
Abstract
Background
Chronic pro-inflammatory signaling propagates damage to neural tissue and affects the rate of disease progression. Increased activation of Toll-like receptors (TLRs), master regulators of the innate immune response, is implicated in the etiology of several neuropathologies including amyotrophic lateral sclerosis, Alzheimer’s disease, and Parkinson’s disease. Previously, we identified that the Bcl-2 family protein BH3-interacting domain death agonist (Bid) potentiates the TLR4-NF-κB pro-inflammatory response in glia, and specifically characterized an interaction between Bid and TNF receptor associated factor 6 (TRAF6) in microglia in response to TLR4 activation.
Methods
We assessed the activation of mitogen-activated protein kinase (MAPK) and interferon regulatory factor 3 (IRF3) inflammatory pathways in response to TLR3 and TLR4 agonists in wild-type (wt) and bid-deficient microglia and macrophages, using Western blot and qPCR, focusing on the response of the E3 ubiquitin ligases Pellino 1 (Peli1) and TRAF3 in the absence of microglial and astrocytic Bid. Additionally, by Western blot, we investigated the Bid-dependent turnover of Peli1 and TRAF3 in wt and bid−/− microglia using the proteasome inhibitor Bortezomib. Interactions between the de-ubiquitinating Smad6-A20 and the E3 ubiquitin ligases, TRAF3 and TRAF6, were determined by FLAG pull-down in TRAF6-FLAG or Smad6-FLAG overexpressing wt and bid-deficient mixed glia.
Results
We elucidated a positive role of Bid in both TIR-domain-containing adapter-inducing interferon-β (TRIF)- and myeloid differentiation primary response 88 (MyD88)-dependent pathways downstream of TLR4, concurrently implicating TLR3-induced inflammation. We identified that Peli1 mRNA levels were significantly reduced in PolyI:C- and lipopolysaccharide (LPS)-stimulated bid-deficient microglia, suggesting disturbed IRF3 activation. Differential regulation of TRAF3 and Peli1, both essential E3 ubiquitin ligases facilitating TRIF-dependent signaling, was observed between wt and bid−/− microglia and astrocytes. bid deficiency resulted in increased A20-E3 ubiquitin ligase protein interactions in glia, specifically A20-TRAF6 and A20-TRAF3, implicating enhanced de-ubiquitination as the mechanism of action by which E3 ligase activity is perturbed. Furthermore, Smad6-facilitated recruitment of the de-ubiquitinase A20 to E3-ligases occurred in a bid-dependent manner.
Conclusions
This study demonstrates that Bid promotes E3 ubiquitin ligase-mediated signaling downstream of TLR3 and TLR4 and provides further evidence for the potential of Bid inhibition as a therapeutic for the attenuation of the robust pro-inflammatory response culminating in TLR activation.
Similar content being viewed by others


Background
Neuroinflammation is a contributing factor to the pathogenesis of multiple diseases of the central nervous system (CNS), including Alzheimer’s disease [1, 2], Amyotrophic Lateral Sclerosis [3, 4], Parkinson’s disease [5], stroke [6], and epilepsy [7]. Attenuating the robust pro-inflammatory response associated with the diseased brain is crucial for preventing neuronal death. Accumulation of activated microglia and astrocytes in areas of localized neurodegeneration has been reported [8], with chronic microglial polarization towards the pro-inflammatory M1 state demonstrated to induce neuronal death [9, 10] and initiate the infiltration of peripheral immune cells, such as macrophages [11].
A plethora of evidence exists for the involvement of TLRs in neuropathogenesis [12–18]. Glial crosstalk potentiates a pro-inflammatory feedback loop, with evidence for Toll-like receptor (TLR) signaling in the recruitment of the adaptive immune response [11]. TLRs are highly conserved master regulators of the cellular innate immune response [19, 20], responsible for the initiation and propagation of the inflammatory cascade in response to bacterial, viral or microbial pathogens, and nucleic acids, known as pathogen-associated molecular patterns (PAMPs) or danger-associated molecular pattern (DAMPs) [21–23].
TLR4-induced activation of multiple sophisticated protein complexes occurs via the MyD88- and TRIF-dependent pathways [24]. Ubiquitin, a 76 amino acid cellular protein which covalently attaches to a target molecule to form ubiquitin chains, facilitates the multifunctional post translational modification termed ubiquitination, resulting in enhanced turnover or altered signal transduction of the target protein [25–27]. Ubiquitination plays a critical role in the orchestration of the innate immune response [28], with downstream signaling from TLRs tightly regulated by a number of E3 ubiquitin ligases, such as Peli1, TRAF3, and TRAF6, which facilitate the polyubiquitination of several critical components mediating the pro-inflammatory response [27, 29, 30]. Peli1 is a positive regulator of both MyD88- and TRIF-dependent TLR4 activation, promoting the assembly of signaling complexes in a K63-ubiquitin-dependent manner, and is identified as a key player in both the MyD88-IRAK1-TAK1-TRAF6 and TRIF-TBK1 axes [31, 32]. TRAF3 has differential roles in the regulation in the MyD88- and TRIF-dependent pathway dictated by the alternative ubiquitination of TRAF3 [33]. TRAF6-mediated K63-linked ubiquitination of the IKK complex subunit, IKKγ, positively activates NF-κB signaling in a MyD88- [34] and TRIF-dependent manner [29, 35]. Additionally, ubiquitination is modulated by a number of de-ubiquitinating enzymes (DUBs), specifically A20 (also known as TNFAIP3), which negatively regulate the TLR-induced response by ensuring the cleavage and removal of polyubiquitin chains from target proteins and thus terminate signaling [36–38]. Dysregulated A20 expression and polymorphisms in tnfaip3 are associated with multiple inflammatory diseases [39, 40], with spontaneous neuroinflammation reported in A20-deficient mice [41]. Furthermore, overexpression of A20 was shown to be protective in epilepsy [42] and cerebral ischemia [43].
BH3-interacting domain death agonist (Bid) is a pro-apoptotic BH3-only domain Bcl2 (B cell lymphoma 2) family member [44], and a key executioner of the intrinsic apoptotic pathway, linking death receptor activation to the mitochondrial apoptosis pathway [45]. Bid is cleaved by caspase-8 to its truncated form tBid, which subsequently leads to outer mitochondrial membrane permeabilization [46], initiating the caspase cascade [47]. Several studies have suggested additional non-apoptotic roles of Bid, including modulation of inflammatory signaling by associating with the IKK complex in astrocytes [48], and NOD2 receptors in intestinal epithelia [49]. Importantly, Bid was shown to modulate the immunological profile in both microglia and macrophages [50].
Previously, we have demonstrated that microglial Bid positively regulates TLR4 signaling by promoting the K63-linked polyubiquitination of TRAF6 [51]. Here, we further elucidated the mechanism of action of Bid-dependent polyubiquitination of several E3-ligases and identified increased associations between the de-ubiquitinase A20 and the E3 ligases TRAF3, TRAF6, and Peli1 in bid-deficient glia and macrophages. Collectively, these results demonstrate that Bid modulates MyD88- and TRIF-dependent signaling by regulating protein interactions and attenuating the cleavage of polyubiquitin chains, thereby facilitating downstream signaling and propagating a chronic pro-inflammatory response.
