
Abstract
Diabetic retinopathy (DR) is a common complication of diabetes and has been recognized as a vascular dysfunction leading to blindness in working-age adults. It becomes increasingly clear that neural cells in retina play an important role in the pathogenesis of DR. Neural retina located at the back of the eye is part of the brain and a representative of the central nervous system. The neurosensory deficits seen in DR are related to inflammation and occur prior to the clinically identifiable vascular complications. The neural deficits are associated with abnormal reactions of retina glial cells and neurons in response to hyperglycemia. Improper activation of the innate immune system may also be an important contributor to the pathophysiology of DR. Therefore, DR manifests characteristics of both vasculopathy and chronic neuroinflammatory diseases. In this article, we attempt to provide an overview of the current understanding of inflammation in neural retina abnormalities in diabetes. Inhibition of neuroinflammation may represent a novel therapeutic strategy to the prevention of the progression of DR.
Similar content being viewed by others


Introduction
Diabetic retinopathy (DR) is a common complication of diabetes and a leading cause of legal blindness in working-age adults in the world [1, 2]. According to the report of World Health Organization (WHO), the prevalence of DR is expected to increase and the number of people at the risk of vision loss is predicted to double by the year 2030 [3]. DR is staged into several levels of severity, including mild, moderate, and severe nonproliferative DR (NPDR), followed by an advanced proliferative DR (PDR), as defined by the presence of retinal neovascularization [4]. In PDR, proliferative neovasculature causes severe complications, such as vitreous hemorrhage, retinal scars, and tractional retinal detachment, all of which may lead to irreversible vision loss.

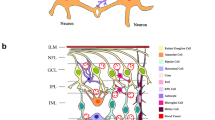
Clinicopathologic characteristics of the neural retina in diabetes. DR manifests characteristics of both vasculopathy and neuroinflammatory diseases. Neural retina including retina glial cells and neurons is involved in the neuroinflammatory responses of DR. NPDR nonproliferative diabetic retinopathy, PDR proliferative diabetic retinopathy, RGCs retinal ganglion cells, IRMA intraretinal microvascular anomalies, DME diabetic macular edema, AION anterior ischemic optic neuropathy, PION posterior ischemic optic neuropathy
The clinical evidence indicates that there is an increased capillary permeability and capillary occlusion in DR. Many DR studies in both clinic and animal models focused on vascular dysfunction, such as impaired endothelial cells, death of pericytes, thickening of retina capillary basement membrane, and altered tight junctions [5, 6]. However, retinal thickness, as measured by the retinal thickness analyzer, has been found to be abnormally diffused in the retina including the areas without clinically apparent retinopathy [7]. Microaneurysms, acellular capillary, and pericyte ghosts are more numerous in the temporal retina than in the nasal retina. However, the change in the thickness of retina capillary basement membrane is similar in all retina areas of retinopathy [8]. Both DR and diabetic nephropathy are considered as microvascular complications of diabetes. However, diabetic microvasculopathy may not explain the susceptibility of peripheral nerves or cerebral complications [9]. Thus, DR may not simply be a vasculopathy.
Recent studies revealed that electroretinogram (ERG) is defective in patients with diabetes who have no clinical retinopathy [10, 11]. The thickness of the nerve fiber layer in retinal superior polar quadrant was significantly reduced in patients with 15-year diabetic history, suggesting a loss of axons in this area [12]. In addition, functional changes in the earliest stages of human diabetic retinopathy were detected prior to the development of vascular dysfunction; therefore, the effect of hyperglycemia may be direct on the neural retina rather than secondary to the breakdown of the blood-retinal barrier [13]. Actually, neural retina located in the back of the eye is the evagination of the brain and a representation of the central nervous system (CNS). It is noted that chronic neurodegeneration is a critical cause of vision loss in DR [14]. The neurosensory deficits in DR occur prior to the clinically identifiable vascular complications [15]. Then, a major question is what links the neural responses of the retina, brain, and peripheral nerves and makes the neural tissues susceptible to hyperglycemia?
Epidemiologic studies have shown an association between the appearance of inflammatory biomarkers and the occurrence of type 2 diabetes mellitus (T2DM) and its complications [16]. Diabetics have increased serum levels of inflammatory markers, including C-reactive protein (CRP), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) [17]. There is increasing evidence that inflammatory processes play a considerable role in the pathogenesis of DR [18]. Leukostasis is particularly increased in the retinas of diabetic mice [19], while leukostasis in rats is associated with retinal endothelial cell injury and death [20]. There is also an association between high levels of proinflammatory cytokines and the development of diabetic retinopathy [21]. Although DR exhibits features of chronic neuroinflammation, the precise relationship between inflammatory alterations in DR and the loss of neural function is currently unknown.
In order to delineate inflammatory processes involved in DR, we will provide an overview of the current understanding of retinal neural abnormalities evoked by inflammation. Although treatment of vascular disorders in later stages of the disease may preserve vision in many DR patients, prevention of the onset of the disease or arresting its progression at early stage of chronic inflammation is highly desired.
The retinal neural system
The neural retina is a highly specialized nervous tissue, a part of the brain. It is divided into nine layers. From developmental perspective, the retina is a heterocellular collection of interacting cellular systems assembled by three distinct neuron-like groups: 1, superset of rod and cone photoreceptors and bipolar cells [22]; 2, superset of amacrine cells, axonal cells, and ganglion cells (GCs) [23]; and 3, the gliaform cell phenotype and the superclass of horizontal cells. In addition, a complete vertebrate retina contains two traditional classes of glial cells: Müller’s cells and astrocytes. Furthermore, functional neuroanatomy contains not only the neuronal architecture for signal processing but also the synaptic connectivity, network topology, and signaling biophysics of retinal networks.
DR involves alterations of all retinal cellular elements, including vascular endothelial cells, pericytes, glial cells (macroglia/microglia), and neurons (photoreceptors, bipolar cells, amacrine cells, and ganglion cells), showing a diffused pathological process.
Clinicopathologic and bioelectrical characteristics of the neural retina in diabetes
DR is characterized by a long period of clinical silence without significant signs and symptoms. However, by the time worsening vision is experienced, pathology may have been significantly advanced. Visual electrophysiology including ERG and visual-evoked potentials (VEPs) can reveal the earliest sign of impairment of retinal and optic nerve function both in diabetic human and model animals [24–26]. Oscillatory potentials (OPs) relating to amacrine cells, GCs and Müller cells [27], are considered as sensitive indicators of DR in diabolic patients and model rats [28–30]. The clinicopathologic and bioelectrical characteristics of retina in diabetes are shown in Table 1.
Retinopathy in diabetes
NPDR stages in patients are determined by the number and severity of microaneurysms, dot-and-blot hemorrhages, hard exudates [31], cotton-wool spots, venous abnormalities, and intraretinal microvascular anomalies (IRMAs) [32]. PDR is a characteristic of neovascularization. The clinicopathologic features of the neural retina in NPDR and PDR are shown in Fig. 1. Clinically, there is a stage called “no apparent retinopathy”, which also can be labeled as NPDR grade 1. During this stage, there is no apparent morphological change. However, multifocal ERG (mfERG) technology reveals that functional changes are measurable in patients with diabetes without classic indicators of retinopathy [33, 34], suggesting that disfunction occurs before the appearance of morphological changes [35]. Local mfERG implicit times are significantly prolonged in the eyes of diabetic subjects without retinopathy [36]. In patients with type 1 diabetes of 3 months without retinopathy, VEP recordings show delayed P100 implicit time but with amplitudes similar to those of control subjects [37].
The neovascularization in PDR, which is initially intraretinal, usually breaks through the internal limiting membrane and lies between the membrane and the vitreous. Neovascularization is eventually accompanied by hemorrhage [38] and retinitis proliferation. Shrinkage of the fibroglial component often leads to neural retinal detachment. In mild background diabetic retinopathy, the reduced amplitudes of pattern electroretinogram (PERG) reveal the presence of cotton-wool spots and angiographic evidence of capillary nonperfusion [25], suggesting that PERG has certain advantages as a screening test when NPDR deteriorates to a PDR stage. Some study considers the amplitude of OPs as the prediction of the progression of eyes with NPDR or mild PDR to severe PDR, and the changes correlate with the grade of DR [28, 39]. The significantly reduced OP amplitudes indicate a high risk to develop proliferative diabetic retinopathy [28]. Moreover, the probability of regression curves based on lower OPs amplitudes, greater retinopathy severity, higher fluorescein leakage, and higher capillary nonperfusion can be used to support clinical decisions concerning the time to perform panretinal laser photocoagulation and how often to follow up [40].
