
Abstract
Background
Lysosomal storage diseases (LSD) are a large family of inherited disorders characterized by abnormal endolysosomal accumulation of cellular material due to catabolic enzyme and transporter deficiencies. Depending on the affected metabolic pathway, LSD manifest with somatic or central nervous system (CNS) signs and symptoms. Neuroinflammation is a hallmark feature of LSD with CNS involvement such as mucolipidosis type IV, but not of others like Fabry disease.
Methods
We investigated the properties of microglia from LSD with and without major CNS involvement in 2-month-old mucolipidosis type IV (Mcoln1−/−) and Fabry disease (Glay/−) mice, respectively, by using a combination of flow cytometric, RNA sequencing, biochemical, in vitro and immunofluorescence analyses.
Results
We characterized microglia activation and transcriptome from mucolipidosis type IV and Fabry disease mice to determine if impaired lysosomal function is sufficient to prime these brain-resident immune cells. Consistent with the neurological pathology observed in mucolipidosis type IV, Mcoln1−/− microglia demonstrated an activation profile with a mixed neuroprotective/neurotoxic expression pattern similar to the one we previously observed in Niemann-Pick disease, type C1, another LSD with significant CNS involvement. In contrast, the Fabry disease microglia transcriptome revealed minimal alterations, consistent with the relative lack of CNS symptoms in this disease. The changes observed in Mcoln1−/− microglia showed significant overlap with alterations previously reported for other common neuroinflammatory disorders including Alzheimer’s, Parkinson’s, and Huntington’s diseases. Indeed, our comparison of microglia transcriptomes from Alzheimer’s disease, amyotrophic lateral sclerosis, Niemann-Pick disease, type C1 and mucolipidosis type IV mouse models showed an enrichment in “disease-associated microglia” pattern among these diseases.
Conclusions
The similarities in microglial transcriptomes and features of neuroinflammation and microglial activation in rare monogenic disorders where the primary metabolic disturbance is known may provide novel insights into the immunopathogenesis of other more common neuroinflammatory disorders.
Trial registration
ClinicalTrials.gov, NCT01067742, registered on February 12, 2010
Similar content being viewed by others


Background
Microglia are the resident myeloid cell population of the central nervous system (CNS) and are involved in the homeostasis and immune protection of the brain [1]. They arise from progenitors in the embryonic yolk sac, migrate into the brain during development, and persist there throughout life [2]. Microglia are involved in healthy neuronal dendritic pruning during brain development and in the removal of apoptotic neurons [2, 3]. Under normal conditions, microglia maintain a highly ramified state, which can change under pathological conditions to an activated state with an amoeboid shape associated with a high phagocytic activity [3]. This switch to an activated phenotype has been observed during the progression of several neurodegenerative diseases [4,5,6,7,8]. Microglial activation can be triggered by signals arising from the death of either surrounding glial or neuronal cells, in response to an infection or exposure to inflammatory cytokines [1, 6].
Neuroinflammation is found in many inherited metabolic disorders and neurodegenerative diseases and has also been described in autism spectrum disorder [4, 5, 9,10,11,12,13,14,15]. In many of these conditions, lysosomal pathways become dysfunctional, which suggests that these pathways may be integral to the pathological neuroinflammation or neurodegeneration [11, 15, 16]. Only lysosomal storage diseases with neurodegeneration are associated with activated microglia (microgliosis) and dysregulated microglia function, which is not described in some other diseases associated with lysosomal dysfunction [5, 17, 18]. To understand the relationship between microglia activation and lysosomal dysfunction, we characterized microglia from two non-overlapping lysosomal storage diseases: Fabry disease (FD) [19] and the early stages of mucolipidosis type IV (MLIV) prior to the onset of neurodegeneration [17, 20, 21].
FD is an X-linked disorder caused by deficiency in the lysosomal enzyme α-galactosidase A [19]. Clinical manifestations in FD patients include renal failure, premature myocardial infarction, and stroke [22]. A mouse model of FD, Gla−/−, displays globotriaosylceramide accumulation in the lysosome; however, no significant neuropathology related to neuronal or glial cells dysfunction has been reported in these mice. In the present work, the FD mouse is used as a model for a lysosomal storage disease with minimal neurodegeneration [19]. In contrast, other lysosomal storage diseases such as MLIV, neuronopathic Gaucher and Niemann-Pick disease, type C1 and NPC1 and NPC2, and cerebroid neuronal lipofuscinosis 3 (CLN3) exhibit significant neuroinflammation and neurodegeneration [5, 23, 24].
MLIV is an autosomal recessive disorder caused by mutations in the MCOLN1 gene encoding the calcium transporter TRPML1 [20, 21, 25, 26]. Clinical manifestations in MLIV patients include cognitive impairment, delayed motor function, and ophthalmologic abnormalities that are accurately recapitulated in the Mcoln1−/− murine model [20, 21, 27]. This model is characterized by early gliosis that decreases with disease progression accompanied by a progressive loss of motor function in the later stages of the disease [17, 20, 28]. Although there are no current medical therapies for patients with MLIV, bone marrow transplantation and N-butyl-deoxynojirimycin (miglustat) have been shown to delay the pathologic progression in the mouse model of MLIV [28, 29].
Herein, we analyzed microglia from FD and MLIV mice and compared the mRNA expression profiles, from 2-month-old animals, with previously published microglia transcriptomes. Our results, as anticipated, show minor changes in the FD mice microglia compared to control microglia, with only the NOD-like signaling pathway altered. In contrast, microglia isolated from 2-month-old MLIV mice display an altered expression pattern similar to other neurodegenerative diseases.