Methods
Antibodies and reagents
All common chemicals were obtained from Sigma-Aldrich (Wicklow, Ireland) unless otherwise stated. Antibodies used for Western blot and co-immunoprecipitation include rabbit anti-Bid (Abcam, ab62469, 1:1000), rabbit anti-Peli1 (Abcam ab199336, 1:1000), rabbit anti-TRAF3 (Abcam, ab76147, 1:500), mouse anti-TRAF6 (Santa Cruz, sc8409, 1:200), rabbit anti-phospho-IRF3 (Cell Signaling, 4947P, 1:500), rabbit anti-phospho-TBK1 (Cell Signaling, 5483P, 1:500), rabbit anti-phospho c-Jun (Cell Signaling, 9261S, 1:1000), rabbit anti-A20/TNFAIP3 (Cell Signaling, 5630, 1:1000), rabbit anti-LC3 (Abcam, ab51520, 1:2000), rabbit anti-FLAG (OctA-Probe D-8, Santa Cruz, sc-807, 1:500), anti- α-Tubulin (Sigma-Aldrich, T6199, 1:5000), anti-β-Actin (Sigma-Aldrich, A3853, 1:5000), and anti-GAPDH (Abcam, ab8245-100,1:5000). Bortezomib was obtained from Millennium Pharmaceuticals (Cambridge, MA, USA). Lipopolysaccharide (LPS) was obtained from (Sigma-Aldrich, L4391), and PolyI:C (31852-29-6) and Pam3CSK4 (112208-00-1) were obtained from InvivoGen.
Preparation of astrocytes and microglia
Mixed glial cultures were obtained from the cortices of P0–P2 wt and bid−/− mice of mixed sexes on a C57BL/6 background, as described previously [51]. Briefly, the cortices were isolated, the meninges were removed, and the tissue was incubated with Trypsin-EDTA at 37 °C for 10 min. The Trypsin-EDTA was removed and replaced with DMEM-F12/L-glutamine (Gibco, Life Technologies) containing Penicillin-Streptomycin (1%, Sigma-Aldrich) and fetal bovine serum (10%, Sigma-Aldrich). The cells were triturated and passed through a 40-μm nylon cell strainer (BD Falcon) before being centrifuged at 300 × g for 5 min. The cells were plated at a density of 2 cortices/T75 flask and cultured for 10 days in the presence of M-CSF (10 ng/ml, R & D Systems) and GM-CSF (20 ng/ml, R & D Systems) in order to promote microglial proliferation [52]. Microglia were isolated from the co-culture, and the remaining cells were passaged twice and cultured in the absence of M-CSF and GM-CSF as astrocyte cultures. Bid−/− mice were generated in the laboratory of Dr. Andreas Strasser, WEHI, Melbourne, Australia [53].
Preparation of macrophages
Macrophages were obtained from the bone marrow of 6-week-old wt and bid−/− mice of mixed sexes on a C57BL/6 background. Briefly, the mouse was euthanized by cervical dislocation, and the femur and tibia were carefully removed. Following the complete removal of attached muscle, the bones were cut using a sterile scalpel in a sterile laminar flow hood, and the bone marrow was flushed out using a 27 G needle containing DMEM (Gibco, Life Technologies) supplemented with penicillin-streptomycin (1%, Sigma-Aldrich) and fetal bovine serum (10%, Sigma-Aldrich). The bone marrow was homogenized and passed through a 40-μm nylon cell strainer (BD Falcon) and centrifuged at 445 × g for 3 min. The cells were resuspended in red blood cell lysis buffer (8.26 g NH4Cl, 1 g KHCO3, 0.037 g EDTA) and incubated for 1 min at room temperature before addition of DMEM (plus penicillin-streptomycin and fetal bovine serum). The cells were centrifuged at 445 × g for 3 min and cultured for 10 days in DMEM plus Pen/Strep, plus FBS, containing 40 ng/ml M-CSF (10 ng/ml, R & D Systems), in order to stimulate macrophage proliferation.
siRNA transfection
Macrophages were transfected (100 μM siRNA/3 × 105 cells) with an siRNA targeting Bid, sequence ACACGACUGUCAACUUUAU, which was designed using an algorithm optimized for siRNA selection [54]. The macrophages were transfected using lipofectamine, and the optimal silencing of bid was determined by qPCR analysis to be 48 h post transfection. A control siRNA consisting of a scrambled nucleotide sequence was also used.
Western blot
The astrocytes, microglia, or macrophages were stimulated with Pam3CSK4 (100 ng/ml), PolyI:C (100 ng/ml), or LPS (100 ng/ml), or Bortezomib (100 μM) in full serum media for the desired time point, and lysed in RIPA buffer, containing protease and phosphatase inhibitors (1:100). The cells were incubated on ice for 20 min, centrifuged at 14,000 rpm for 15 min, and the protein concentration was determined by BCA assay (Micro BCA protein determination kit, Thermo Scientific). Following the addition of 1 × Laemmli Buffer, the samples were boiled for 5 min and loaded onto 10, 12, or 15% polyacrylamide gels as appropriate. The transfer was carried out using semi-dry transfer apparatus and PVDF membrane for 1.5 h at 18 V, with the membranes exposed to Ponceau S and blocked in 3% milk for 1 h post transfer. The membranes were incubated with the primary antibodies in 3% milk either overnight at 4 °C or 2 h at room temperature, were washed in TBS-Tween-20 (0.05%), and were incubated in 3% blocking solution containing the appropriate secondary antibody (peroxidase-conjugated anti-mouse IgG, anti-rabbit IgG, or anti-goat IgG, Sigma, 1:5000, as appropriate) for 2 h at room temperature. The membrane was washed 3 times for 5 min in TBS-Tween20, exposed to ECL Chemilluminescent Reagent (Millipore) for 5 min before being imaged on a LAS-3000 Imager (Fuji, Sheffield, UK), and the quantification of protein levels were calculated using the optical density measurements from Western blot experiments and further normalized to respective loading control (α-tubulin, β-actin, or GAPDH).
qPCR analysis
RNA was extracted from each sample using the Qiagen RNAeasy kit, and 0.5 μg RNA was used for cDNA synthesis, using random primers and Superscript RT-II (Invitrogen). Two microliters of each cDNA sample and 18 μl of Mastermix (1 μl 10 μM primer (forward and reverse), 10 μl SYBRgreen PCR Mix, 7 μl RNase-free H20) were added to give a total volume of 20 μl per capillary tube. The following cycle parameters were applied; 95 °C for 15 min, 94 °C for 15 s, 57 °C for 25 s, and 72 °C for 30 s. gapdh was used as an internal control for each sample analyzed. Primers were designed using Primer3 (http://biotools.umassmed.edu/bioapps/primer3_www.cgi), and are between 150 and 250 base pairs, optimized for SYBR detection, and obtained from Sigma-Aldrich. The following sequences were used: gapdh (mouse) forward 5’ AACTTTGGCATTGTGGAAGG 3’, reverse 5’ ACACATTGGGGGTAGGAACA 3’; Peli1 forward 5’ TGCCGAAATCAATCAATCAA 3’, reverse 5’ CAATGGAGTGTCACTGGGTG 3’. qPCR analysis was carried out on a Roche Lightcycler 2.0 using SYBRgreen (Quantitect SYBRgreen kit, Qiagen).