Diabetic macular edema
Clinically diabetic macular edema (DME) is the most important single cause of vision impairment in diabetic patients. Morphologic evidence suggests that macular edema may be caused by functional damage to the retinal vascular endothelium, resulting from intracellular fluid collection in Müller cells. Excessive swelling and rupture or death of Müller cells produce pockets of fluid and cell debris. Extracellular fluid mainly in the outer plexiform and the inner nuclear layers of the central part of the retina may also result in macular edema [41], causing similar changes in adjacent neurons. mfERG may evaluate the macular function with high sensitivity [42, 43]. The response of the positive wave (P1) in macular and paramacular areas tends to decrease in latency and increase in amplitude 3 months after vitrectomy of diabetic macular edema [44]. Macular OPs are reduced in diabetic maculopathy, leaving the a- and b-waves intact, suggesting macular OPs can also be a sensitive indicator to assess the macular function of DME [45].
Diabetic optic neuropathy
Diabetes is a known risk factor for the development of ischemic optic neuropathy, particularly non-arteritic anterior ischemic optic neuropathy (NA-AION) and posterior ischemic optic neuropathy (PION) [46]. It is characterized by optic disc swelling caused by vascular leakage and axonal edema in and around the optic nerve head [47]. VEP supports the diagnosis of the AION, which causes optic disc edema in type-I diabetes [48]. Increased VEP latency is statistically correlated with the changes of the glucose level in the blood [26].
The role of inflammatory mediators and adhesion molecules in the pathogenesis of DR
A considerable body of evidence from animal models and patients shows that DR is a chronic low-grade inflammatory disorder with participation of inflammatory mediators [49, 50]. Cytokines, chemokines, adhesion molecules, prostaglandins, and inflammatory cells including macrophages and neutrophils participate in a complex chain of events [41, 51, 52]. The inflammatory responses of specific cell types in DR are shown in Table 2.
Vascular endothelial growth factor (VEGF) plays a fundamental role in angiogenesis, and its concentration in the vitreous in patients with DR is significantly increased [53]. Although initially considered as a vascular permeability factor, VEGF is recognized as an important contributor to the progression of DR. VEGF also promotes the expression of intercellular cell adhesion molecule-1 (ICAM-1) and initiates early diabetic retinal leukocyte adhesion [54, 55]. Leukocyte adhesion to the vascular endothelium is a necessary first step in DR, mediated by chemokines and adhesion molecules including monocyte chemoattractant protein-1 (MCP-1), chemokine (C-C motif) ligand 2 (CCL2), ICAM-1, and vascular cell adhesion molecule-1 (VCAM-1) [51, 56, 57]. The levels of ICAM-1, VCAM-1, and E-selectin in vitreous are significantly higher in eyes with PDR [58]. ICAM-1 and VCAM-1 are also upregulated in the conjunctiva of diabetic patients with or without retinopathy [51]. TNF-α, together with diabetic duration, remains a single, persistent, independent, and determinant inflammatory marker for PDR [59]. The levels of IL-6, IL-8, IL-5, and IL-10 in the vitreous of patients with PDR are also increased [60]. It appears that inflammatory mediators and adhesion molecules dominate the pathogenesis of DR. However, other mediators that closely related to inflammatory mediator may also be important in the pathophysiology of the disorder. Nuclear factor kappa B (NF-κB) activation was observed in epiretinal membranes of patients with PDR, and selective inhibition of NF-κB reduces the expression of ICAM-1 and VEGF in vivo [61, 62]. Metalloproteinase (MMP)-9 in vitreous is elevated in diabetic patients with retinopathy [63]. Moreover, the level of high-mobility group box-1 (HMGB1) is increased in epiretinal membranes and vitreous fluid from patients with PDR and in the diabetic retina, suggesting HMGB1 is involved in inflammatory and angiogenic signaling pathways in diabetic retina through its putative receptor termed receptor for advanced glycation end products (RAGE) [64].
The effect of inflammation on diabetic neurosensory retina
Retina glial cells in neuroinflammation
Retinal glia are classified into two groups: macroglia (Müller cells and astrocytes) and microglia. Each glial subtype differs markedly in distribution, morphology, and pathophysiology. Some studies reported that glial fibrillary acidic protein (GFAP), a glial cell marker, is a sensitive indicator of CNS injury. GFAP is increased in glial cells in patients after 1 to 3 months of uncontrolled diabetes, with pathological potential reduction after longer durations of the disease [65, 66]. Pathological changes in retina glial cells in DR are shown in Table 3.
Müller cells
Müller cells are the major glial cell type in mammalian retina, which span the entire depth of the neural retina. Müller cell somata are located in the inner nuclear layer (INL), from which two major trunks extend in opposite directions. The outer trunk forms a network of adherent junctions known as the outer limiting membrane between Müller cells and photoreceptors [67]. In vascularized retina, the end feet contact and surround blood vessels within the retina. The secondary processes branching from the main trunk of Müller cells form extensive sheaths that surround neuronal cell bodies, dendrites, and the axons of ganglion cells [68]. In the normal retina, Müller cells limit the spread of excitatory neurotransmitters such as glutamate, provide metabolic support for a subset of inner retinal cells, and maintain the stability of the extracellular environment [68, 69]. In diseases, Müller cells possess a marked capacity to respond to a wide variety of environmental insults with pathophysiologic and biosynthetic changes [69].
The density of Müller cells is significantly increased at 4 weeks of diabetic rats. The expression of GFAP in Müller cells is not detectable at 4 weeks (early stage) but the expression becomes prominent at 12 weeks. It is noteworthy that hyperplasia of Müller cells precedes GFAP overexpression in the diabetic retina [70]. On electron microscopy, Müller cells in diabetic rats exhibit dispersion of nuclear chromatin and electrondense nuclear granulations, with the presence of increased glycogen, dense bodies, and lysosomes in the cytoplasm [71]. Diabetic retina shows edematous Müller cell end feet in the nerve fiber layer, ganglion cell loss, intercellular space increase in the inner and outer nuclear layers, and outer retina degeneration due to apoptotic cell death as a result of overexpression of caspase-3 [72]. Müller cells are major sources of inflammatory mediators [73] and become “activated” or “reactive” in response to virtually all pathological changes in the retina [74]. By using high-throughput techniques, diabetes-induced alteration of gene expression profile in Müller cells reveals that among 78 altered genes, one third are associated with inflammation [75], suggesting that Müller cells contribute to inflammatory responses during the development of DR. VEGF is rapidly released from Müller cells in early DR, enhancing perfusion by locally increased permeability of blood vessels with concomitant decrease in anti-angiogenic pigment epithelium-derived factor [76, 77]. In VEGF knockout mice, diabetes-induced retinal inflammation, vascular leakage, and vascular degeneration exhibit a significant reduction [76]. In Müller cells cultured in high glucose, the levels of histone acetylation at histone H3 (AcH3K9), AcH3K18, AcH2BK5, and AcH4K8 are increased, with upregulated mRNA of inflammatory genes, such as VEGFR1, IL1-β, ICAM-1, TNF-α, and MCP-1 (CCL2) [78]. These findings suggest that elevation of histone acetylations in Müller cells plays an important regulating role in the inflammatory response under diabetic conditions. The expression of VEGF and pigment epithelium-derived factor (PEDF) in Müller cells is disregulated in high glucose concentration, which contributes to retinal neovascularization in DR [79]. The anti-angiogenic P60, a PEDF derivative, reduces vascular leakage by increasing tight junction proteins in retina vessels through Müller glia signaling and by reducing the levels of inflammatory cytokines that promote vessel abnormalities. The neuroprotective P78, another PEDF derivative, is more effective in the prevention of cell dropout and inner plexiform layer (IPL) thinning with reduction of vitreous levels of TNF-α and IL-2 and activation of the PI3K/AKT pathway in Müller glia [80].
In an STZ-induced diabetic mouse model, disruption of β-catenin in Müller cells attenuates the overexpression of inflammatory cytokines and ameliorates pericyte dropout in the retina. Thus, Müller cell-derived β-catenin is an important contributor to retinal inflammation in DR, and the Wnt/β-catenin pathway is activated in DR model mice [81].
Müller cells produce IL-1 and exert an inhibitory activity on Ag- and IL-2-driven proliferation of T helper cell lines. Under conditions where the inhibitory capacity of Müller cells is suppressed, the cells display APC function to show a dual effect on autoimmune T helper lymphocytes [82]. Müller cells have been reported to produce increased amount of IL-1β when exposed to high glucose in vitro [83], in which caspase-1/IL-1β signaling plays an important role in diabetes-induced retinal pathology [19]. IL-1β has also been reported to induce IL-6 production by Müller cells predominantly through the activation of p38 MAPK/NF-κB signaling pathway [74].
Studies of our laboratory have shown that hyperglycemia induced the overexpression and activation of HMGB1 in Müller cells. HMGB1 mediates toll-like receptor 4 (TLR4)-dependent angiogenesis [84]. The expression of TLR4 was markedly increased in fibrovascular membranes from DR patients and in retinal vascular endothelial cells of diabetic mice [85]. We therefore speculate that Müller cells are involved in inflammation-driven angiogenesis.