Materials and methods
Mouse models
All mouse experiments were approved by the NICHD Animal Care and Use Committee. BALB/c-Npc1+/− and BALB/c-Npc2+/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME). C57BL6-Cln3+/− mice were obtained from Dr. Steven Walkley with permission provided by Dr. Susan Cotman. Fabry (C57BL6-Glay/−) mice were provided by Dr. Ashok Kulkarni [19]; mucolipidosis type IV (C57BL6-Mcoln1−/−) mice were obtained from Dr. James Pickel (20); and Pompe (C57BL6-Gaa−/−) mice were obtained from Dr. Nina Raben [30]. Both the Gla and Gaa mutations were maintained as homozygotes, whereas Cln3, Mcoln1, Npc1, and Npc2 were maintained as heterozygotes and intercrossed to obtain mutant and control littermates. The age of each group of animals and matched controls is specified in the corresponding figures or figure legends. Sera from 2-hydroxy-propyl-β-cyclodextrin (HPβCD)-treated animals were collected in a previous study [24].
Human biospecimens
Serum samples were obtained from MLIV patients as part of natural history studies conducted at the NIH Clinical Center (NCT01067742). The MLIV study was approved by the NINDS Institutional Review Board. Informed consent was obtained from guardians or participants. Assent was obtained when applicable. The study was conducted in accordance with the Declaration of Helsinki.
Microglia isolation and flow cytometry analyses
Microglia were isolated and analyzed as previously described [24, 31, 32]. Mice were euthanized using CO2, before cardiac perfusion with 25 mL of ice-cold PBS. Brains were removed without the spinal cord and suspended and triturated in ice-cold FACS Buffer (PBS containing 0.5% BSA (Sigma-Aldrich, St. Louis, MO, USA)) using a syringe plunger. Microglia were separated from neurons using a discontinuous Percoll gradient 30–70% (GE healthcare, Chicago, IL, USA). Microglia at the 70% interphase were collected, washed twice in FACS Buffer, and stained with fixable LIVE/DEAD fluorescent or pacific blue dye (UV-blue L-23105 or Violet L-34955, Thermo Fisher Scientific, Waltham, MA, USA)) for 10 min (1:500) in PBS at 4 °C and then incubated with rat anti-mouse CD16/32 (2.4G2; BD Biosciences, San Jose, CA, USA) for 10 min (1:100) in FACS buffer at 4 °C to block Fc receptors. For the staining of surface antigens, cells were incubated with fluorochrome-conjugated (FITC, PE, PE-Cy7, allophycocyanin [APC], APC-Cy7, APC-eFluor 780, Alexa Fluor 700, eFluor 450, eFluor 605 NC, or PerCP-Cy5.5) antibodies against mouse CD45 ((Ly-5), Biolegend, San Diego, CA, USA), F4/80 ((BM8), San Diego, CA, USA), CD11b ((M1/70), San Diego, CA, USA ), MHCII (M5/114.15.2), and CX3CR1 (FAB5825P, R&D System, Minneapolis, MN, USA). After three washes with FACS buffer, stained cells were fixed using 2% paraformaldehyde prior to analysis. Stained samples were analyzed using an LSR Fortessa (BD) and the data analyzed using FlowJo software (TreeStar, Ashland, OR, USA). Only single cells were analyzed; cell numbers were quantified using PE-conjugated fluorescent counting beads (Spherotech, Lake Forest, IL, USA). Additional file 1: Figure S1a shows the gating strategy used and Additional file 1: Figure S1b the indirect evaluation of cell purity based on the presence of non-microglial cell markers.
RNA extraction and sequencing
CX3CR1+, CD45+, and CD11b+ triple-positive live cells were sorted using a FACS Aria 3 (BD) from four 2-month-old mice per condition. Cells were collected in 200 μL of FACS buffer, centrifuged at 1000×g for 10 min at 4 °C and the cell pellet was frozen in 700 μL TRIzol. The samples were thawed at room temperature, and 200 μL Chloroform (Sigma) was added and mixed for 15 s. The aqueous phase, after 15 min of centrifugation at 12,000×g at 4 °C, was mixed with anhydrous ethanol. The solution was loaded onto RNeasy micro plus columns (Qiagen, Hilden, Germany) and the total RNA was purified according to the manufacturer’s protocol. The concentration and quality of the eluted RNA was evaluated using qubit and PicoChip Bioanalyzer. RNA-Seq libraries were constructed using SMARTer Stranded Total RNA-Seq Kit - Pico Input Mammalian (Takara Bio USA, Inc. Mountain View, USA). In brief, 2 ng of total RNA from each sample was fragmented and reverse-transcribed to cDNA, and then adapters for Illumina sequencing (Illumina, San Diego, CA, USA) (with specific barcodes) were added through PCR. The PCR products were purified, and then ribosomal cDNA was depleted using probes specific to mammalian rRNA. The cDNA fragments were further amplified with primers universal to all libraries. These libraries were pooled together and sequenced using the Illumina HiSeq 2500 to generate approximately 40 million 2 × 100 paired-end reads for each sample. The raw data was demultiplexed and analyzed further. Sequence alignment was performed using RNA-STAR [33], quantitation was performed with subread featureCounts [34], and comparisons of expression was performed with DESeq2 [35]. The raw data is available under the GEO accession number GSE112895.
Measurement of reactive oxidative stress
2′,7′-Dichlorofluorescin diacetate (DCFDA; Sigma-Aldrich, St. Louis, MO, USA) staining was performed according to the manufacturer’s instructions. In brief, 10,000 FACS-sorted microglia per well were seeded just after isolation and allowed to attach for 2 h to the bottom of fluorescence 96 well plates (Sigma-Aldrich, St. Louis, MO, USA). The wells were washed twice with 100 μL PBS and stained for 30 min with 20 μM DCFDA solution per well before reading with an automated plate reader with Ex/Em 488/520 nm (BMG Labtech, Ortenberg, Germany).