Co-immunoprecipitation
Overexpression of TRAF6-FLAG, Smad6-FLAG, or Ubiquitin-HA was carried out by transfection of 2.5 μg plasmid/9 × 105 cells in a T75 flask, using lipofectamine. Twenty hours post transfection, the cells were stimulated with Pam3CSK4 (100 ng/ml), PolyI:C (100 ng/ml), or LPS (100 ng/ml) for 1 h before lysis in RIPA buffer (Tris 50 mM, NaCl 150 mM, SDS 0.1%, Sodium-deoxycholate 0.5%, Triton X-100 or NP-40 1%, plus 1:100 Protease Inhibitor, Sigma). Co-immunoprecipitation or pull-down experiments were carried out using DynaBeads Protein G [35 μl of Dynabeads®/sample (100–250 μg protein), Life Technologies, 10007D] and a magnetic rack (Life Technologies). Briefly, the beads were washed in RIPA buffer and incubated with 5 μg primary antibody (in 200 μl PBS for 1 h rotating at room temperature) and washed 3 times for 5 min in PBS buffer before equal amounts of protein (100–250 μg) were incubated for 2 h at room temperature (in a total volume of 750 μl). Elution of the samples from the beads using the magnetic rack, where the protein was denatured in RIPA buffer plus 1 × Laemmli buffer by incubating for 10 min at 70 °C, was followed by gel electrophoresis.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA), and the results are represented as mean ± SD. Statistical significance and p values were determined using the tests as detailed in the figure legends (* denotes p ≤ 0.05).
Results
Bid-dependent activation of IRF3 and JNK pathways in response to TLR3 and TLR4 stimulation
Previously, we have demonstrated that bid deficiency in astrocytes [48] and microglia [51] results in reduced TLR4-induced NF-κB activation, which was identified to occur via Bid-induced TRAF6 polyubiquitination. TRAF6 mediates NF-κB signaling [55] and additionally facilitates the activation of mitogen-activated protein kinases (MAPK) and interferon regulatory factor 3 (IRF3) [56]. TRIF-dependent signaling requires the phosphorylation of TANK-binding kinase 1 (TBK1) and the subsequent association with the IKK subunit IҡB kinase ε (IKKε) to activate the transcription factor IRF3 [57]. Moreover, transforming growth factor β-activated kinase 1 (TAK1)-mediated phosphorylation of Jun N-terminal kinase (JNK) is proposed to induce IRF3 activation, thus suggesting that crosstalk between the MyD88-independent and -dependent pathways with respect to IRF3 activation is mediated via JNK activation [58]. Here, we further investigated the Bid-dependent effects on the regulation of TLR4 downstream signaling, focusing on IRF3 activation and JNK-mediated MAPK activation. We observed reduced phosphorylation of TBK1 (Fig. 1a, b, 4.59 fold ± 3.6 increase vs. 2.85 fold ± 3.0 increase ± SD; Fig. 1e, f, 11.4 fold ± 7.3 increase vs. 5.6 fold ± 4.8 increase ± SD), IRF3 (Fig. 1c, d, 1.7 fold ± 0.73 increase vs. 1.0 fold ± 0.65 increase ± SD), and c-Jun (Fig. 1i, j, 3.9 fold ± 2.8 increase vs. 1.5 fold ± 1.4 increase ± SD; Fig. 1k, l, 5.3 fold ± 3.5 increase vs. 3.0 fold ± 3.0 increase ± SD) in bid−/− microglia and macrophages compared with their wt counterparts. Additionally, silencing of bid using an siRNA showed a tendency towards lower levels of TLR4-induced phosphorylation of TBK1 in wt macrophages compared with scrambled siRNA control-transfected macrophages (Fig. 1g, h, 0.71 ± 0.29 fold decrease ± SD, Con LPS vs. siBid LPS).

Reduced phosphorylation of TBK1, IRF3, and c-Jun in bid-deficient glia and macrophages compared with wt. a, b wt and bid-deficient microglia were stimulated with LPS (100 ng/ml) for 1 h and lysed in RIPA buffer, and phosphorylated TBK1 levels were determined by Western blot (n = 6–7, 6–7 cultures from 6 separate platings, p = 0.0068, Kruskall-Wallis, Dunn’s non-parametric multiple comparison post hoc test). c, d wt and bid-deficient microglia were stimulated with LPS (100 ng/ml) for 5, 15, and 30 min or 1 h and lysed in RIPA buffer, and phosphorylated-IRF3 levels were determined by Western blot (n = 3, 3 cultures from 3 separate platings, p = 0.041, paired two-tailed t test, wt Veh vs. LPS 1 h). e, f wt and bid−/− macrophages were stimulated with PolyI:C (100 ng/ml) for 30 min or 1 h and lysed in RIPA buffer, and phosphorylated-TBK1 levels were determined by Western blot (n = 4–6, 4–6 cultures from 5 separate platings, p = 0.0006 one-way ANOVA, Tukey’s multiple comparison post hoc test). g, h wt macrophages were transfected with an siRNA targeting Bid. Forty-eight hours post transfection, when Bid is optimally silenced, the cells were stimulated with LPS (100 ng/ml) for 1 h and lysed in RIPA buffer, and phosphorylated-TBK1 levels were determined by Western blot (n = 3, 3 cultures from 2 separate platings, p = 0.0588, paired two-tailed t test siControl LPS 1 h vs. siBid LPS 1 h). i, j wt and bid-deficient microglia were stimulated with LPS (100 ng/ml) for 5, 15, and 30 min or 1 h and lysed in RIPA buffer, and phosphorylated c-Jun levels were determined by Western blot (n = 5–7, 5–7 cultures from 6 separate platings, p < 0.0001, one-way ANOVA, Tukey’s multiple comparison post hoc test). k, l wt and bid-deficient macrophages were stimulated with LPS (100 ng/ml) for 30 min or 1 h and lysed in RIPA buffer, and phosphorylated c-Jun levels were determined by Western blot (n = 5–7, 5–7 cultures from 5 separate platings, p = 0.0017 one-way ANOVA, Tukey’s multiple comparison post hoc test). All samples were lysed in RIPA buffer containing protease and phosphatase inhibitors. The protein levels were quantified using optical densities. The data are represented as mean ± SD
Differential regulation of TRAF3 and Peli1 in TLR4-induced microglia, astrocytes, and macrophages
The critical role of the E3 ubiquitin ligase Peli1 in TLR signaling [59, 60] and in microglial-mediated CNS inflammation has been highlighted previously [61]. Peli1 facilitates TRAF6-mediated downstream signaling to MAPK [61]. Additionally, Peli1-mediated K63-linked polyubquitination of TBK1 is induced upon LPS stimulation activating IRF3 [32], with subsequent bidirectional signaling leading to the TBK1/IKKε-induced phosphorylation of Peli1 [62].