Retinal Müller cells (rMC-1) cultured in high glucose increase their production of nitric oxide (NO) and prostaglandin E2 (PGE2) as well as the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2. In vitro results suggest that hyperglycemia-induced increase in NO in retinal Müller cells promotes the production of cytotoxic prostaglandins via COX-2. iNOS appears to account for the increased production of NO by Müller cells [86].
Exposure of Müller cells to high glucose also induces their expression of RAGE and S100B. RAGE signaling via MAPK pathway was linked to cytokine production. Blockade of RAGE prevents cytokine production induced by high glucose and S100B in Müller cells [87].
Müller cells regulate the level of substances in the neuronal microenvironment. One of the most characterized functions of Müller cells is the regulation of K+ in the retina [88]. The accumulation of K+ in extracellular space leads to changes in neuronal excitability. Müller cells may also control neuronal activity more directly. When sufficiently depolarized, glutamate uptake by salamander Müller cells is reversed and glutamate is released into extracellular space [89]. Additionally, glycogen stores in the retina are restricted to Müller cells. Furthermore, Müller cells also regulate blood flow in retinal vessels in response to the changes in neuronal activity.
Astrocytes
Astrocytes are the primary glia in the brain, constituting approximately one third of the brain mass [90]. Astrocytes in the retina show a stellate morphology, with somata located in the ganglion cell layer and nerve fiber layer (NFL). In the monkey retina, GFAP-positive astrocytes are found ubiquitously in the NFL. Astrocytes are absent in avascular foveal region. The concurrence of retinal astrocytes and intraretinal vascularization may be a common feature for many mammalian species [91]. Despite the fact that astrocytes are far less pervasive in the retina than in the brain, these cells play an important role in the development and maintenance of retinal neurons and blood vessels. They provide energy substrates to neurons and regulate the production of trophic factors and antioxidants in retinal microenvironment [92].
Astrocytes show opposite reactions as compared with Müller cells in response to hyperglycemia. The density of Müller cells is increased, whereas the number of astrocytes is decreased in diabetic retinas. In 4-week diabetic rat retina, astrocyte density is significantly reduced in the peripapillary region and in the far periphery [70]. Astrocytic profiles, notably the processes investing axonal bundles, are scanty in rat diabetic tissue, and the starlike cell bodies are irregularly distributed [70]. In addition, recent study demonstrates that exosomes from retinal astrocytes contain multiple anti-angiogenic components that inhibit laser-induced choroidal neovascularization in model mice [93].
Astrocytes are the major cell population in the optic nerve head and are responsible for the remodeling of the lamina cribrosa structure [94]. Astrocytes are important in stress over-activation of inflammatory responses in glaucoma that leads to local axonal damage within the optic nerve head [95]. Astrocytes have the potential to secrete a wide array of mediators [96]. COX-2 can be constitutively produced by astrocytes and is generally considered as an “immediate early response gene” following damage to the CNS [97]. As an acute phase gene, COX-2 is readily induced in a variety of cells by inflammatory and mitogenic stimuli, including cytokines and growth factors [98]. Overexpression of transforming growth factor-alpha (TGF-α) and epidermal growth factor receptor (EGFR) occurs in active astrocytes [99]. EGFR-dependent induction of COX-2 occurs early in astrocytes following optic nerve injury [100]. COX-2 and COX-2-induced PGE2 participate in DR and regulate the expression of VEGF [101]. In human diabetic retina, COX-2 is induced in astrocytes and contributes markedly to preretinal neovascularization in ischemic retinopathies. This effect appears to be PGE2-mediated mostly via prostaglandin E receptor 3 (EP3) implicating a new interaction through thrombospondin-1 (TSP-1) and CD36 [102].
IL-1β induces its own synthesis in the retinal vascular endothelial cells, Müller cells, and astrocytes. The combination of high glucose stimulation and the upregulation of IL-1β in the diabetic retina is responsible for sustained IL-1β overexpression in astrocytes [103].
Microglia
Microglia are bone marrow-derived mononuclear phagocytes, representing the major component of the innate immune cells in the retina [104]. Like macrophages in the rest of the body, microglia use phagocytic and cytotoxic mechanisms to destroy foreign materials. However, microglia differ from macrophages in that they are much more tightly regulated spatially and temporally to maintain proper immune responses in the eye. The size of microglia is small relative to macroglia (such as astrocytes), with changing shapes and oblong nuclei. Microglia together with invading choroidal macrophages significantly contribute to chronic para-inflammation present in several aging retinal pathologies [105].
Microglia in human diabetic retinopathy
Microglia settle into the plexiform layers of the retina and gain a highly branched morphology with small cell bodies and long protrusions that may span the complete nuclear layers [106]. In human DR, perivascular microglia in the background form are moderately increased in numbers and are hypertrophic in the inner retinal layers, extending from internal to middle limiting membranes. Hypertrophic microglia in the preproliferative form cluster around cotton-wool spots and infiltrate into optic nerve region. Dilated new vessels in proliferative retinopathy are heavily surrounded by microglia, featuring microglial perivasculitis [107].
Microglia in animal models of diabetic retinopathy
Retinal microglia are activated, and the morphology is changed at 4–8 weeks of animal diabetic models [70, 108, 109]. The number of microglia is increased in the outer plexiform layer at 4-month diabetic models. Reactive microglia at 14 to 16-month diabetic models are detected in the outer nuclear and photoreceptor layer [110]. Active Iba1-positive microglia with retracted and swollen processes are present in insulin-2 Akita (Ins2Akita/t) mice after 8 weeks of hyperglycemia [111].
Minocycline, an antibiotic that inhibits microglia, decreases diabetes-induced inflammatory cytokine production and reduces the release of cytotoxins from activated microglia as well as the activity of caspase-3 in rodent retina [112]. Therefore, activated microglia are considered as a major source of proinflammatory and neurotoxic mediators. These cells are also recognized as a potential culprit contributing to the early inflammatory outcome in DR [107, 113].
Advanced glycation end products (AGEs) may act directly on microglia to initiate DR and promote its advancement. AGEs increases the expression of TNF-α in cultured rat retinal microglia, thereby trigging infiltration of leukocytes to the site of vascular injury and causing vascular inflammation [114]. Increased levels of AGEs also lead to the formation of reactive oxygen species (ROS) and ERK/P38 activation during microglial activation in diabetes [109, 115]. Inhibition of the production of NO and other free radicals by glial cells with intracellularly acting antioxidants may imply their ability to reduce AGE-induced neuroinflammatory processes [114]. Identification of the redox-active signal transduction pathways involved in microglial activation and the chemical structures of the responsible AGEs and AGE receptors/binding proteins will provide additional molecular targets for the treatment of AGE-associated inflammatory conditions [114].
NF-κB is activated in pericytes, vascular endothelial cells, macrophages, and microglia in hypoxia-induced C57BL/6N mouse model of neovascularization [116]. NF-κB activation is required for retinal angiogenesis and inhibition of NF-κB ameliorates neuronal cell death in PDR [116]. It is well known that IL-1, IL-6, IFN-γ, and TNF-α activate microglia in vitro [117]. STZ induces a rapid and sustained increase in glycemia and causes microglial activation along with increased levels of TNF-α and IL1-β during a very short period of time [118]. In STZ-induced diabetic rats, TNF-α colocalizes with ionized calcium binding adaptor molecule-1 (Iba-1+) in microglia but not in Müller cells or astrocytes. TNF-α production induced by glycated albumin was blocked by ERK and p38 MAPK inhibitors [119]. Molecules released by activated retinal microglia include glutamate, proteases, leukotrienes, IL-1β, IL-3, IL-6, TNF-α, VEGF, lymphotoxin, macrophage inflammatory protein 1 (MIP-1), and MMPs [120]. Purinergic P2 receptors in high glucose-cultured rat microglia are upregulated, eliciting calcium influx and release of proinflammatory mediators [121]. Furthermore, in Ins2Akita/t mice, a PEDF peptide PEDF78-121 (P78) is effective in preventing cell dropout and IPL thinning, presumably due to its inhibition of microglia activation. P78 also inhibits the activation of PI3K/AKT pathway in Müller glia and reduces vitreous levels of TNF-α and IL-2 in vitro [80].
Neurons in neuroinflammation
Neurons in retina include photoreceptors, bipolar cells, amacrine cells, and retinal ganglion cells (RGCs). Diabetes affects both neurites (axons and dendrites) and cell bodies of retinal neurons, as evidenced by neuritic swellings [122].
RGCs
Diabetes impairs axonal retrograde transport in large- and medium-sized RGCs in type 1 but not type 2 diabetic rats [123]. The total number of RGC bodies is reduced in type 2 diabetic rats [14]. There is a reduction in the overall thickness of inner layers of the retina, accompanied by diminished number of RGCs in rat retinas after long term experimental diabetes [124]. After 22 weeks of hyperglycemia, there is a 23.4 % reduction in the number of cell bodies in the RGC layer in Ins2Akita mice [125]. In diabetic patients, the number of RGCs is also reduced [124, 126]. Consistent with this, scanning laser polarimetry revealed a reduced thickness of the nerve fiber layer in diabetic patients [127]. The morphology of a subset of RGCs is altered in diabetes, including axonal swellings with associated constriction, enlarged cell bodies, and increased dendritic branches and terminals [127].