Measurement of energy metabolism
Real-time measurements of extracellular acidification rates, a measure of lactate production, were performed on an XF96e Seahorse extracellular flux analyzer (Agilent Technology, Santa Clara, CA, USA) according to the manufacturer’s instructions. Measurement was performed on 10,000 FACS-sorted microglia.
ELISA and western blots
Total protein was extracted from FACS-sorted microglia using radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl, pH 7.4, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, 40 mM NaF, 0.2 mM Na3VO4) with complete protease inhibitor cocktail (Roche, Basel, Switzerland). Protein amounts were determined using the DC assay (Bio-Rad Laboratories, Hercules, USA). Protein lysates (typically 5–20 μg) were loaded onto a 4–12% gradient NuPAGE Gel, and electrophoresis was carried out at a constant 120 V. Protein transfer was performed using the iBLOT2 dry transfer setup (25 V, 12 min). Following transfer, nitrocellulose membranes were incubated in a 2% BSA and PBS 0.1% Tween 20 blocking buffer for 1 h at room temperature, followed by incubation with the primary antibody at 4 °C overnight. After the initial incubation, the membrane was rinsed and incubated with the appropriate secondary antibody for 1 h at room temperature. Membrane development was carried out using the Bio-Rad Chemiluminescence Detection Kit (Bio-Rad Laboratories). Primary antibodies used include ERK1/2 (1/1000, Cell Signaling Technology (CST), Denver, USA), Phospho-ERK1/2 (1/1000, CST), HIF1α (1/800, Sigma), iNOS (1/800, Sigma), β-tubulin (1/2000, Sigma), anti-mouse HRP, anti-goat HRP or anti-rabbit HRP (1/20,000, Sigma), CTSD (1/500, Santa Cruz Biotechnology, USA), GAPDH (1/2000, CST), DAP12 (1/1000, Thermo Fisher Scientific, Waltham, USA), and APOE (1/500, Biodesign, Saco, USA). ELISA anti-CCL5 human or mouse was performed according to the manufacturer’s instructions (Thermo Fisher Scientific). VTS-270-treated mouse sample was collected in a previous study [24].
Immunofluorescence
Mice were euthanized by CO2 asphyxiation and transcardially perfused with PBS followed by ice-cold 4% paraformaldehyde in PBS, pH 7.4. The brains were postfixed in 4% paraformaldehyde solution for 24 h and then cryoprotected in 30% sucrose (Sigma-Aldrich, St. Louis, MO, USA). Cerebellar tissues were cryostat-sectioned parasagittally (20 μm) and floating sections were collected in PBS with 0.25% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA). Sections were incubated overnight at 4 °C with rabbit anti-IBA1 (1/200, Wako, Richmond, VA, USA), rat anti-CD68-PE (1/100, FA-11, Biolegend, San Diego, CA, USA), and chicken anti-GFAP (1/500, nbp1-05198, Novus, Centennial, CO, USA). Secondary antibodies prepared in goat and conjugated with Alexa-488, Alexa-594, or Alexa-647 and Hoechst3342 (1/1000 and 1/5000, respectively; Thermo Fisher Scientific, Waltham, MA, USA) were incubated for 1 h at room temperature. Images were taken using a Zeiss Axio Observer Z1 microscope fitted with an automated scanning stage, Colibri II LED illumination and Zeiss ZEN2 software using a high-res AxioCam MRm camera and a 20× objective. Each fluorophore channel was pseudo-colored in ZEN2, exported as CZI, and analyzed using ImageJ [36].
Datasets, pathway, and statistical analysis
Published datasets used for comparison in our study include those from App, Sod1G93A, Ercc1, and aged mice [15] and Niemann-Pick type C1 [24]. Results are presented as mean ± SD unless otherwise specified. A Mann-Whitney U test was used to compare differences between groups. Differences were considered significant if the p value was *p < 0.05, **p < 0.01, and ***p < 0.001. Statistical calculations were performed using GraphPad Prism software version 5 (San Diego, USA). Heatmap and clustering were performed with Morpheus https://software.broadinstitute.org/morpheus/. Pathway analysis was performed using WebGSALT [37], g:Profiler [38], STRING [39], and ConsensusPathDB [40]. Default setting was used for all four web resources. A significant enrichment for a pathway was set at p < 0.05 and the fold difference cutoff was set at twofold change for up- and downregulated genes.
Results
Microglia phenotypes are lysosomal storage disease-specific and mirror the presence of corresponding neurological manifestations
Mcoln1−/− mice exhibit early gliosis, which decreases over time without reaching control levels [17], unlike the aged-matched Glay/− mice that do not present with gliosis and do not exhibit neurological deficits at 2 months of age [24]. We previously showed that Mcoln1−/− microglia had normal granularity and size at 4 months of age [24]. However, analysis of microglia and myeloid lineage markers (CX3CR1, CD11b (Fig. 1a)) and activation markers (CD86, MHCII (Fig. 1b)) showed significant changes in Mcoln1−/− (MLIV) when compared to either littermate controls (WT) or Glay/− (FD) mice. Although with a decreasing trend between 2 and 8 months of age, CD86 surface expression was significantly elevated relative to control mice at all ages (Fig. 1a), supporting prior observations [17]. A time course analysis of CX3CR1 and CD11b surface expression showed significantly decreased levels at 2 months of age, with increases toward control levels between 2 and 8 months of age (Fig. 1a). Analysis of MHCII expression showed significantly increased expression only in 2-month-old Mcoln1−/− animals with normalization thereafter. Analogous investigations, performed on brains from other LSD mouse models (Gaucher [41], Sandhoff [42], NPC1 [13], and NPC2 and CLN3 (unpublished data)) also displayed an altered morphology and surface marker expression levels on their microglia.