MyD88-dependent signal transduction to TAK1 requires the dissociation of the TRAF6-Peli1-IRAK complex from the membrane-bound receptor complex [33]. It is proposed that the cytoplasmic translocation of this complex, regulated by Peli1-mediated degradation of TRAF3, is essential for MAPK activation [33, 61]. In this way, TRAF3 negatively regulates MyD88 signaling, with the induction of pro-inflammatory genes restored in Peli1-deficient mice by the depletion of TRAF3 [61] and positively regulates the TRIF-dependent pathway mediating IRF3 activation, by executing K63-linked autoubiquitination on interaction with TRIF, which does not require cIAP1 or cIAP2 [33].
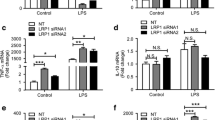
Previously, we have identified a differential TLR4-induced response in Peli1 levels in wt and bid-deficient microglia [51]. We therefore also examined the levels of Peli1 upon both TLR3 and TLR4 activation and observed a lack of Peli1 induction in bid−/− macrophages (Fig. 2a, b, 1.54 fold ± 0.22 increase vs. 0.65 fold ± 0.12 decrease ± SD) and astrocytes (Fig. 2c, d, 1.47 fold ± 0.9 increase vs. 0.75 fold ± 0.5 decrease ± SD). Additionally, increased TRAF3 levels were identified in bid−/− microglia (Fig. 2e, f, 1.65 fold ± 1.3 increase vs. 3.5 fold ± 1.7 increase ± SD).

Differential induction of Peli1 and TRAF3 degradation in TLR3- and TLR4-stimulated bid-deficient glia. a, b wt and bid−/− macrophages were stimulated with PolyI:C (100 ng/ml) for 1 h, lysed in RIPA buffer, and prepared for Western blot where Peli1 levels were determined (n = 5, 5 cultures from 4 separate platings, p = 0.004 one-way ANOVA, Tukey’s post hoc test). c, d wt and bid-deficient astrocytes were stimulated with LPS (100 ng/ml) for 5, 15, and 30 min and 1 h before being lysed in RIPA buffer and prepared for Western blot analysis of Peli1 levels (n = 4–5, 4–5 cultures from 3 separate platings, p = 0.031, Kruskall-Wallis, Dunn’s non-parametric multiple comparison post hoc test). e, f wt and bid-deficient microglia were stimulated with LPS (100 ng/ml) for 1 h and lysed in RIPA buffer, and TRAF3 levels were determined by Western blot (n = 3–4, 3–4 cultures from 4 separate platings, p = 0.019 one-way ANOVA, Tukey’s multiple comparison post hoc test). All samples were lysed in RIPA buffer containing protease and phosphatase inhibitors. The protein levels were quantified using optical densities. The data are represented as mean ± SD
Bid promotes Peli1 transcription in response to TLR3 and TLR4 activation
Peli1 is a target gene of IRF3 [62]. We next investigated Peli1 mRNA levels by qPCR and observed that Peli1 transcription was reduced in bid-deficient microglia, compared with wt, following TLR3 activation (Fig. 3a, 4.2 fold ± 3.3 vs. 2.3 fold ± 1.5 increase ± SD) or TLR4 activation (Fig. 3b, 5.4 fold ± 2.6 increase vs. 4.3 fold ± 2.5 increase ± SD), suggesting Bid-dependent activation of the TRIF-IRF3 signaling pathway.

Bid-dependent induction of Peli1 in TLR3- and TLR4-activated microglia. a wt and bid-deficient microglia were stimulated with PolyI:C (100 ng/ml) for 1 h, lysed in RLT buffer, and prepared for qPCR analysis where Peli1 mRNA was quantified using the LightCycler (n = 6–7, 6–7 cultures from 6 separate platings, p = 0.0143, Kruskall-Wallis, Dunn’s non-parametric multiple comparison post hoc test). b wt and bid-deficient microglia were stimulated with LPS (100 ng/ml) for 1 h, lysed in RLT buffer, and prepared for qPCR analysis where Peli1 mRNA was quantified using the LightCycler (n = 5–7, 5–7 cultures from 3 separate platings, p = 0.0025 one-way ANOVA, Tukey’s multiple comparison post hoc test). c, d wt and bid-deficient microglia were treated with Bortezomib (100 μM) for 1, 2, or 6 h, before being lysed in RIPA buffer, and Peli1 levels were determined by Western blot (n = 3–4, 3–4 cultures from 4 separate platings, p = 0.072, one-way ANOVA). e, f wt and bid-deficient microglia were treated with Bortezomib (100 μM) for 1, 2, or 6 h, before being lysed in RIPA buffer, and TRAF3 levels were determined by Western blot (n = 3, 3 cultures from 3 separate platings, p = 0.123, one-way ANOVA). All samples were lysed in RIPA buffer containing protease and phosphatase inhibitors. The protein levels were quantified using optical densities. The data are represented as mean ± SD
Additionally, we assessed the levels of Peli1 and TRAF3 upon inhibition of the proteasome in wt and bid-deficient microglia. Accumulation of TRAF3 in Peli1-deficient mice was identified to contribute to inflammatory pathogenesis in experimental autoimmune encephalitis (EAE) [31]. Peli1 is phosphorylated, ubiquitinated, and sumoylated [63, 64], and although Peli1 is degraded via the proteasome, it is unclear to date what targets Peli1 for degradation. Interestingly, when we examined the post translational effects of Bid, we demonstrated a tendency towards differential regulation of both Peli1 and TRAF3 in bid-deficient microglia, with a lack of accumulation of Peli1 (Fig. 3c, d, 1.65 fold ± 0.8 increase vs. 0.53 fold ± 0.71 fold decrease ± SD), and TRAF3 (Fig. 3e, f, 2.73 fold ± 1.5 increase vs. 0.74 fold ± 0.82 decrease ± SD) following 1 h Bortezomib treatment, suggesting a Bid-dependent regulation of E3 ligase stability.
Increased associations of A20 with TRAF3 and TRAF6 in bid-deficient glia and macrophages are facilitated by Smad6-A20 complex recruitment
Following the identification of a Bid-dependent dysregulated proteasomal degradation of Peli1 and TRAF3, we subsequently focused on the deubiquitinase A20. Inflammatory regulation in response to TLR4 activation is mediated by A20 which restricts signaling cascades by cleaving K48- and K64-linked polyubiquitin chains [38], thereby providing a negative feedback loop. A20 regulates TLR-induced autophagy by cleaving K63 polyubiquitin chains on Beclin 1, opposing the action of TRAF6 and limiting autophagy [65]. Therefore, we examined the role of Bid in autophagosome formation. However, we failed to observe differential regulation of LC3 lipidation in bid-deficient microglia stimulated with LPS, suggesting that Bid does not directly affect autophagy induction (Fig. 4a, b, 1.97 ± 1.18 vs. 1.7 ± 1.02 fold change ± SD).