The proteinase-activated receptor-2 (Par2) is recognized for its marked proangiogenic properties in the retina [128]. Par2 mRNA in cultured retinal neuronal cells (RGC-5) is increased by IL-1β [129]. Par2 stimulation activates several downstream effector events, including Ca2+ mobilization and MAPK [130, 131]. RGC-5 cells treated with SLIGRL exhibit increased MAPK signaling, including Erk1/2, Jnk, and p38 phosphorylation [129].
Regeneration of injured RGCs is supported by Müller cell-derived neurotrophic/protective factors [132]; among those are VEGF [133], ciliary neurotrophic factor (CNTF) [134], and PEDF [135]. PEDF activates NF-κB in RGC. Addition of NF-κB inhibitor (SN50) to PEDF-treated RGC reduces their survival. Thus, NF-κB activation in RGC is critically involved in the effect of Müller cell-derived PEDF on maintaining neuronal survival [136].
Recent research demonstrates that hyperglycemia causes succinate accumulation and G protein-coupled receptor 91 (GPR91) activation in RGC, which mediate VEGF-induced retinal vascular change via the ERK1/2/COX-2/PGE2 pathway [137]. Proinflammatory and proapoptotic thioredoxin-interacting protein (TXNIP) has a causative role in the development of diabetes [138, 139]. TXNIP expression is increased in the brain of diabetic rats [140] and plays a role in RGC injury in glaucoma [141, 142]. Blocking the expression of TXNIP in diabetic rat retinas results in the inhibition of its target genes COX-2 and FN thus demonstrating TXNIP’s role in aberrant gene induction in early DR. RNAi silencing TGS of TXNIP abolishes diabetes-induced retinal gliosis and ganglion injury [143].
Other neurons
Amacrine cells are the third-order retinal interneurons, projecting their processes into the IPL and contribute to the most of the synapses in the inner plexiform layer and mediate visual information input from bipolar cells onto retinal ganglion cells [144]. Mammalian AII retinal amacrine cells are arrow-field, multistratified glycinergic neurons best known for its capacity to collect scotopic signals from rod bipolar cells and distribute the signals to ON and OFF cone pathways across the network [145].
There are three classes of photoreceptors: rods, long-wave system (LWS) cones, and short-wave system 1 (SWS1) cones. Each class displays a distinct morphology as well as visual pigment. Because photoreceptors are especially vulnerable to hypoxia [146], diabetes may also affect the function of photoreceptors. It has been reported that there are foveal cone photopigment bleaching abnormalities in patients with diabetes [147]. There are decreases in the sensitivity parameter (log S) for both rod-isolated and cone-isolated ERG a-wave responses in patients with DR. Moreover, rod and cone b-wave changes in DR patients, including changes in both amplitude and implicit time [148]. However, the function of these neurons in DR remains unclear.
Immuno-inflammatory response in DR
It has recently been recognized that the pathology of diabetic retinopathy has strong immunological underpinnings [149]. T cell abnormalities are believed to be the major cause of autoimmune disease in type 1 diabetes, leading to the destruction of pancreatic islets. In type 2 diabetes, inflammation and activation of monocytes are important for enhancing insulin resistance and may contribute to the loss of insulin secretory function of islet cells [150]. In many diabetic complications, there is dysregulation of innate immunity associated with increased inflammatory responses [151]. Improper activation of the innate immune system may result in DR. TLR4 is an important mediator of innate immunity, and genetic alterations of TLR4 is associated with inflammation in the hyperglycemic condition [152].
Resident microglia are regarded as immunological watchdogs in the brain and retina. These cells are active sensors of neuronal microenvironment and rapidly respond to insults with morphological and functional transformation into reactive phagocytes [153]. Inflammation in diabetes activates microglia, stimulates a cascade of inflammation that recruits leukocytes, causes vascular breakdown, and directly induces glial dysfunction and neuronal cell death through the release of cytotoxic substances. Increased levels of sICAM-1 and sVCAM-1 as well as high concentrations of vitreous IL-6 and TNF-α in patients with PDR appear to confirm the inflammatory-immune nature of PDR [154].
DR is a result of systemic neuroinflammation
CNS inflammation in diabetes
Diabetes causes chronic inflammatory complications in the peripheral and CNS. Amylin deposition is promoted by chronic hyperamylinemia, which is common in humans with pre-diabetic insulin resistance. The majority of patients with T2DM have abundant amylin amyloid deposition in the pancreas [155]. A recent study indicates that chronic hyperamylinemia promotes the accumulation of oligomerized amylin in the brain, which may trigger inflammatory responses and lead to neurological defects [156]. β-amyloid deposition around brain microvessels can cause direct toxicity to microvascular endothelial cells (BMVECs). Impaired clearance of β-amyloid across the blood brain barrier (BBB), aberrant angiogenesis, and senescence of the cerebrovascular system may initiate neurovascular uncoupling, brain hypoperfusion, and neurovascular inflammation [157].
T1DM is also associated with increased expression of proinflammatory mediators, such as IL-1β, IL-2, IL-6, TNF-α, and NF-κB, compared to age matched control brains [21, 158]. In addition, TNF-α and IL-1β induce COX-2 activity in perivascular macrophages of BBB and generate prostaglandin E2, which enters the brain and stimulates paraventricular nucleus (PVN) neurons to release adrenocorticotropic hormone (ACTH). Increased expression of Ang II, ICAM-1, lymphocyte function-associated antigen-1 (LFA-1) and CD8 positive cells are found in diverse zones of the cerebrum and cerebellum in STZ-induced diabetic rats [159]. Local Ang II increases vascular permeability by promoting the secretion of VEGF [160]. Ang II also contributes to the recruitment of inflammatory cells into tissues by stimulating the production of cytokines and chemokines.
Systemic inflammation in diabetes
Connections of neuropathy with the bone marrow (BM), CNS, and peripheral nervous system may exist. Systemic hyperglycemia-induced inflammation in diabetes may result in BM neuropathy by enhancing the generation of inflammatory cells and lead to vascular complications such as DR by reducing the production of endothelial progenitors, which maintain the endothelial function and renewal. [21]. Endothelial progenitor cells (EPCs) arising from BM circulate in the bloodstream and traffic to areas of injury to orchestrate vascular repair [161, 162]. Diabetic individuals have fewer EPCs in the circulation with decreased migratory and reparative potential. Acellular capillaries in the retina in type 2 diabetic rats are observed at the precise time when there is denervation of the BM and reduction in peripheral clock gene expression. The resultant acellular capillaries appear at 4 months of diabetes, due to the loss of proper EPC reparative function and the failure of circadian EPC release secondary to diabetes-associated denervation of BM [163]. Therefore, BM neuropathy precedes the development of DR. The decrease in circulating EPCs reduces the repair of injured retinal vessels in diabetes and leads to the development of acellular capillaries.
BM-derived cells such as leukocytes play a critical role in the development of diabetic retinopathy in animals [164]. Diabetes-induced inflammatory changes, superoxide production, and degeneration of retinal capillaries are inhibited in diabetic mice in which inflammatory proteins (iNOS and PARP-1) are deleted from BM cells [165].
Intervention of neuroinflammation in DR
Pharmacologic interventions are available to reduce neural inflammatory response in patients with DR [21], in particular in patients who fail to respond to anti-VEGF therapy. Neuroprotection as a new approach to the treatment of early stage DR has been emphasized. Neuroprotection effect is based on administering natural protective factors that may downregulate inflammatory responses in the diabetic retina. The factors such as pigment epithelial growth factor, somatostatin, corstistatin, and neurotrophins are abundant in the physiological retinas. Therefore, administration of these factors could be considered as replacement treatments [166].
Hesperetin
Hesperetin (3′,5,7-trihydroxy-4-methoxyflavanone) is a member of the flavanone subclass of flavonoids. It is a potential anti-inflammatory agent with potent inhibition of LPS-induced expression of the COX-2 gene in RAW 264.7 macrophage cell line [167]. Hesperetin also inhibits the appearance of oxidative stress biomarkers, such as thiobarbituric acid-reactive substance (TBARS) and carbonyl content. Moreover, hesperetin activates catalase and total superoxide dismutase (SOD) in mice [168]. Thus, hesperetin may be neuroprotective as shown by the fact that hesperetin-treated retina reduces the expression of caspase-3, GFAP, and AQP4, which are increased in diabetic rat retina [72].