Analysis of the microglia in Fabry disease and mucolipidosis type IV mice. Mean fluorescence intensity (MFI) of the surface expression of microglial lineage markers a CX3CR1 and CD11b and microglial activation markers. N ≥ 5; *p < 0.05, **p < 0.01 Mann-Whitney U test vs. WT. b CD86 and MHCII. WT, control; FD, 2-month-old Fabry disease; MLIV, 2-, 4-, and 8-month-old mucolipidosis type IV. N ≥ 5; *p < 0.05, **p < 0.01 Mann-Whitney U test vs. WT. c DCFDA analysis of free radical production by 10,000 microglial cells. 1 μM H2O2 treatment was used as a positive control for free radical production. N = 5, n ≥ 3; *p < 0.05, **p < 0.01 Mann-Whitney U test vs. WT. d Western blot analysis iNOS, P-ERK1/2, ERK1/2, and HIF1α and β-tubulin on 100,000 microglia lysates. N = 4, *p < 0.05 Mann-Whitney U test. e Basal extracellular acidification rate (ECAR) in mpH.minutes−1 of unbuffered seahorse media by 10,000 cells. N = 5, n ≥ 3; *p < 0.05 Mann-Whitney U test vs. WT
We previously reported increased free radicals and a switch to glycolytic metabolism in activated NPC1 microglia [24]. Similar to what we observed in NPC1 mice, the free radical content was significantly increased in microglia isolated from 2-month-old MLIV mice (Fig. 1c). As expected, based on the absent microglial activation observed in FD mice, the free radical content was not increased in Glay/− microglia (Fig. 1c). Consistent with these results, we also observed increased expression of iNOS in MLIV, but not FD, microglia (Fig. 1d). Glycolytic metabolism was evaluated by measuring extracellular medium acidification rate (ECAR), which was significantly increased in microglia obtained from MLIV mice, but not FD microglia, indicative of a shift to glycolytic metabolism (Fig. 1e). The metabolic shift measured by the increased ECAR in MLIV microglia was supported by the observation of increased expression of HIF1α and pERK1/2 (Fig. 1d), two key effector pathways in the regulation of energy metabolism [43].
Microglia transcriptomic changes are lysosomal storage diseases specific
In order to better understand the phenotypic changes observed in MLIV microglia, we performed RNA sequencing of microglia isolated from Mcoln1−/− brain. The transcriptomic profile was then compared to the transcriptomes of microglia isolated from wild-type and FD mouse brain. Transcriptome analysis of Glay/− microglia identified few genes with significantly differential expression relative to age-matched wild-type control microglia (i.e., 44 upregulated and 42 downregulated genes (Additional file 5: Table S1, Additional file 1: Figure S1)). The small number of differentially expressed genes between wild-type and Glay/− microglia is in support of the absent gliosis and lack of a neurodegenerative phenotype in 2-month-old FD mice. Pathway analysis using four different software programs consistently identified the upregulation of only the NOD-like receptor signaling pathway in Glay/− microglia compared to aged-matched wild-type control microglia (Additional file 1: Figure S1). In addition, in agreement with the lack of significant neuroinflammation in Glay/− mice, analysis of common markers of neuroinflammation and microglial activation revealed only minor changes (Fig. 2a).

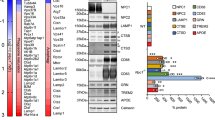
Microglia transcriptome analysis. a Heatmap of the microglia/macrophage activation markers in MLIV and FD expressed as Log2 fold change relative to control levels. b Heatmap of neurotoxic and neuroprotective markers in MLIV and FD expressed as Log2 fold change relative to control levels. The genes in this list are based upon [16, 24, 44]. c Top 50 most differentially overexpressed genes in MLIV compared to control microglia. Interferon signaling genes are boxed in red. Data underlying the heatmaps are provided in Additional file 5: Table S1 and Additional file 6: Table S2
In contrast to Glay/− microglia, transcriptomic analysis of microglia isolated from MLIV animals showed major expression differences compared to microglia from littermate wild-type control animals. Specifically, we found 386 significantly upregulated and 44 significantly downregulated genes in MLIV relative to wild-type microglia (Additional file 6: Table S2, Additional file 2: Figure S2). Pathway analysis performed on these differentially expressed genes demonstrated changes in the expression of genes related to lysosomal function, activation of immune cells (i.e., complement, antigen presentation and processing, and rheumatoid arthritis) and neurodegeneration (Alzheimer’s, Parkinson’s, and Huntington’s diseases, Additional file 2: Figure S2). The RNA expression data (Fig. 2a) are indicative of microglial activation, in support of our phenotypic analyses that showed increased expression of CD86 (Fig. 1b) and HIF1α (Fig. 1d). In addition, consonant with the increased expression of Hif1a/HIF1α and the functional shift to glycolytic metabolism, we observed significantly increased expression of the known HIF1 target genes Aldoa, Bnip3, Egln3, Igf1, Nampt1, Pgk1, and Vegfa (Additional file 6: Table S2) [45].
The RNA expression profile in MLIV microglia showed a mixed neuroprotective/neurotoxic pattern (Fig. 2b), similar to what was previously observed in NPC1 microglia, amyotrophic lateral sclerosis (ALS), and glioblastoma [16, 24, 44]. Specifically, there was a significant increase in expression of neurotoxic-related genes such as Cd22, Il1b, and Cybb, while at the same time, there was a significant increase in expression of genes generally regarded as neuroprotective such as Ccl5, Igf1, and Gpnmb (Fig. 2b). The expression of genes associated with both neurotoxic and neuroprotective function suggests a complex role played by microglia in MLIV. Of note, advances in single-cell transcriptomic analyses have shown that in both ALS and Alzheimer’s disease (AD) (SOD1G93A and 5xFAD models, respectively), a continuum of ramified to amoeboid microglial cells is present in the mouse brain with “disease-associated microglia” (DAM) present in the brain areas affected by pathology [46]. Indeed, we found the DAM marker Cd11c/Itgax to be significantly overexpressed in MLIV microglia (Fig. 2a, Additional file 6: Table S2)
Of the 50 most significantly upregulated genes in MLIV microglia, 15 (30%) are related to interferon signaling (Fig. 2c). From our in silico analysis, most of these genes are regulated by five different interferon regulatory factors: IRF1 (9 out of 50), IRF2 (5 out of 50), IRF3 (7 out of 50), IRF7 (7 out of 50), and IRF9 (10 out of 50). It has been suggested that the transcription factor EB (TFEB) could potentially be important in MLIV pathology [47]; however, it does not appear to regulate the 50 most significantly overexpressed genes in Mcoln1−/− microglia in our analysis.