Bid deficiency does not alter autophagosome formation. a, b wt and bid-deficient microglia were stimulated with LPS (100 ng/ml) for 30 min or 1 h and lysed in RIPA buffer, and anti-LC3-II levels were determined by Western blot (n = 3, cultures from 3 separate platings, one-way ANOVA, p = 0.43). The data are represented as mean ± SD
Ubiquitin linkage to the target protein is a highly precise process, with the specificity of the substrate ensured by the E3 ubiquitin ligase [66], and each lysine residue (K6, K11, K27, K29, K33, K48, or K63) eliciting different linkage types leading to distinct functional outcomes [28]. TRAF6 undergoes K63-linked autoubiquitination and target-induced polyubiquitination [29, 34], whereas both TRAF3 [33, 61] and Peli1 [67, 68] accept K48- and K63-linked ubiquitin chains. A20 has been shown to cleave both K48- and K63-linked ubiquitin chains [38]. We next determined the interactions between A20 and the E3 ligases TRAF3 and TRAF6, determined by pull-down and co-immunoprecipitation assays, in the presence and absence of Bid, in order to determine a Bid-dependent effect on A20 interactions following TLR3 or TLR4 activation. A20-TRAF6 interactions were increased in LPS-stimulated bid-deficient glia upon TRAF6-FLAG overexpression (Fig. 5a), with a similar pattern observed in ubiquitin-HA (Ub-HA) overexpressing bid-deficient glia at rest and upon LPS activation (Additional file 1: Figure S1A). Additionally, TRAF6-Peli1 interactions were elevated in TRAF6-FLAG overexpressing LPS-induced wt glia, which was not observed in Ub-HA overexpressing bid-deficient glia at rest and upon LPS activation (Additional file 1: Figure S1B). Similarly, A20-TRAF3 associations were increased in Ub-HA overexpressing bid-deficient glia stimulated with the TLR2 agonist Pam3CSK4 (Pam), but not in Poly-I:C stimulated samples (Fig. 5b). Additionally, we observed increased A20-Peli1 interactions in bid-deficient macrophages upon LPS stimulation (Additional file 1: Figure S1C).

Increased A20-TRAF6, A20-TRAF3, and Smad6-A20 interactions in bid-deficient glia. a wt and bid−/− mixed glia were transfected with TRAF6-FLAG and stimulated for 1 h with LPS (100 ng/ml) 20 h post transfection. The cells were lysed in RIPA buffer, and anti-FLAG was immunoprecipitated from the samples. A20-TRAF6-FLAG interaction was detected by Western blotting using an anti-A20 antibody (n = 2 separate experiments, repeated with similar results). b wt and bid−/− glia were transfected with ubiquitin-HA for 20 h and subsequently stimulated with Pam3CSK4 (Pam, 100 ng/ml) or PolyI:C (100 ng/ml) for 1 h before being lysed in RIPA buffer. Anti-TRAF3 was co-immunoprecipitated from each of the samples, and TRAF3-A20 interactions were determined by Western blotting using anti-A20 (n = 1 experiment). c wt and bid-deficient mixed glia were transfected with Smad6-FLAG, or control vector pcDNA4.1, 20 h prior to stimulation with PolyI:C (100 ng/ml) or LPS (100 ng/ml) in full serum media. Following a 1-h stimulation, the cells were lysed in RIPA buffer, containing protease and phosphatase inhibitors, and the protein concentration was assessed using BCA assay. Anti-FLAG was immunoprecipitated from each of the samples, and the samples were prepared for Western blot analysis. The membrane was exposed to anti-A20 to determine the Smad6-FLAG-A20 interactions (n = 2 experiments, repeated with similar results). All samples were lysed in RIPA buffer containing protease inhibitors, and protein concentrations were determined by BCA assay. Anti-IgG was used as co-immunoprecipitation and pull-down controls for each experiment. d Schematic showing the mechanism of action of Bid in response to TLR2 and − 3 and − 4 stimulation in glia and macrophages. Bid sequesters Smad6 and A20 thereby preventing Smad6-mediated recruitment of A20 to the E3 ligases, resulting in a lack of polyubiquitin chain cleavage and promotes pro-inflammatory signaling
Smad6 mediates the recruitment of A20 to E3 ubiquitin ligases in response to TLR activation, allowing for the subsequent negative feedback in the form of polyubiquitin chain cleavage [69]. Smad6-A20 interactions were examined by FLAG pull-down in Smad6-FLAG overexpressing wt and bid-deficient mixed glia simulated with PolyI:C and LPS. The association between A20 and Smad6 was increased in both PolyI:C and LPS-stimulated bid-deficient compared with wt controls (Fig. 5c).
Discussion
Ubiquitination plays a pivotal role in a plethora of signaling pathways, including apoptotic and inflammatory processes, with dysregulated ubiquitin activity described in multiple diseases (reviewed in [70]). Here, we show that the absence of bid in glia and macrophages results in increased interactions between A20 and the E3 ubiquitin ligases TRAF3 and TRAF6 in a Smad6-dependent manner, facilitating the cleavage of K48 and K63-linked ubiquitin chains, thereby inhibiting downstream signaling to NF-κB, MAPK, and IRF3, upon TLR3 and TLR4 activation.
TRAF6 was previously shown to promote the TAK1- and JNK-mediated phosphorylation of TBK1 and subsequently IRF3 activation [58]. We propose that the absence of Bid leads to constrained TRAF3 K48-linked degradation in microglia resulting in the attenuation of TRAF6 K63-linked signaling to TAK1 and therefore reduced phosphorylation of JNK. In addition, TRAF6 interacts with IRAK and Peli1 to form a complex that becomes dissociated from MyD88 and the TLR4 receptor, translocating to the cytoplasm where further downstream signaling occurs [33, 59]. Peli1 mediates TRAF6 K63-linked polyubiquitination of cIAP2 which induces the degradation of TRAF3 via the K48-linked ubiquitination of TRAF3 upon MyD88 signal induction [61]. Our data also demonstrates reduced phosphorylation of TBK1 in bid-deficient glia accompanied by reduced Peli1 transcription, suggesting IRF3 activation is compromised by diminished MyD88 and TRIF pathway crosstalk. Peli1 mediates TRAF6- and TAK1 K63-linked polyubquitination in macrophages [32, 59]. Furthermore, Peli1 promotes the phosphorylation of TBK1 resulting in the enhanced activation of Peli1 [32, 62]. We propose that Bid-dependent regulation of Peli1 levels upon LPS stimulation may affect the interaction of Peli1 with the TBK1-IKKε (IκB kinase ε) complex, thus adding to the complexity to the role of Bid in TLR activation. Furthermore, we demonstrate that the Bid-dependent positive regulation of TLR4 signaling is not restricted to NF-κB, but also spans IRF3 activation and JNK-mediated MAPK activation, as demonstrated by the reduced levels of phosphorylated TBK1 in bid-deficient microglia and macrophages.