Minocycline and doxycycline
Minocycline (MINO) and doxycycline (DOXY) derived from tetracycline show neuroprotection in animal models of ischemia [169–171]. MINO exerts anti-inflammatory effect on microglia by inhibiting the production of inflammatory mediators, such as NO, cyclooxygenases, prostaglandins, IL-1β, and TNF-α, while DOXY downregulates NO and IL-1β [169, 172–174]. MINO inhibits hyperglycemia-induced histone acetylations, Müller cell activation and upregulation of inflammatory mediators [175]. MINO also inhibits the formation of acellular capillaries in the retina of diabetic and galactosemic mice. The activation of caspase-1 and caspase-3 by high glucose and subsequent neuronal apoptosis in retina Müller cells and microglia are also inhibited by MINO [19, 112, 176]. In the clinic, MINO improves visual acuity and neuropathic pain resulted from inflammation in diabetic patients [21]. These findings support that MINO is a novel promising therapeutic drug for DR [21].
VEGF
VEGF is an angiogenic and vessel-permeability factor [177]. Anti-VEGF therapy in the management of PDR and DME has shown beneficial effects [178, 179]. Antibodies Ranibizumab (Lucentis®) [180, 181], Bevacizumab (Avastin®) [182, 183], and Aflibercept (Eylea®) [184], which inhibit VEGF isoforms, are currently used in the clinic. Most studies focus on inhibition of vascular permeability and endothelial cell proliferation stimulated by VEGF [185]. However, further research suggests that VEGF also has neurotrophic and neuroprotective activity [186]. The hypoxia-induced neuroprotective effects sequentially require the activation of VEGF/VEGFR-2 and Akt/PKB phosphorylation, indicating that VEGF is a hypoxia-induced neurotrophic factor [187]. VEGF also has a protective effect on hippocampal neurons against glutamate-mediated toxicity, and this effect is dependent on PI3-K/Akt and MEK/ERK signaling pathways mediated primarily through Flk-1 receptor [188].
Therefore, VEGF may have a dual role: neuroprotection and neovascularization in hypoxic regions of the tissues. Its effect on angiogenesis and vascular permeability appears paradoxical versus the neuroprotective activation [14]. Patients who failed in anti-VEGF therapy may be due to inhibition of its neuroprotective function. Moreover, diabetic patients may be at higher risk for both systemic and ocular complications, such as cardiovascular and renal diseases, susceptibility to infection, endophthalmitis, retinal detachment, and intraocular hemorrhage [189–192]. Thus, although anti-VEGF therapy is helpful, complications of this treatment should not be overlooked.
Other novel therapeutic medicine
There are novel therapies focusing on inflammation and neurodegeneration to mitigate retinal damage associated with diabetes. Cannabidiol (CBD) is a non-psychoactive component considered to have the properties of anti-inflammation and anti-oxidation [193, 194]. CBD attenuates high glucose-induced NF-κB activation in human coronary endothelial cells (HCAECs) in vitro. It also attenuates high glucose-induced iNOS expression and 3-nitrotyrosine (3-NT) formation in endothelial cells [195]. Moreover, CBD decreases the incidence of diabetes possibly through an immunomodulatory mechanism that induces regulatory Th2 responses [196]. It is reported that CBD reduces neurotoxicity, inflammation, and BRB breakdown in STZ-induced diabetic rats by blocking activation of microglia and p38 MAP kinase, a downstream molecule of proinflammatory cytokines and oxidative stress [197]. Thus, CBD is a promising candidate for anti-inflammatory and neuroprotective therapy for DR.
Resveratrol is a natural polyphenol found in grapes and red wine. Resveratrol has protective effects on atherosclerosis and cardiovascular diseases through reduction in oxidative stress [198–200]. Further research demonstrates that resveratrol protects diabetic neuropathy by improving motor nerve conduction velocity and nerve blood flow, as well as reduction in nociception [199]. A recent study indicates that resveratrol inhibits the activation of NF-κB and TNF-α and reduces apoptotic cells in the retina of type 2 diabetic rats [201]. Resveratrol also exerts its neuroprotective effect on RGCs by activating the sirtuin 1 pathway in an optic nerve transection rat model [202].
The non-steroidal anti-inflammatory drugs (NSAIDs) have been used to treat DME by inhibiting prostaglandin biosynthesis [203, 204]. Injection of intravitreal diclofenac (IVD) sodium, as a potent NSAIDs, has been used in the treatment of macular edema of many etiologies such as uveitic CME, diabetic macular edema, and retinal vein occlusions [203]. A randomized double-masked clinical trial demonstrated that the effect of injection of IVD was superior to intravitreal injection of bevacizumab (IVB) in the treatment of naïve DME [205]. Therefore, using IVD as an adjunct or alternative treatment may enhance the functional outcome of naive DME.
Conclusions
DR manifests characteristics of chronic neuroinflammation. Neurosensory retina including retina glial cells and neurons are involved in neuroinflammatory responses of DR. A caveat that should be kept in mind is that most DR pathogenic studies are conducted on animals, but none of the animal models may replicate all features of human disease possibly due to different anatomic characteristics of retinal structure. Nevertheless, animal models remain necessary tools to study the pathogenesis of diseases and have provided useful information for better understanding of the pathogenesis of neuroinflammation in DR. Many features simulate the process of human disease and may aid the development of more efficient therapeutic strategies against human DR.
Abbreviations
- AGE:
-
Advanced glycation end products
- AION:
-
anterior ischemic optic neuropathy
- BM:
-
bone marrow
- CNS:
-
central nervous system
- COX:
-
cyclooxygenase
- DME:
-
diabetic macular edema
- DOXY:
-
doxycycline
- DR:
-
diabetic retinopathy
- ERG:
-
electroretinogram
- GFAP:
-
glial fibrillary acidic protein
- HMGB1:
-
high-mobility group box-1
- ICAM-1:
-
intercellular cell adhesion molecule-1
- IL:
-
interleukin
- IRMAs:
-
intraretinal microvascular anomalies
- MINO:
-
minocycline
- NF-κB:
-
nuclear factor kappa B
- OPs:
-
oscillatory potentials
- PION:
-
posterior ischemic optic neuropathy
- RGCs:
-
retinal ganglion cells
- T2DM:
-
type 2 diabetes mellitus
- TNF-α:
-
tumor necrosis factor-alpha
- VCAM-1:
-
vascular cell adhesion molecule-1
- VEGF:
-
vascular endothelial growth factor
- VEPs:
-
visual-evoked potentials
References
Klein BE. Overview of epidemiologic studies of diabetic retinopathy. Ophthalmic Epidemiol. 2007;14:179–83.
Yau JW, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35:556–64.
Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–53.
Stitt AW, Lois N, Medina RJ, Adamson P, Curtis TM. Advances in our understanding of diabetic retinopathy. Clin Sci (Lond). 2013;125:1–17.
Roy S, Tonkiss J, Roy S. Aging increases retinal vascular lesions characteristic of early diabetic retinopathy. Biogerontology. 2010;11:447–55.
Li W, Yanoff M, Liu X, Ye X. Retinal capillary pericyte apoptosis in early human diabetic retinopathy. Chin Med J (Engl). 1997;110:659–63.
Yanoff M, Sassani JW. Ocular pathology. 6th ed. Edinburgh: Mosby; 2009.
Ashton N. Vascular basement membrane changes in diabetic retinopathy. Montgomery lecture, 1973. Br J Ophthalmol. 1974;58:344–66.
Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31:425–45.
Tyrberg M, Lindblad U, Melander A, Lovestam-Adrian M, Ponjavic V, Andreasson S. Electrophysiological studies in newly onset type 2 diabetes without visible vascular retinopathy. Doc Ophthalmol. 2011;123:193–8.
Juen S, Kieselbach GF. Electrophysiological changes in juvenile diabetics without retinopathy. Arch Ophthalmol. 1990;108:372–5.
Lopes de Faria JM, Russ H, Costa VP. Retinal nerve fibre layer loss in patients with type 1 diabetes mellitus without retinopathy. Br J Ophthalmol. 2002;86:725–8.
Lieth E, Gardner TW, Barber AJ, Antonetti DA, Penn State Retina Research G. Retinal neurodegeneration: early pathology in diabetes. Clin Experiment Ophthalmol. 2000;28:3–8.
Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:283–90.
Jackson GR, Barber AJ. Visual dysfunction associated with diabetic retinopathy. Curr Diab Rep. 2010;10:380–4.
Lontchi-Yimagou E, Sobngwi E, Matsha TE, Kengne AP. Diabetes mellitus and inflammation. Curr Diab Rep. 2013;13:435–44.
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801.
Rajab HA, Baker NL, Hunt KJ, Klein R, Cleary PA, Lachin J, Virella G, Lopes-Virella MF, Investigators DEGo. The predictive role of markers of Inflammation and endothelial dysfunction on the course of diabetic retinopathy in type 1 diabetes. J Diabetes Complications. 2015;29:108–14.
Vincent JA, Mohr S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes. 2007;56:224–30.
Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–52.
Yellowlees Douglas J, Bhatwadekar AD, Li Calzi S, Shaw LC, Carnegie D, Caballero S, et al. Bone marrow-CNS connections: implications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res. 2012;31:481–94.
Morgan JL, Dhingra A, Vardi N, Wong RO. Axons and dendrites originate from neuroepithelial-like processes of retinal bipolar cells. Nat Neurosci. 2006;9:85–92.