Elevated CCL5 in microglia and serum of MLIV mice
We observed the chemokine Ccl5 (RANTES) to be highly overexpressed, second only to Ifit2, in microglia isolated from MLIV mice (Fig. 2c). Increased expression of Ccl5 has been reported in other mouse models with neurodegeneration and neuroinflammation including Gaucher [23], NPC1 [23], Sandhoff [23], Krabbe [23], AD [15, 48], and ALS [16], as well as during aging [15] (Fig. 3a). Whole brain immunofluorescence staining showed strong CCL5 signal in the hippocampus, but not other brain areas, of 2-month-old MLIV mice. Co-immunostaining with the microglial marker IBA1 indicated CCL5 expression within microglia (Fig. 3b, Additional file 2: Figure S2). Micsenyi et al. previously reported pathological defects in the hippocampus of MLIV mice [20]. In Npc1 mutant mice, CCL5 immunostaining was observed in the cerebellar tissue (Additional file 3: Figure S3), a brain region extensively documented to have neuronal loss and microgliosis. The brain region-specific expression of CCL5 within microglia suggests that CCL5 may be a functional role in the pathological process. Given that CCL5 is a secreted protein, we measured its level in the serum of MLIV mice and observed a significant increase at 2 and 4 months, but not at 8 months, when compared to levels found in wild-type control mice (Fig. 3c). This observation is in agreement with the decrease in gliosis occurring in the MLIV mouse brain between 4 and 8 months (Fig. 1b) [17]. Given that Ccl5 expression is increased in other disorders of neuroinflammation [23, 41, 49, 50], we measured serum CCL5 levels in other mouse models of lysosomal storage and neurodegenerative diseases. Significant increases in CCL5 serum levels were observed in ALS, CLN3, NPC1, and NPC2 compared to wild-type control mice (Fig. 3b). The progressive increase in CCL5 in NPC1 and CLN3 with disease progression, which mirrors the MLIV pattern, supports the previously reported increase inflammation over time in these pathologies [5, 24, 51]. Consistent with the lack of neuroinflammation, CCL5 serum levels were not increased in either Pompe or FD mice (Fig. 3c). To determine if reducing neuroinflammation correlated with a change in sera concentration of CCL5, we measured CCL5 levels in the serum of NPC1 mice subcutaneously injected with the most potent therapeutic compound in this disease model: 2-hydroxy-propyl-β-cyclodextrin (HPβCD/VTS-270) at 4 g/kg [24], and observed a significant decrease toward normal (Fig. 3c).

Analysis of CCL5 expression in microglia. a Ccl5 Log2 fold change and Log10 (pAdj) in 2-month-old Fabry disease (FD), 7-week-old Niemann-Pick type C1 (NPC1), 3-month-old amyotrophy lateral sclerosis (SOD1), 16-month-old App-Ps1 (AD), and 2-month-old mucolipidsosis type IV (MLIV) microglia isolated from mouse brain b IBA1 and CCL5 double staining in hippocampi. The scale bar is 10 μm. c CCL5 quantification by enzyme-linked immunosorbent assay (ELISA) in mouse serum. Npc1−/− N = 7, MLIV N = 8, FD N = 16, 2-month-old Gaa−/− (Pompe disease) N = 10, 3-month-old Npc2−/− N = 7, Sod1tgG93A N = 4, 2-, 3-, and 6-month-old Cln3−/− N = 4–6. HPβCD/VTS-270-treated animals were subcutaneously injected every other day from day 7 to 49 [24], N = 4 and 5 for control and Npc1−/−, respectively. d CCL5 ELISA from control and MLIV patient’s serum. Ncontrol = 14, NMLIV = 48
Given the temporally related changes in serum CCL5 levels observed in NPC1, CLN3, and MLIV mutant mice, we wondered whether serum CCL5 levels could be used as a biomarker in MLIV patients. A decreased CCL5 concentration in the serum of 48 MLIV patients compared to control individuals (Fig. 3d) did not support the animal results. Nonetheless, with respect to its utility as a biomarker, serum CCL5 levels did not appear to correlate with either patient age or disease severity [52].