We propose the Bid-dependent attenuation of A20-mediated polyubiquitin chain cleavage accounts for the differential regulation of Peli1 and TRAF3 following LPS stimulation. A20, a ubiquitously expressed cytoplasmic protein, is a potent negative regulator of ubiquitin-dependent signaling, with perinatal lethality of A20-deficient mice accompanied by multi-organ inflammation [71]. Characterization of A20 identified ubiquitin ligase domains [38]. In addition, A20 has been shown to cleave unanchored K48-linked polyubiquitin chains [72], further adding to its mechanism of preventing degradation of proteins such as TRAF3.
A20 regulates LPS-induced autophagy by cleaving K63-linked polyubiquitin chains on Beclin-1 [73]. However, our data shows that the formation of LPS-induced autophagosomes is unaffected by bid deficiency in microglia, suggested a specificity of Bid-dependent A20 activity which may be cell type specific or limited to inflammatory signaling. Furthermore, as evidenced by the response of bid-deficient microglia to Bortezomib treatment, the absence of Bid may lead to sensitization towards autophagic rather than proteasomal degradation upon proteasomal inhibition.
Here, we have demonstrated that bid-deficient glia and macrophages have increased A20-E3 ligase interactions and increased Smad6-A20 interactions, specifically TLR4-induced TRAF6-A20 associations and Pam2CSK4-induced TRAF3-A20 associations. Additionally, reduced Peli1-TRAF6 interactions were observed in TLR4-stimulated bid-deficient glia, further implicating Bid-dependent modulation of ubiquitin ligase complexes that facilitate TLR-induced inflammatory signaling. Moreover, we previously demonstrated that Bid associates with TRAF6 in both unstimulated and LPS-stimulated conditions [51], and this study strengthens our previous finding by demonstrating A20-TRAF6 interactions are increased in both basal and LPS-activated bid-deficient glia.
We propose that Bid primarily regulates A20-E3 ligase and A20-Smad6 interactions in a MyD88-dependent manner, exerting its effects on TRIF signaling through TRIF-MyD88 crosstalk facilitated by phosphorylated JNK and the TBK1 complex. Delayed kinetics of the LPS-induced TRIF-dependent pathway has been described, highlighting dependence on the MyD88 pathway [74–76]. Furthermore, our data shows that Bid impairs Smad6-A20 complex formation, suggesting increased degradation of polyubiquitin chains in the MyD88 pathway in the absence of Bid. This is of key importance, not only in regulating innate immune signaling, but also in both the initiation of the adaptive immune response [77, 78], and TLR-mediated immune regulation in CD4+ T cells [79, 80] and dendritic cells [81].
In addition to the deubiquitinase activity of A20, interactions with E1 and E2 ubiquitin -activating enzymes demonstrate the central role of A20 in ubiquitin regulation [38]. Interestingly, associations between E2 and E3 ligases are disrupted by A20, specifically due to interactions between A20 and the E2 ubiquitin ligase Ubc13 [82]. Ubc13 is required for the formation of K63-linked polyubiquitin chains [83, 84], and Ubc13-deficient mice display a blunted LPS-induced NF-κB activation [85].
The role of Bid-dependent regulation of deubiquitination in TLR activation may also encompass NOD receptor signaling, as A20 facilitates the deubiquitination of proteins in both pathways [86], further elucidating previous studies implicating Bid and IKK complex association [49].
Targeting ubiquitin signaling is a promising mechanism to negatively regulate chronic inflammation in neurodegeneration and cancer. Therapeutics targeting ubiquitin include the NF-κB inhibitor, BAY 11-7802, which was recently been shown to interact with E2 ubiquitin ligases upon TLR4 activation, thus preventing K63-linked ubiquitin chain formation [87]. As BAY 11-7802 has multiple targets [88], a more specific ubiquitin-editing mechanism would be beneficial as an anti-inflammatory therapeutic. A20 is a highly potential target for ubiquitin-modulation, with astrocytic A20 shown to ameliorate EAE in mice [89]. Moreover, targeting A20 has potential for cancer therapeutics; however, disease specificity has been reported due to the pleiotropic nature of A20. Recently, a study demonstrated potent anti-inflammatory responses using Smad6 peptides, specifically by targeting Peli1 at the membrane-bound complex, thereby inhibiting downstream pro-inflammatory signaling [90]. This study therefore demonstrates that peptides targeting ubiquitination pathways may inhibit TLR responses.
Conclusions
Collectively, our data demonstrates the potential of the inhibition of Bid as a co-therapeutic regulator of inflammatory pathways, specifically targeting TLR-induced ubiquitin signaling. Targeting innate immune signaling in the CNS is critical for the attenuation of adaptive immune cell infiltration and a chronic pro-inflammatory response. This study further highlights the immunoregulatory role of Bid and demonstrates a central role of Bid in modulating protein interactions, specifically the A20-mediated deubiquitination of E3 ubiquitin ligases in TLR-induced pro-inflammatory pathways.
Abbreviations
- Bid:
-
BH3-interacting domain death agonist
- CNS:
-
Central nervous system
- DAMPs:
-
Danger-associated molecular pattern
- DUBs:
-
De-ubiquitinating enzymes
- IKKε:
-
IҡB kinase ε
- IRF3:
-
Interferon regulatory factor 3
- JNK:
-
Jun N-terminal kinases
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- MyD88:
-
Myeloid differentiation primary response 88
- NF-ҠB:
-
Nuclear factor ҠB
- NOD:
-
Nucleotide-binding oligomerization domain-containing protein
- PAMPs:
-
Pathogen-associated molecular patterns
- Peli1:
-
Pellino 1
- TAK1:
-
Transforming growth factor β-activated kinase 1
- TBK1:
-
Tank-binding kinase 1
- TLR:
-
Toll-like receptor
- TRAF3/6:
-
TNF receptor associated factor 3/6
- TRIF:
-
TIR-domain-containing adapter-inducing interferon-β
References
Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28:8354–60.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, HILTUNEN M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, De Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9.
Hensley K, Abdel-Moaty H, Hunter J, Mhatre M, Mou S, Nguyen K, Potapova T, Pye QN, Qi M, Rice H, Stewart C, Stroukoff K, West M. Primary glia expressing the G93A-SOD1 mutation present a neuroinflammatory phenotype and provide a cellular system for studies of glial inflammation. J Neuroinflammation. 2006;3:2.
Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol. 1998;24:104–17.
Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909.
Lima RR, Santana LN, Fernandes RM, Nascimento EM, Oliveira AC, Fernandes LM, dos Santos EM, Tavares PA, dos Santos IR, Gimaraes-Santos A, Gomes-Leal W. Neurodegeneration and glial response after acute striatal stroke: histological basis for neuroprotective studies. Oxidative Med Cell Longev. 2016;2016:3173564.
Devinsky O, Vezzani A, Najjar S, de Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36:174–84.
Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140:691–707.
Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69.