Albert DM, Miller JW, Azar DT. Albert & Jakobiec’s principles and practice of ophthalmology. 3rd ed. Philadelphia: Saunders/Elsevier; 2008.
Li Q, Zemel E, Miller B, Perlman I. Early retinal damage in experimental diabetes: electroretinographical and morphological observations. Exp Eye Res. 2002;74:615–25.
Arden GB, Hamilton AM, Wilson-Holt J, Ryan S, Yudkin JS, Kurtz A. Pattern electroretinograms become abnormal when background diabetic retinopathy deteriorates to a preproliferative stage: possible use as a screening test. Br J Ophthalmol. 1986;70:330–5.
Ghita AM, Parvu D, Sava R, Georgescu L, Zagrean L. Electrophysiological changes in optic neuropathy of streptozotocin induced diabetic rats. J Med Life. 2013;6:340–8.
Wachtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res. 1998;17:485–521.
Bresnick GH, Korth K, Groo A, Palta M. Electroretinographic oscillatory potentials predict progression of diabetic retinopathy. Preliminary report Arch Ophthalmol. 1984;102:1307–11.
Hancock HA, Kraft TW. Oscillatory potential analysis and ERGs of normal and diabetic rats. Invest Ophthalmol Vis Sci. 2004;45:1002–8.
Vadala M, Anastasi M, Lodato G, Cillino S. Electroretinographic oscillatory potentials in insulin-dependent diabetes patients: a long-term follow-up. Acta Ophthalmol Scand. 2002;80:305–9.
Jeon S, Lee WK. Effect of intravitreal bevacizumab on diabetic macular edema with hard exudates. Clin Ophthalmol. 2014;8:1479–86.
Bresnick GH, Davis MD, Myers FL, de Venecia G. Clinicopathologic correlations in diabetic retinopathy. II. Clinical and histologic appearances of retinal capillary microaneurysms. Arch Ophthalmol. 1977;95:1215–20.
Bronson-Castain KW, Bearse Jr MA, Neuville J, Jonasdottir S, King-Hooper B, Barez S, et al. Early neural and vascular changes in the adolescent type 1 and type 2 diabetic retina. Retina. 2012;32:92–102.
Chan HH, Chu PH, Lung JC, Ho WC, Ting PW, Sum RW, et al. Detection of early functional changes in diabetic retina using slow double-stimulation mfERG paradigm. Br J Ophthalmol. 2011;95:1560–3.
Lung JC, Swann PG, Wong DS, Chan HH. Global flash multifocal electroretinogram: early detection of local functional changes and its correlations with optical coherence tomography and visual field tests in diabetic eyes. Doc Ophthalmol. 2012;125:123–35.
Bearse Jr MA, Adams AJ, Han Y, Schneck ME, Ng J, Bronson-Castain K, et al. A multifocal electroretinogram model predicting the development of diabetic retinopathy. Prog Retin Eye Res. 2006;25:425–48.
Parisi V, Uccioli L. Visual electrophysiological responses in persons with type 1 diabetes. Diabetes Metab Res Rev. 2001;17:12–8.
Levin AV. Retinal hemorrhages: advances in understanding. Pediatr Clin North Am. 2009;56:333–44.
Aylward GW. The scotopic threshold response in diabetic retinopathy. Eye (Lond). 1989;3(Pt 5):626–37.
Bresnick GH, Palta M. Predicting progression to severe proliferative diabetic retinopathy. Arch Ophthalmol. 1987;105:810–4.
Ascaso FJ, Huerva V, Grzybowski A. The role of inflammation in the pathogenesis of macular edema secondary to retinal vascular diseases. Mediators Inflamm. 2014;2014:432685.
Brodie SE, Naidu EM, Goncalves J. Combined amplitude and phase criteria for evaluation of macular electroretinograms. Ophthalmology. 1992;99:522–30.
Bearse Jr MA, Ozawa GY. Multifocal electroretinography in diabetic retinopathy and diabetic macular edema. Curr Diab Rep. 2014;14:526.
Ma J, Yao K, Jiang J, Wu D, Gao R, Yin J, et al. Assessment of macular function by multifocal electroretinogram in diabetic macular edema before and after vitrectomy. Doc Ophthalmol. 2004;109:131–7.
Miyake Y. Macular oscillatory potentials in humans. Macular OPs. Doc Ophthalmol. 1990;75:111–24.
Reddy D, Rani PK, Jalali S, Rao HL: A study of prevalence and risk factors of diabetic retinopathy in patients with non-arteritic anterior ischemic optic neuropathy (NA-AION). Semin Ophthalmol 2013.
Giuliari GP, Sadaka A, Chang PY, Cortez RT. Diabetic papillopathy: current and new treatment options. Curr Diabetes Rev. 2011;7:171–5.
Brogelli S, Valentini G. Anterior ischemic optic neuropathy in type I diabetes. Metab Pediatr Syst Ophthalmol. 1986;9:90–3.
Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86:363–5.
Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–2.
Khalfaoui T, Lizard G, Ouertani-Meddeb A. Adhesion molecules (ICAM-1 and VCAM-1) and diabetic retinopathy in type 2 diabetes. J Mol Histol. 2008;39:243–9.
Grant MB, Afzal A, Spoerri P, Pan H, Shaw LC, Mames RN. The role of growth factors in the pathogenesis of diabetic retinopathy. Expert Opin Investig Drugs. 2004;13:1275–93.
Duh E, Aiello LP. Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes. 1999;48:1899–906.
Lu M, Perez VL, Ma N, Miyamoto K, Peng HB, Liao JK, et al. VEGF increases retinal vascular ICAM-1 expression in vivo. Invest Ophthalmol Vis Sci. 1999;40:1808–12.
Joussen AM, Poulaki V, Qin W, Kirchhof B, Mitsiades N, Wiegand SJ, et al. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160:501–9.
Lee WJ, Kang MH, Seong M, Cho HY. Comparison of aqueous concentrations of angiogenic and inflammatory cytokines in diabetic macular oedema and macular oedema due to branch retinal vein occlusion. Br J Ophthalmol. 2012;96:1426–30.
Sampson MJ, Davies IR, Brown JC, Ivory K, Hughes DA. Monocyte and neutrophil adhesion molecule expression during acute hyperglycemia and after antioxidant treatment in type 2 diabetes and control patients. Arterioscler Thromb Vasc Biol. 2002;22:1187–93.
Limb GA, Hickman-Casey J, Hollifield RD, Chignell AH. Vascular adhesion molecules in vitreous from eyes with proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 1999;40:2453–7.
Gustavsson C, Agardh E, Bengtsson B, Agardh CD. TNF-alpha is an independent serum marker for proliferative retinopathy in type 1 diabetic patients. J Diabetes Complications. 2008;22:309–16.
Koskela UE, Kuusisto SM, Nissinen AE, Savolainen MJ, Liinamaa MJ. High vitreous concentration of IL-6 and IL-8, but not of adhesion molecules in relation to plasma concentrations in proliferative diabetic retinopathy. Ophthalmic Res. 2013;49:108–14.
Harada C, Harada T, Mitamura Y, Quah HM, Ohtsuka K, Kotake S, et al. Diverse NF-kappaB expression in epiretinal membranes after human diabetic retinopathy and proliferative vitreoretinopathy. Mol Vis. 2004;10:31–6.
Nagai N, Izumi-Nagai K, Oike Y, Koto T, Satofuka S, Ozawa Y, et al. Suppression of diabetes-induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor-kappaB pathway. Invest Ophthalmol Vis Sci. 2007;48:4342–50.
Jin M, Kashiwagi K, Iizuka Y, Tanaka Y, Imai M, Tsukahara S. Matrix metalloproteinases in human diabetic and nondiabetic vitreous. Retina. 2001;21:28–33.
Abu El-Asrar AM, Mohammad G, Nawaz MI, Siddiquei MM. High-mobility group box-1 modulates the expression of inflammatory and angiogenic signaling pathways in diabetic retina. Curr Eye Res. 2014;1–12.
Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Invest Ophthalmol Vis Sci. 2000;41:3561–8.
Masser DR, VanGuilder Starkey HD, Bixler GV, Dunton W, Bronson SK, Freeman WM. Insulin treatment normalizes retinal neuroinflammation but not markers of synapse loss in diabetic rats. Exp Eye Res. 2014;125:95–106.
Coorey NJ, Shen W, Chung SH, Zhu L, Gillies MC. The role of glia in retinal vascular disease. Clin Exp Optom. 2012;95:266–81.
Newman E, Reichenbach A. The Muller cell: a functional element of the retina. Trends Neurosci. 1996;19:307–12.
Bringmann A, Reichenbach A. Role of Muller cells in retinal degenerations. Front Biosci. 2001;6:E72–92.
Rungger-Brandle E, Dosso AA, Leuenberger PM. Glial reactivity, an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2000;41:1971–80.
Schellini SA, Gregorio EA, Spadella CT, Machado JL, de-Moraes-Silva MA. Muller cells and diabetic retinopathy. Braz J Med Biol Res. 1995;28:977–80.