Comparison of microglia transcriptomes from mice with different neuroinflammatory syndromes
Pathway analysis of differentially expressed genes in MLIV microglia showed an overlap with other neurodegenerative disorders including Parkinson’s, Alzheimer’s, and Huntington’s diseases (Additional file 2: Figure S2). To further investigate this observation, we extracted microglia transcriptomes from Gene Expression Omnibus [53]. Among the extracted transcriptomes, five are from mouse models known to have significant neuroinflammation: these include 18-month-old and Ercc1Δ/KO mice [54], which represent aging models, and three from NPC1, AD (amyloid precursor protein model), and ALS (SOD1G93A) diseases (Fig. 4a). Analysis of the significantly modified genes found in NPC1, AD, and ALS transcriptomes together with the MLIV microglia transcriptome revealed 100 genes that were significantly modified and followed the same expression pattern in all four disease models (Fig. 4b, c; Additional file 4: Figure S4); 13 genes were downregulated and 87 were upregulated among these 100 genes. Notably, 14 of the overexpressed genes (Fig. 4c, Additional file 4: Figure S4) have been associated with a DAM signature of microglia, previously described in both the 5xFAD mouse model of AD and the SOD1G93A mouse model of ALS [46]. Hence, independent of the underlying pathological condition, these genes appear to follow the same pattern with microglia from the two aging models (Fig. 4b) and none of these genes exhibited the DAM pattern in microglia isolated from FD mice. To validate this observation, we evaluated the protein expression level of APOE, GAPDH, CTSD, CD68, and DAP12. In our experiment, we were not able to measure the statistical difference in the expression level of GAPDH between the different groups. Significantly increased expression of HIF1a (Fig. 1), DAP12 (Additional file 4: Figure S4b), and APOE (Additional file 4: Figure S4b) was measured by western blot when comparing ML4 to WT (p < 0.05, N = 4, Mann-Whitney test). CD68 and CTSD expression levels were evaluated by double immunofluorescence with IBA1 (Additional file 4: Figure S4c), and positive double staining was observed in the hippocampus of 2-month-old ML4 mice.

Comparison of microglia transcriptomes in diseases with neuroinflammation. a Volcano plot differentially expressed genes X = Log2 fold change vs control, Y = − Log10(p). The data are supplied in Additional file 7: Table S3. The dashed lines are set at the pAdj of 0.05 and Log2 fold change of – 1 and 1 on Y- and X-axis, respectively. b Venn diagram of the significantly differentially expressed genes (pAdj < 0.05, Log2FC > 1) genes in ALS (SOD1), AD (APP), NPC1, and MLIV disease mouse models. c Heatmap of the 100 modified genes AD, ALS, MLIV, and NPC1 diseases across aging (aged and ERCC1) and FD datasets. Yellow: DAM markers. Orange: HIF1 signaling. Blue: lysosome. The heatmap with all genes labeled is provided in Additional file 4: Figure S4a
Discussion
Neuroinflammation is a common feature shared by several neurodegenerative diseases and a subset of rare LSD [16, 55]. Most of the data available have focused on the neurotoxicity or protective function of the microglia in the context of the studied pathology. Transcriptomic analysis of whole brain-isolated microglia revealed no major differences between healthy and non-reactive microglia in the context of FD. A similar lack of activation of the microglia also occurs in Pompe disease, a lysosomal glycogen storage disease (unpublished observation). The absence of activation is supported by functional analysis of microglia metabolism and free radical production which are unmodified.
One limitation of the current manuscript is that we have not established that the pattern of microglia activation observed in the MLIV mouse model is applicable to the human disorder. Future studies could explore expression of DAM markers in human brain tissue. In addition, in the present work, we report Ccl5/CCL5 as one of the most significantly overexpressed gene in mouse models of lysosomal storage diseases with neuroinflammation correlating with the microgliosis. This observation is not restricted to lysosomal storage diseases but did not translate to human, limiting its potential as a biomarker in MLIV. The use of CCL5 as a biomarker in neurodegenerative diseases has been controversial with conflicting results depending on the study [49, 56].
A similar upregulation of genes in IFN pathway has been observed in microglia from neuronopathic Gaucher and Krabbe disease mice [23], but not in NPC1 or Sandhoff disease animals [23]. Thus, activation of interferon signaling may only occur in a specific subset of LSD. However, Vitner et al. [23] showed that genetic ablation of Ifnar1, a key effector receptor of type I interferon signaling, in Gaucher mice had no impact on survival. Therefore, it is unclear how interferon signaling is involved in the pathological cascade of these LSD, including in MLIV; future studies examining genetic or pharmacological ablation of type I interferon signaling in MLIV mice will be required to shed light into this important question.
Functional and transcriptomic analysis supports the premise that microglia activation is driven by surrounding cells, “death,” rather than the sole loss of lysosomal protein or their functions. Comparison of transcriptomes shows similarities between neurodegenerative disease, AD, aging, ALS, MLIV, and NPC1. Based on this observation, treatments reducing microglial activation in the first three diseases and reducing disease burden should be evaluated in MLIV. Work by Boudewyn et al. [29] have shown that miglustat, a drug which decreases microgliosis in NPC1 mice, is effective in MLIV. Based on these observations, we could expect the inhibition of receptor-interacting protein kinase 1 or 3, which have been suggested to reduce the neuroinflammation in lysosomal storage diseases [57, 58] and could be considered for therapeutic intervention in MLIV.
Among the 87 genes with increased expression in the four disorders with neuroinflammation, 11 encode for lysosomal proteins, indicating that lysosomes may be involved in microglial dysregulation in neurodegenerative diseases and, to a lesser extent, in aging. The last enriched gene set includes HIF1-responsive genes (5/87), a pathway involved in microglia activation and energy metabolism [59]. This pathway or its target genes have previously been shown to be overexpressed in microglia from AD, ALS, and NPC1 mice [16, 24, 60].
The microglial transcriptomic signature analysis of disease state suggests a strong overlap between disease and aging. A subset (14%) of the shared modified genes in neurodegenerative disease and aging is DAM-associated markers, suggesting these cells might be participating in the pathology of MLIV. Our results cannot define if the presence of microglia in the pathologic areas is beneficial or deleterious; however, they strengthen the relevance of studying these specific microglia subpopulations. The recent demonstration of anti-CD22 treatment effectiveness to improve memory function in aged mice [61] suggests this approach could be applied to MLIV which like NPC1 and aged animals display a significant overexpression of this gene.
Conclusions
The results presented in this paper for MLIV microglia, along with our prior characterization of NPC1 microglia, suggest that microglia from common and rare neurodegenerative diseases share similarities in their activation pattern. Specifically, transcriptome and functional analysis indicate that both NPC1 and MLIV microglia resemble microglia isolated from AD and ALS mouse models. Our results thus strengthen the concept of a disease-associated microglia population in these disorders with neuroinflammation. Based on these observed homologies, therapeutic approaches modulating microglia function may be applicable to both common and rare disorders.