Murdock BJ, Bender DE, Segal BM, Feldman EL. The dual roles of immunity in ALS: injury overrides protection. Neurobiol Dis. 2015;77:1–12.
Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–24.
Hovden H, Frederiksen JL, Pedersen SW. Immune system alterations in amyotrophic lateral sclerosis. Acta Neurol Scand. 2013;128:287–96.
Trudler D, Farfara D, Frenkel D. Toll-like receptors expression and signaling in glia cells in neuro-amyloidogenic diseases: towards future therapeutic application. Mediat Inflamm. 2010;2010
Letiembre M, Liu Y, Walter S, Hao W, Pfander T, Wrede A, Schulz-Schaeffer W, Fassbender K. Screening of innate immune receptors in neurodegenerative diseases: a similar pattern. Neurobiol Aging. 2009;30:759–68.
Jin JJ, Kim HD, Maxwell JA, LI L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation. 2008;5:23.
Koedel U, Merbt UM, Schmidt C, Angele B, Popp B, Wagner H, Pfister HW, Kirschning CJ. Acute brain injury triggers MyD88-dependent, TLR2/4-independent inflammatory responses. Am J Pathol. 2007;171:200–13.
Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RC, Bonder C, James WG, Robbins SM, Kubes P. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol. 2004;173:7070–7.
Nguyen MD, D'aigle T, Gowing G, Julien JP, Rivest S. Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2004;24:1340–9.
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7.
Scheffel J, Regen T, Van Rossum D, Seifert S, Ribes S, Nau R, Parsa R, Harris RA, Boddeke HW, Chuang HN, Pukrop T, Wessels JT, Jurgens T, Merkler D, Bruck W, Schnaars M, Simons M, Kettenmann H, Hanisch UK. Toll-like receptor activation reveals developmental reorganization and unmasks responder subsets of microglia. Glia. 2012;60:1930–43.
Hennessy EJ, Parker AE, O'neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293–307.
Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–30.
Moynagh PN. TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. 2005;26:469–76.
Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–7.
Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–86.
Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–42.
Wertz IE, Dixit VM. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. 2010;2:a003350.
Zinngrebe J, Montinaro A, Peltzer N, Walczak H. Ubiquitin in the immune system. EMBO Rep. 2014;15:28–45.
Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem. 2007;282:4102–12.
Schauvliege R, Janssens S, Beyaert R. Pellino proteins are more than scaffold proteins in TLR/IL-1R signalling: a role as novel RING E3-ubiquitin-ligases. FEBS Lett. 2006;580:4697–702.
Chang M, Jin W, Sun SC. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol. 2009;10:1089–95.
Murphy M, Xiong Y, Pattabiraman G, Qiu F, Medvedev AE. Pellino-1 positively regulates Toll-like receptor (TLR) 2 and TLR4 signaling and is suppressed upon induction of endotoxin tolerance. J Biol Chem. 2015;
Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol. 2010;11:70–5.
Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and Toll-like receptor-mediated NF-kappaB activation. Mol Cell Biol. 2008;28:3538–47.
Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y. TRAF6 autoubiquitination-independent activation of the NFkappaB and MAPK pathways in response to IL-1 and RANKL. PLoS One. 2008;3:e4064.
Edelmann MJ, Iphofer A, Akutsu M, Altun M, Di Gleria K, Kramer HB, Fiebiger E, Dhe-Paganon S, Kessler BM. Structural basis and specificity of human otubain 1-mediated deubiquitination. Biochem J. 2009;418:379–90.
Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–63.
Wertz IE, O'rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–9.
Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, Shifrin N, Petri MA, Kamboh MI, Manzi S, Seldin MF, Gregersen PK, Behrens TW, Ma A, Kwok PY, Criswell LA. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40:1062–4.
Tejasvi T, Stuart PE, Chandran V, Voorhees JJ, Gladman DD, Rahman P, Elder JT, Nair RP. TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis. J Invest Dermatol. 2012;132:593–600.
Guedes RP, Csizmadia E, Moll HP, Ma A, Ferran C, Da Silva CG. A20 deficiency causes spontaneous neuroinflammation in mice. J Neuroinflammation. 2014;11:122.
Sunaga K, Sugaya E, Kajiwara K, Tsuda T, Sugaya A, Kimura M. Molecular mechanism of preventive effect of peony root extract on neuron damage. J Herb Pharmacother. 2004;4:9–20.
Yu L, Miao H, Hou Y, Zhang B, Guo L. Neuroprotective effect of A20 on TNF-induced postischemic apoptosis. Neurochem Res. 2006;31:21–32.
Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–69.
Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6.
Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501.
Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–64.
Konig HG, Coughlan KS, Kinsella S, Breen BA, Prehn JH. The BCL-2 family protein Bid is critical for pro-inflammatory signaling in astrocytes. Neurobiol Dis. 2014;70:99–107.
Yeretssian G, Correa RG, Doiron K, Fitzgerald P, Dillon CP, Green DR, Reed JC, Saleh M. Non-apoptotic role of BID in inflammation and innate immunity. Nature. 2011;474:96–9.
Mayo L, Levy A, Jacob-Hirsch J, Amariglio N, Rechavi G, Stein R. Bid regulates the immunological profile of murine microglia and macrophages. Glia. 2011;59:397–412.
Kinsella S, Konig HG, Prehn JH. Bid promotes K63-linked polyubiquitination of tumor necrosis factor receptor associated factor 6 (TRAF6) and sensitizes to mutant SOD1-induced proinflammatory signaling in microglia. eNeuro. 2016;3 https://doi.org/10.1523/ENEURO.0099-15.2016.
Suzumura A, Sawada M, Yamamoto H, Marunouchi T. Effects of colony stimulating factors on isolated microglia in vitro. J Neuroimmunol. 1990;30:111–20.
Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK, Johnstone RW, Dixit VM, Strasser A. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–33.
Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–30.
Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–6.
Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. BioEssays. 2003;25:1096–105.
Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887–94.
Zhang B, Li M, Chen L, Yang K, Shan Y, Zhu L, Sun S, Li L, Wang C. The TAK1-JNK cascade is required for IRF3 function in the innate immune response. Cell Res. 2009;19:412–28.
Jiang Z, Johnson HJ, Nie H, Qin J, Bird TA, Li X. Pellino 1 is required for interleukin-1 (IL-1)-mediated signaling through its interaction with the IL-1 receptor-associated kinase 4 (IRAK4)-IRAK-tumor necrosis factor receptor-associated factor 6 (TRAF6) complex. J Biol Chem. 2003a;278:10952–6.
Moynagh PN. The Pellino family: IRAK E3 ligases with emerging roles in innate immune signalling. Trends Immunol. 2009;30:33–42.
Xiao Y, JIN J, Chang M, Chang JH, Hu H, Zhou X, Brittain GC, Stansberg C, Torkildsen O, Wang X, Brink R, Cheng X, Sun SC. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat Med. 2013;19:595–602.