Kumar B, Gupta SK, Srinivasan BP, Nag TC, Srivastava S, Saxena R, et al. Hesperetin rescues retinal oxidative stress, neuroinflammation and apoptosis in diabetic rats. Microvasc Res. 2013;87:65–74.
Mizutani M, Gerhardinger C, Lorenzi M. Muller cell changes in human diabetic retinopathy. Diabetes. 1998;47:445–9.
Liu X, Ye F, Xiong H, Hu DN, Limb GA, Xie T, et al. IL-1beta Induces IL-6 production in retinal Muller cells predominantly through the activation of P38 MAPK/NF-kappaB signaling pathway. Exp Cell Res. 2014.
Gerhardinger C, Costa MB, Coulombe MC, Toth I, Hoehn T, Grosu P. Expression of acute-phase response proteins in retinal Muller cells in diabetes. Invest Ophthalmol Vis Sci. 2005;46:349–57.
Wang J, Xu X, Elliott MH, Zhu M, Le YZ. Muller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes. 2010;59:2297–305.
Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008;27:331–71.
Wang LL, Chen H, Huang K, Zheng L. Elevated histone acetylations in Muller cells contribute to inflammation: a novel inhibitory effect of minocycline. Glia. 2012;60:1896–905.
Mu H, Zhang XM, Liu JJ, Dong L, Feng ZL. Effect of high glucose concentration on VEGF and PEDF expression in cultured retinal Muller cells. Mol Biol Rep. 2009;36:2147–51.
Liu Y, Leo LF, McGregor C, Grivitishvili A, Barnstable CJ, Tombran-Tink J. Pigment epithelium-derived factor (PEDF) peptide eye drops reduce inflammation, cell death and vascular leakage in diabetic retinopathy in Ins2(Akita) mice. Mol Med. 2012;18:1387–401.
Zhou KK, Benyajati S, Le Y, Cheng R, Zhang W, Ma JX. Interruption of Wnt signaling in Muller cells ameliorates ischemia-induced retinal neovascularization. PLoS One. 2014;9, e108454.
Roberge FG, Caspi RR, Nussenblatt RB. Glial retinal Muller cells produce IL-1 activity and have a dual effect on autoimmune T helper lymphocytes. Antigen presentation manifested after removal of suppressive activity. J Immunol. 1988;140:2193–6.
Yego EC, Vincent JA, Sarthy V, Busik JV, Mohr S. Differential regulation of high glucose-induced glyceraldehyde-3-phosphate dehydrogenase nuclear accumulation in Muller cells by IL-1beta and IL-6. Invest Ophthalmol Vis Sci. 2009;50:1920–8.
He C, Sun Y, Ren X, Lin Q, Hu X, Huang X, et al. Angiogenesis mediated by toll-like receptor 4 in ischemic neural tissue. Arterioscler Thromb Vasc Biol. 2013;33:330–8.
Lin Q, Yang XP, Fang D, Ren X, Zhou H, Fang J, et al. High-mobility group box-1 mediates toll-like receptor 4-dependent angiogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1024–32.
Du Y, Sarthy VP, Kern TS. Interaction between NO and COX pathways in retinal cells exposed to elevated glucose and retina of diabetic rats. Am J Physiol Regul Integr Comp Physiol. 2004;287:R735–741.
Zong H, Ward M, Madden A, Yong PH, Limb GA, Curtis TM, et al. Hyperglycaemia-induced pro-inflammatory responses by retinal Muller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia. 2010;53:2656–66.
Newman EA. Acid efflux from retinal glial cells generated by sodium bicarbonate cotransport. J Neurosci. 1996;16:159–68.
Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–6.
Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–15.
Schnitzer J. Astrocytes in the guinea pig, horse, and monkey retina: their occurrence coincides with the presence of blood vessels. Glia. 1988;1:74–89.
Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53.
Hajrasouliha AR, Jiang G, Lu Q, Lu H, Kaplan HJ, Zhang HG, et al. Exosomes from retinal astrocytes contain antiangiogenic components that inhibit laser-induced choroidal neovascularization. J Biol Chem. 2013;288:28058–67.
Hernandez MR. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog Retin Eye Res. 2000;19:297–321.
Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–37.
Panenka W, Jijon H, Herx LM, Armstrong JN, Feighan D, Wei T, et al. P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci. 2001;21:7135–42.
Zhang X, Neufeld AH. Signal transduction pathways for epidermal growth factor stimulated cyclooxygenase-2 induction in astrocytes. Exp Eye Res. 2007;85:280–8.
Huh YH, Kim SH, Kim SJ, Chun JS. Differentiation status-dependent regulation of cyclooxygenase-2 expression and prostaglandin E2 production by epidermal growth factor via mitogen-activated protein kinase in articular chondrocytes. J Biol Chem. 2003;278:9691–7.
Junier MP. What role(s) for TGFalpha in the central nervous system? Prog Neurobiol. 2000;62:443–73.
Zhang X, Neufeld AH. Activation of the epidermal growth factor receptor in optic nerve astrocytes leads to early and transient induction of cyclooxygenase-2. Invest Ophthalmol Vis Sci. 2005;46:2035–41.
Ayalasomayajula SP, Amrite AC, Kompella UB. Inhibition of cyclooxygenase-2, but not cyclooxygenase-1, reduces prostaglandin E2 secretion from diabetic rat retinas. Eur J Pharmacol. 2004;498:275–8.
Sennlaub F, Valamanesh F, Vazquez-Tello A, El-Asrar AM, Checchin D, Brault S, et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation. 2003;108:198–204.
Liu Y, Biarnes Costa M, Gerhardinger C. IL-1beta is upregulated in the diabetic retina and retinal vessels: cell-specific effect of high glucose and IL-1beta autostimulation. PLoS One. 2012;7, e36949.
Karlstetter M, Ebert S, Langmann T. Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology. 2010;215:685–91.
Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28:348–68.
Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: just bystander or target for therapy? Prog Retin Eye Res. 2015;45:30–57.
Zeng HY, Green WR, Tso MO. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. 2008;126:227–32.
Kezic JM, Chen X, Rakoczy EP, McMenamin PG. The effects of age and Cx3cr1 deficiency on retinal microglia in the Ins2(Akita) diabetic mouse. Invest Ophthalmol Vis Sci. 2013;54:854–63.
Ibrahim AS, El-Remessy AB, Matragoon S, Zhang W, Patel Y, Khan S, et al. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes. 2011;60:1122–33.
Cunha-Vaz JG. Diabetic retinopathy. Hackensack, NJ: World Scientific; 2011.
Barber AJ, Antonetti DA, Kern TS, Reiter CE, Soans RS, Krady JK, et al. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005;46:2210–8.
Krady JK, Basu A, Allen CM, Xu Y, LaNoue KF, Gardner TW, et al. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes. 2005;54:1559–65.
Yang LP, Sun HL, Wu LM, Guo XJ, Dou HL, Tso MO, et al. Baicalein reduces inflammatory process in a rodent model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009;50:2319–27.
Wong A, Dukic-Stefanovic S, Gasic-Milenkovic J, Schinzel R, Wiesinger H, Riederer P, et al. Anti-inflammatory antioxidants attenuate the expression of inducible nitric oxide synthase mediated by advanced glycation endproducts in murine microglia. Eur J Neurosci. 2001;14:1961–7.
Wang AL, Yu AC, He QH, Zhu X, Tso MO. AGEs mediated expression and secretion of TNF alpha in rat retinal microglia. Exp Eye Res. 2007;84:905–13.
Yoshida A, Yoshida S, Ishibashi T, Kuwano M, Inomata H. Suppression of retinal neovascularization by the NF-kappaB inhibitor pyrrolidine dithiocarbamate in mice. Invest Ophthalmol Vis Sci. 1999;40:1624–9.
Liou GI. Diabetic retinopathy: role of inflammation and potential therapies for anti-inflammation. World J Diabetes. 2010;1:12–8.
Kermorvant-Duchemin E, Pinel AC, Lavalette S, Lenne D, Raoul W, Calippe B, et al. Neonatal hyperglycemia inhibits angiogenesis and induces inflammation and neuronal degeneration in the retina. PLoS One. 2013;8, e79545.
Ibrahim AS, El-Shishtawy MM, Pena Jr A, Liou GI. Genistein attenuates retinal inflammation associated with diabetes by targeting of microglial activation. Mol Vis. 2010;16:2033–42.
Grigsby JG, Cardona SM, Pouw CE, Muniz A, Mendiola AS, Tsin AT, et al. The role of microglia in diabetic retinopathy. J Ophthalmol. 2014;2014:705783.
Pereira Tde O, da Costa GN, Santiago AR, Ambrosio AF, dos Santos PF. High glucose enhances intracellular Ca2+ responses triggered by purinergic stimulation in retinal neurons and microglia. Brain Res. 2010;1316:129–38.