Availability of data and materials
The sequencing data are available on Gene Expression Omnibus under the accession number GSE112895. Mouse models are available from the Jackson Laboratory.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- CLN3:
-
Neuronal ceroid lipofuscinosis 3
- DAM:
-
Disease-associated microglia
- DCFDA:
-
2′,7′-Dichlorofluorescin diacetate
- FD:
-
Fabry disease
- HIF1:
-
Hypoxia inducible factor 1
- IRF:
-
Interferon regulatory factor
- MLIV:
-
Mucolipidosis type IV
- WT:
-
Wild type
References
Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300–12.
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51.
Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45.
Pressey SN, Smith DA, Wong AM, Platt FM, Cooper JD. Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol Dis. 2012;45(3):1086–100.
Xiong J, Kielian T. Microglia in juvenile neuronal ceroid lipofuscinosis are primed toward a pro-inflammatory phenotype. J Neurochem. 2013;127(2):245–58.
Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90.
German DC, Liang CL, Song T, Yazdani U, Xie C, Dietschy JM. Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience. 2002;109(3):437–50.
Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7(4):354–65.
Takano T. Role of Microglia in Autism: Recent Advances. Dev Neurosci. 2015;37(3):195–202.
Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci. 2014;8:315.
Vincenti JE, Murphy L, Grabert K, McColl BW, Cancellotti E, Freeman TC, et al. Defining the microglia response during the time course of chronic neurodegeneration. J Virol. 2015;90(6):3003–17.
Qian L, Flood PM. Microglial cells and Parkinson’s disease. Immunol Res. 2008;41(3):155–64.
Peake KB, Campenot RB, Vance DE, Vance JE. Niemann-Pick type C1 deficiency in microglia does not cause neuron death in vitro. Biochim Biophys Acta. 2011;1812(9):1121–9.
Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):594–604.
Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. 2015;3:31.
Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4(2):385–401.
Grishchuk Y, Sri S, Rudinskiy N, Ma W, Stember KG, Cottle MW, et al. Behavioral deficits, early gliosis, dysmyelination and synaptic dysfunction in a mouse model of mucolipidosis IV. Acta Neuropathol Commun. 2014;2:133.
Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005;128(4):413–31.
Ohshima T, Murray GJ, Swaim WD, Longenecker G, Quirk JM, Cardarelli CO, et al. alpha-Galactosidase A deficient mice: a model of Fabry disease. Proc Natl Acad Sci U S A. 1997;94(6):2540–4.
Micsenyi MC, Dobrenis K, Stephney G, Pickel J, Vanier MT, Slaugenhaupt SA, et al. Neuropathology of the Mcoln1(-/-) knockout mouse model of mucolipidosis type IV. J Neuropathol Exp Neurol. 2009;68(2):125–35.
Venugopal B, Browning MF, Curcio-Morelli C, Varro A, Michaud N, Nanthakumar N, et al. Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipidosis type IV. Am J Hum Genet. 2007;81(5):1070–83.
Kolodny E, Fellgiebel A, Hilz MJ, Sims K, Caruso P, Phan TG, et al. Cerebrovascular involvement in Fabry disease: current status of knowledge. Stroke. 2015;46(1):302–13.
Vitner EB, Farfel-Becker T, Ferreira NS, Leshkowitz D, Sharma P, Lang KS, et al. Induction of the type I interferon response in neurological forms of Gaucher disease. J Neuroinflammation. 2016;13(1):104.
Cougnoux A, Drummond RA, Collar AL, Iben JR, Salman A, Westgarth H, et al. Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum Mol Genet. 2018;27(12):2076–89.
Bassi MT, Manzoni M, Monti E, Pizzo MT, Ballabio A, Borsani G. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am J Hum Genet. 2000;67(5):1110–20.
Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, et al. Identification of the gene causing mucolipidosis type IV. Nat Genet. 2000;26(1):118–23.
Boudewyn LC, Walkley SU. Current concepts in the neuropathogenesis of mucolipidosis type IV. J Neurochem. 2019;148(5):669–89.
Walker MT, Montell C. Suppression of the motor deficit in a mucolipidosis type IV mouse model by bone marrow transplantation. Hum Mol Genet. 2016;25(13):2752–61.
Boudewyn LC, Sikora J, Kuchar L, Ledvinova J, Grishchuk Y, Wang SL, et al. N-butyldeoxynojirimycin delays motor deficits, cerebellar microgliosis, and Purkinje cell loss in a mouse model of mucolipidosis type IV. Neurobiol Dis. 2017;105:257–70.
Raben N, Lu N, Nagaraju K, Rivera Y, Lee A, Yan B, et al. Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene in the GAA knockout mice: implications for therapy. Hum Mol Genet. 2001;10(19):2039–47.
Lionakis MS, Lim JK, Lee CC, Murphy PM. Organ-specific innate immune responses in a mouse model of invasive candidiasis. Journal of innate immunity. 2011;3(2):180–99.
Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, et al. CARD9(+) microglia promote antifungal immunity via IL-1beta- and CXCL1-mediated neutrophil recruitment. Nat Immunol. 2019;20(5):559–70.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82.
Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33(Web Server issue):W741-W748.
Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35(Web Server issue):W193-W200.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52.
Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB--a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37(Database issue):D623–8.
Vitner EB, Farfel-Becker T, Eilam R, Biton I, Futerman AH. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain : a journal of neurology. 2012;135(Pt 6):1724–35.
Wu YP, Proia RL. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc Natl Acad Sci U S A. 2004;101(22):8425–30.
Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41(4):518–28.
Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015;10(2):e0116644.
Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Research. 2009;37(14):4587–602.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–90 e17.
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17(3):288–99.