Smith H, Liu XY, Dai L, Goh ET, Chan AT, Xi J, Seh CC, Qureshi IA, Lescar J, Ruedl C, Gourlay R, Morton S, Hough J, Mciver EG, Cohen P, Cheung PC. The role of TBK1 and IKKepsilon in the expression and activation of Pellino 1. Biochem J. 2011;434:537–48.
Butler MP, Hanly JA, Moynagh PN. Kinase-active interleukin-1 receptor-associated kinases promote polyubiquitination and degradation of the Pellino family: direct evidence for PELLINO proteins being ubiquitin-protein isopeptide ligases. J Biol Chem. 2007;282:29729–37.
Moynagh PN. The roles of Pellino E3 ubiquitin ligases in immunity. Nat Rev Immunol. 2014;14:122–31.
Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010b;3:ra42.
Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434.
Ordureau A, Smith H, Windheim M, Peggie M, Carrick E, Morrice N, Cohen P. The IRAK-catalysed activation of the E3 ligase function of Pellino isoforms induces the Lys63-linked polyubiquitination of IRAK1. Biochem J. 2008;409:43–52.
Smith H, Peggie M, Campbell DG, Vandermoere F, Carrick E, Cohen P. Identification of the phosphorylation sites on the E3 ubiquitin ligase Pellino that are critical for activation by IRAK1 and IRAK4. Proc Natl Acad Sci U S A. 2009;106:4584–90.
Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem. 1996;271:18068–73.
Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20:1242–53.
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4.
Lin SC, Chung JY, Lamothe B, Rajashankar K, Lu M, Lo YC, Lam AY, Darnay BG, Wu H. Molecular basis for the unique deubiquitinating activity of the NF-kappaB inhibitor A20. J Mol Biol. 2008;376:526–40.
Shi CS, Kehrl JH. Traf6 and A20 differentially regulate TLR4-induced autophagy by affecting the ubiquitination of Beclin 1. Autophagy. 2010a;6:986–7.
Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BR, Li X. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem. 2003b;278:16713–9.
Palsson-McDermott EM, O'neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–62.
Roy S, Karmakar M, Pearlman E. CD14 mediates Toll-like receptor 4 (TLR4) endocytosis and spleen tyrosine kinase (Syk) and interferon regulatory transcription factor 3 (IRF3) activation in epithelial cells and impairs neutrophil infiltration and Pseudomonas aeruginosa killing in vivo. J Biol Chem. 2014;289:1174–82.
Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol Today. 2000;21:141–7.
Olson JK, Girvin AM, Miller SD. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler’s virus. J Virol. 2001;75:9780–9.
Fukata M, Breglio K, Chen A, Vamadevan AS, Goo T, Hsu D, Conduah D, Xu R, Abreu MT. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180:1886–94.
Hammond T, Lee S, Watson MW, Flexman JP, Cheng W, Fernandez S, Price P. Toll-like receptor (TLR) expression on CD4+ and CD8+ T-cells in patients chronically infected with hepatitis C virus. Cell Immunol. 2010;264:150–5.
Re F, Strominger JL. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. 2001;276:37692–9.
Parvatiyar K, Harhaj EW. Regulation of inflammatory and antiviral signaling by A20. Microbes Infect. 2011;13:209–15.
Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–61.
Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–53.
Fukushima T, Matsuzawa S, Kress CL, Bruey JM, Krajewska M, Lefebvre S, Zapata JM, Ronai Z, Reed JC. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc Natl Acad Sci U S A. 2007;104:6371–6.
Hitotsumatsu O, Ahmad RC, Tavares R, Wang M, Philpott D, Turer EE, Lee BL, Shiffin N, Advincula R, MALYNN BA, Werts C, Ma A. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28:381–90.
Strickson S, Campbell DG, Emmerich CH, Knebel A, Plater L, Ritorto MS, Shpiro N, Cohen P. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem J. 2013;451:427–37.
Lee J, Rhee MH, Kim E, Cho JY. BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediat Inflamm. 2012;2012:416036.
Wang X, Deckert M, Xuan NT, Nishanth G, Just S, Waisman A, Naumann M, Schluter D. Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF-kappaB- and STAT1-dependent chemokine production in astrocytes. Acta Neuropathol. 2013;126:711–24.
Lee YS, Park JS, Jung SM, Kim SD, Kim JH, Lee JY, Jung KC, Mamura M, Lee S, Kim SJ, Bae YS, Park SH. Inhibition of lethal inflammatory responses through the targeting of membrane-associated Toll-like receptor 4 signaling complexes with a Smad6-derived peptide. EMBO Mol Med. 2015;7:577–92.
Acknowledgements
The authors wish to thank James Grehan and Ina Woods for their technical assistance in animal breeding.
Funding
This work was supported by the BioAnalysis and Therapeutics PhD scholars’ program under the Programme for Research in Third Level Institutions (PRTLI) Cycle 5, co-funded through the European Regional Development Fund (ERDF) as part of the European Union Structural Funds Programme 2007–2013, and by a Science Foundation Ireland award (14/IA/2582) to JHMP.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.
Author information
Authors and Affiliations
Contributions
SK, JHMP, and HGK conceived and designed the study. SK and JHMP wrote the manuscript. SK, MF, and OW generated and analyzed Western Blot and Immunoprecipitation data. SK carried out mRNA expression data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures involving the preparation of cultures from mice were conducted under a license from the Department of Health and Children and the Health Products Regulatory Authority (HPRA) in Ireland. Additionally, the procedures were reviewed by the Ethics Committee of the Royal College of Surgeons in Ireland.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Increased A20-TRAF3, A20-Peli1, and TRAF6-Peli1 interactions in TLR3- and TLR4-stimulated bid-deficient glia and macrophages, compared with wt. (A) wt and bid−/− mixed glia were transfected with Ubiquitin-HA and stimulated for 1 h with LPS (100 ng/ml) 20 h following transfection. Co-immunoprecipitation of anti-TRAF6 was carried out for each sample, and TRAF6-A20 interactions were determined by Western blot using an anti-A20 antibody. TRAF6-A20 interactions were quantified using optical density (n = 1 experiment). (B) wt and bid-deficient mixed glia were transfected with TRAF6-FLAG and stimulated for 1 h with LPS (100 ng/ml) 20 h post transfection. The cells were lysed in RIPA buffer, and anti-FLAG was immunoprecipitated from each of the samples. Peli1 was detected by Western blot, indicating the interaction between TRAF6-FLAG and Peli1 (n = 1 experiment). (C) wt and bid-deficient macrophages were stimulated with PolyI:C (100 ng/ml) or LPS (100 ng/ml) for 1 h and lysed in RIPA buffer. Anti-Peli1 was co-immunoprecipitated from each sample, and Peli1-A20 interactions were determined by Western blot using an anti-A20 antibody (n = 1 experiment). (TIFF 1770 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kinsella, S., Fichtner, M., Watters, O. et al. Increased A20-E3 ubiquitin ligase interactions in bid-deficient glia attenuate TLR3- and TLR4-induced inflammation. J Neuroinflammation 15, 130 (2018). https://doi.org/10.1186/s12974-018-1143-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-018-1143-3