiopoietin-1 production in cultured rat astrocytes: Implications on astrocytic proliferation. Glia. 2012;60:1954–63.
Dusaban SS, Chun J, Rosen H, Purcell NH, Brown JH. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J Neuroinflammation. 2017;14(1):111.
López-Valdés HE, Clarkson AN, Ao Y, Charles AC, Carmichael ST, Sofroniew MV, Brennan KC. Memantine enhances recovery from stroke. Stroke. 2014;45:2093–100.
Zhang N, Song C, Zhao B, Xing M, Luo L, Gordon ML, Cheng Y. Neovascularization and synaptic function regulation with memantine and rosuvastatin in a rat model of chronic cerebral hypoperfusion. J Mol Neurosci. 2017;63:223–32.
Parsons CG, Stöffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system - too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699–723.
Acknowledgements
Not applicable
Funding
This study was supported by the Seed Fund for Basic Research, University Research Committee, The University of Hong Kong (grant number 201611159217).
Author information
Authors and Affiliations
Contributions
LWY conceived, designed, and performed all experiments and wrote the manuscript. CHT contributed to beam walking test and tissue preparation. OKFM contributed to beam walking test. JSCK contributed to IgG preparation and purification. KHC conceived the study, arranged patients for blood taking, and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent for using serum/plasma samples in the study was obtained from all study subjects. The experiments on C57BL/6 mice were approved by the Committee on the Use of Live Animals in Teaching and Research of The University of Hong Kong.
Consent for publication
All authors read and approved the final manuscript.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Preventive memantine at 60 mg/kg/day, but not 20 mg/kg/day ameliorates human AQP4-IgG-induced motor impairments in mice. Mice were pretreated with CFA and PTx. From day 0, IgG(AQP4+) mice received daily i.p. injection of purified IgG from AQP4-IgG-seropositive NMOSD patients and oral gavage of vehicle, memantine at 20 mg/kg/day (Mem 20) or 60 mg/kg/day (Mem 60) till 7 dpi. Beam walking test was performed at 8 dpi. a-b Time required a and number of paw slips b during walking across a 1.2 x 80 cm (width x length) beam in beam walking test on IgG(AQP4+) mice treated with vehicle, Mem 20 or Mem 60 at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. c-d Time required c and number of paw slips d during walking across a 0.6 x 80 cm (width x length) beam in beam walking test on mice in different groups. n = 8 per group. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; *p < 0.05; ***p < 0.001. e H&E staining of longitudinal sections of hind limb muscle of IgG(con) mice, and IgG(AQP4+) mice treated with vehicle or Mem 60 at 8 dpi. Scale bars = 100 μm.
Additional file 2.
Infiltration of human IgG in different animal groups. a Representative photomicrographs of cross sections showing the white matter in ventrolateral region of cervical spinal cord. Immunofluorescence staining of human IgG in the spinal cords of IgG(AQP4+) mice treated with preventive memantine or vehicle at 1, 4, 8 dpi; or with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. Sections were counterstained with DAPI. b Lower magnification photomicrographs of spinal cord cross sections showing human IgG immunofluorescence. c-d Representative photomicrographs of brain stem (area postrema) cross sections c and optic nerve longitudinal sections d showing human IgG immunofluorescence. Scale bars = 100 μm.
Additional file 3.
Histopathology of spinal cords of mice in different groups. Pictures are representative photomicrographs of cross sections showing the white matter in ventrolateral region of cervical spinal cord. Luxol fast blue and H&E staining of the spinal cords of IgG(AQP4+) mice treated with preventive memantine or vehicle at 1, 4, 8 dpi; or with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. Scale bar = 100 μm.
Additional file 4.
No T cells infiltration to the spinal cord. a Immunofluorescence staining of CD4 (T helper cell marker) and CD8 (T cytotoxic cell marker) in the spinal cords of IgG(AQP4+) mice treated with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. Spleens were used as a positive staining control. Sections were counterstained with DAPI. Photomicrographs are representatives of 5 animals from each group. Scale bar = 100 μm.
Additional file 5.
Memantine decreases AQP4 and GFAP loss in the area postrema and optic nerves of IgG(AQP4+) mice. a Representative photomicrographs of brain stem cross sections showing the area postrema. Double immunofluorescence staining of AQP4 and GFAP in IgG(AQP4+) mice treated with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. b Relative intensity of AQP4 and GFAP immunofluorescence in the area postrema. c Representative photomicrographs of longitudinal sections of the optic nerve region proximal to the optic chiasm. Double immunofluorescence staining of AQP4 and GFAP in IgG(AQP4+) mice treated with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. d Relative intensity of AQP4 and GFAP immunofluorescence in the optic nerve. Dotted lines demarcate the areas where quantifications of fluorescence intensities were performed. n = 3 per group. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; ***p < 0.001. Scale bar = 100 μm.
Additional file 6.
Memantine reduces demyelination and axonal loss in the area postrema and optic nerves of IgG(AQP4+) mice. a Representative photomicrographs of brain stem cross sections showing the area postrema. Double immunofluorescence staining of MBP and NF-H in IgG(AQP4+) mice treated with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. b Relative intensity of MBP and NF-H immunofluorescence in the area postrema. c Representative photomicrographs of longitudinal sections of the optic nerve region proximal to the optic chiasm. Double immunofluorescence staining of MBP and NF-H in IgG(AQP4+) mice treated with therapeutic memantine or vehicle at 8 dpi. IgG(con) mice treated with vehicle were used as a sham control. d Relative intensity of MBP and NF-H immunofluorescence in the optic nerve. n = 3 per group. Dotted lines demarcate the areas where quantifications of fluorescence intensities were performed. Data are mean ± SEM. One-way ANOVA with Tukey-Kramer post hoc test. ns, not significant; ***p < 0.001. Scale bar = 100 μm.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yick, LW., Tang, CH., Ma, O.KF. et al. Memantine ameliorates motor impairments and pathologies in a mouse model of neuromyelitis optica spectrum disorders. J Neuroinflammation 17, 236 (2020). https://doi.org/10.1186/s12974-020-01913-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-020-01913-2