Haskins M, Jones TE, Lu Q, Bareiss SK. Early alterations in blood and brain RANTES and MCP-1 expression and the effect of exercise frequency in the 3xTg-AD mouse model of Alzheimer’s disease. Neurosci Lett. 2016;610:165–70.
Tripathy D, Thirumangalakudi L, Grammas P. RANTES upregulation in the Alzheimer’s disease brain: a possible neuroprotective role. Neurobiol Aging. 2010;31(1):8–16.
Lopez ME, Klein AD, Scott MP. Complement is dispensable for neurodegeneration in Niemann-Pick disease type C. J Neuroinflamm. 2012;9.
Parviainen L, Dihanich S, Anderson GW, Wong AM, Brooks HR, Abeti R, et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol Com. 2017;5.
Smith D, Wallom KL, Williams IM, Jeyakumar M, Platt FM. Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol Dis. 2009;36(2):242–51.
Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 2002;30(1):207–10.
Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, et al. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7(6):427–39.
Lim MJ, Alexander N, Benedict JW, Chattopadhyay S, Shemilt SJ, Guerin CJ, et al. IgG entry and deposition are components of the neuroimmune response in Batten disease. Neurobiol Dis. 2007;25(2):239–51.
Kester MI, van der Flier WM, Visser A, Blankenstein MA, Scheltens P, Oudejans CB. Decreased mRNA expression of CCL5 [RANTES] in Alzheimer’s disease blood samples. Clin Chem Lab Med. 2012;50(1):61–5.
Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, et al. RIPK3 as a potential therapeutic target for Gaucher's disease. Nat Med. 2014;20(2):204–8.
Cougnoux A, Clifford S, Salman A, Ng SL, Bertin J, Porter FD. Necroptosis inhibition as a therapy for Niemann-Pick disease, type C1: Inhibition of RIP kinases and combination therapy with 2-hydroxypropyl-beta-cyclodextrin. Mol Genet Metab. 2018;125(4):345–50.
Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126(5):855–67.
Kamphuis W, Kooijman L, Schetters S, Orre M, Hol EM. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim Biophys Acta. 2016;1862(10):1847–60.
Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature. 2019;568(7751):187–92.
Acknowledgements
The authors wish to thank Dr. Nina Raben (NIAMS, Bethesda, USA) for providing the Gaa−/− mice. We would like to thank Dr. Susan Cotman (Massachusetts General Hospital, Harvard Medical School, Boston, USA) and Dr. Steven Walkley (Albert Einstein College of Medicine, New York, USA) for providing the Cln3+/− mice. We thank Dr. Steve Walkley who read and provided valuable input on the manuscript. We would like to thank the NHLBI flow cytometry core for assistance with the microglia isolation and the NICHD Molecular Genomics Core for library preparation and sequencing. We would also like to thank the clinical teams and the dedicated individuals who contributed to the clinical studies. Finally, we would like to express our appreciation to the patients and their family members for their participation in our clinical trials.
Funding
This work was supported by the intramural research programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (ZIA HD000139) and the National Institute of Allergy and Infectious Diseases (ZIA AI001175).
Author information
Authors and Affiliations
Contributions
AC, RAD, MSL, and FDP designed the study. AC and RAD performed most of the experiment. MF, JI, ALC, and FN supported the experiments and collected data. JP and ABK provided the animals used in this study. RS and FDP provided the clinical samples. FDP and MSL acquired the funding and supervised the experiments. AC, RAD, MSL, and FDP wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All mouse experiments were approved by the NICHD Animal Care and Use Committee. Collection of human biomaterial from MLIV study participants was approved by the NINDS Institutional Review Board. Informed consent was obtained.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1
. Gating strategy and cell purity evaluation. a, Representative FACS plot of 2-month-old wild type, FD and ML4 (from left to right) microglia. b, Bar graph of the Read counts in each replicate for monocyte/macrophage specific (Pf4), oligodendrocyte (Mog and Sox10), Neuron (Map2, Snap25, Isl2 and Mnx2), astrocyte (Aldh1a1 and S100b1) and microglia (Cx3cr1, Tmem119 and P2ry12) markers.
Additional file 2: Figure S2
. Transcriptomic analysis of 2-month-old Fabry Disease and Mucolipidosis type IV mice microglia. Differential expression was set at + or – 1Log2-fold divergence vs. control with pAdj<0.05. *p<0.01, **p<0.001. Pathway analysis performed with gProfiler (A), ConsensusPathDB (B), STRING (C) and WebGSALT default settings. Data are in Additional file 5: Table S1 and Additional file 6: Table S2.
Additional file 3: Figure S3
. Double immunostaining for IBA1 and CCL5 in cerebellar section from 2-month-old wild type (WT), FD, MLIV and NPC1 mice. The scale bar is 20 μm.
Additional file 4: Figure S4
. Expanded Fig. 4c. a Enlarged heatmap with all genes labeled. b Western blot analysis of GAPDH, B-Actin, DAP12, APOE on 100,000 microglia lysates. N=4. c representative IBA1/CD68 immunostaining of WT, FD and ML4 mice hippocampus (DG). d representative IBA1/CTSD immunostaining of WT, FD and ML4 mice hippocampus (DG). Scale bare is 10 μm.
Additional file 5: Table S1
. List of genes expressed in Fabry disease mice microglia with expression as log2 fold change vs. WT.
Additional file 6: Table S2
. List of genes expressed in mucolipidosis type IV mice microglia with expression as log2 fold change vs. WT.
Additional file 7: Table S3
. raw data used to build Fig. 4a with expression as log2 fold change vs. controls.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cougnoux, A., Drummond, R.A., Fellmeth, M. et al. Unique molecular signature in mucolipidosis type IV microglia. J Neuroinflammation 16, 276 (2019). https://doi.org/10.1186/s12974-019-1672-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-019-1672-4