Abcouwer SF, Gardner TW. Diabetic retinopathy: loss of neuroretinal adaptation to the diabetic metabolic environment. Ann N Y Acad Sci. 2014;1311:174–90.
Zhang L, Ino-ue M, Dong K, Yamamoto M. Retrograde axonal transport impairment of large- and medium-sized retinal ganglion cells in diabetic rat. Curr Eye Res. 2000;20:131–6.
Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–91.
Barile GR, Pachydaki SI, Tari SR, Lee SE, Donmoyer CM, Ma W, et al. The RAGE axis in early diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:2916–24.
Gillies MC, Su T, Stayt J, Simpson JM, Naidoo D, Salonikas C. Effect of high glucose on permeability of retinal capillary endothelium in vitro. Invest Ophthalmol Vis Sci. 1997;38:635–42.
Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol. 2008;586:4401–8.
Uusitalo-Jarvinen H, Kurokawa T, Mueller BM, Andrade-Gordon P, Friedlander M, Ruf W. Role of protease activated receptor 1 and 2 signaling in hypoxia-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27:1456–62.
Sitaras N, Rivera JC, Noueihed B, Bien-Aime M, Zaniolo K, Omri S, et al. Retinal neurons curb inflammation and enhance revascularization in ischemic retinopathies via proteinase-activated receptor-2. Am J Pathol. 2014.
Rothmeier AS, Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol. 2012;34:133–49.
Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol. 2010;160:191–203.
Hauk TG, Muller A, Lee J, Schwendener R, Fischer D. Neuroprotective and axon growth promoting effects of intraocular inflammation do not depend on oncomodulin or the presence of large numbers of activated macrophages. Exp Neurol. 2008;209:469–82.
Kilic U, Kilic E, Jarve A, Guo Z, Spudich A, Bieber K, et al. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating ERK-1/2 and Akt pathways. J Neurosci. 2006;26:12439–46.
Wen R, Tao W, Li Y, Sieving PA. CNTF and retina. Prog Retin Eye Res. 2012;31:136–51.
Unterlauft JD, Eichler W, Kuhne K, Yang XM, Yafai Y, Wiedemann P, et al. Pigment epithelium-derived factor released by Muller glial cells exerts neuroprotective effects on retinal ganglion cells. Neurochem Res. 2012;37:1524–33.
Unterlauft JD, Claudepierre T, Schmidt M, Muller K, Yafai Y, Wiedemann P, et al. Enhanced survival of retinal ganglion cells is mediated by Muller glial cell-derived PEDF. Exp Eye Res. 2014;127:206–14.
Li T, Hu J, Du S, Chen Y, Wang S, Wu Q. ERK1/2/COX-2/PGE2 signaling pathway mediates GPR91-dependent VEGF release in streptozotocin-induced diabetes. Mol Vis. 2014;20:1109–21.
Chen J, Hui ST, Couto FM, Mungrue IN, Davis DB, Attie AD, et al. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 2008;22:3581–94.
Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes. 2008;57:938–44.
Lappalainen Z, Lappalainen J, Oksala NK, Laaksonen DE, Khanna S, Sen CK, et al. Diabetes impairs exercise training-associated thioredoxin response and glutathione status in rat brain. J Appl Physiol (1985). 2009;106:461–7.
Munemasa Y, Ahn JH, Kwong JM, Caprioli J, Piri N. Redox proteins thioredoxin 1 and thioredoxin 2 support retinal ganglion cell survival in experimental glaucoma. Gene Ther. 2009;16:17–25.
Caprioli J, Munemasa Y, Kwong JM, Piri N. Overexpression of thioredoxins 1 and 2 increases retinal ganglion cell survival after pharmacologically induced oxidative stress, optic nerve transection, and in experimental glaucoma. Trans Am Ophthalmol Soc. 2009;107:161–5.
Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010;1, e65.
Balasubramanian R, Gan L. Development of retinal amacrine cells and their dendritic stratification. Curr Ophthalmol Rep. 2014;2:100–6.
Marc RE, Anderson JR, Jones BW, Sigulinsky CL, Lauritzen JS. The AII amacrine cell connectome: a dense network hub. Front Neural Circuits. 2014;8:104.
Wachtmeister L. Basic research and clinical aspects of the oscillatory potentials of the electroretinogram. Doc Ophthalmol. 1987;66:187–94.
Elsner AE, Burns SA, Lobes Jr LA, Doft BH. Cone photopigment bleaching abnormalities in diabetes. Invest Ophthalmol Vis Sci. 1987;28:718–24.
Holopigian K, Greenstein VC, Seiple W, Hood DC, Carr RE. Evidence for photoreceptor changes in patients with diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38:2355–65.
Perez VL, Caspi RR. Immune mechanisms in inflammatory and degenerative eye disease. Trends Immunol. 2015.
King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol. 2008;79:1527–34.
Graves DT, Kayal RA. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008;13:1227–39.
Singh K, Kant S, Singh VK, Agrawal NK, Gupta SK, Singh K. Toll-like receptor 4 polymorphisms and their haplotypes modulate the risk of developing diabetic retinopathy in type 2 diabetes patients. Mol Vis. 2014;20:704–13.
Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: just bystander or target for therapy? Prog Retin Eye Res. 2015;45:30–57.
Adamiec-Mroczek J, Oficjalska-Mlynczak J. Assessment of selected adhesion molecule and proinflammatory cytokine levels in the vitreous body of patients with type 2 diabetes--role of the inflammatory-immune process in the pathogenesis of proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2008;246:1665–70.
Hoppener JW, Ahren B, Lips CJ. Islet amyloid and type 2 diabetes mellitus. N Engl J Med. 2000;343:411–9.
Srodulski S, Sharma S, Bachstetter AB, Brelsfoard JM, Pascual C, Xie XS, et al. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol Neurodegener. 2014;9:30.
Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–36.
Sima AA, Zhang W, Muzik O, Kreipke CW, Rafols JA, Hoffman WH. Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-peptide. Rev Diabet Stud. 2009;6:211–22.
Vargas R, Rincon J, Pedreanez A, Viera N, Hernandez-Fonseca JP, Pena C, et al. Role of angiotensin II in the brain inflammatory events during experimental diabetes in rats. Brain Res. 2012;1453:64–76.
Kitayama H, Maeshima Y, Takazawa Y, Yamamoto Y, Wu Y, Ichinose K, et al. Regulation of angiogenic factors in angiotensin II infusion model in association with tubulointerstitial injuries. Am J Hypertens. 2006;19:718–27.
Grant MB, May WS, Caballero S, Brown GA, Guthrie SM, Mames RN, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–12.
Thomas HE, Redgrave R, Cunnington MS, Avery P, Keavney BD, Arthur HM. Circulating endothelial progenitor cells exhibit diurnal variation. Arterioscler Thromb Vasc Biol. 2008;28:e21–22.
Busik JV, Tikhonenko M, Bhatwadekar A, Opreanu M, Yakubova N, Caballero S, et al. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206:2897–906.
Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–58.
Li G, Veenstra AA, Talahalli RR, Wang X, Gubitosi-Klug RA, Sheibani N, et al. Marrow-derived cells regulate the development of early diabetic retinopathy and tactile allodynia in mice. Diabetes. 2012;61:3294–303.
Simo R, Hernandez C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog Retin Eye Res. 2015.
Hirata A, Murakami Y, Shoji M, Kadoma Y, Fujisawa S. Kinetics of radical-scavenging activity of hesperetin and hesperidin and their inhibitory activity on COX-2 expression. Anticancer Res. 2005;25:3367–74.
Choi EJ, Ahn WS. Neuroprotective effects of chronic hesperetin administration in mice. Arch Pharm Res. 2008;31:1457–62.
Lai AY, Todd KG. Hypoxia-activated microglial mediators of neuronal survival are differentially regulated by tetracyclines. Glia. 2006;53:809–16.
Jantzie LL, Cheung PY, Todd KG. Doxycycline reduces cleaved caspase-3 and microglial activation in an animal model of neonatal hypoxia-ischemia. J Cereb Blood Flow Metab. 2005;25:314–24.
Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–500.
Suk K. Minocycline suppresses hypoxic activation of rodent microglia in culture. Neurosci Lett. 2004;366:167–71.
Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95:15769–74.
Wang AL, Yu AC, Lau LT, Lee C, Wu le M, Zhu X, et al. Minocycline inhibits LPS-induced retinal microglia activation. Neurochem Int. 2005;47:152–8.
Kadiyala CS, Zheng L, Du Y, Yohannes E, Kao HY, Miyagi M, et al. Acetylation of retinal histones in diabetes increases inflammatory proteins: effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J Biol Chem. 2012;287:25869–80.
Mohr S. Potential new strategies to prevent the development of diabetic retinopathy. Expert Opin Investig Drugs. 2004;13:189–98.
Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–12.
Martinez-Zapata MJ, Marti-Carvajal AJ, Sola I, Pijoan JI, Buil-Calvo JA, Cordero JA, et al. Anti-vascular endothelial growth factor for proliferative diabetic retinopathy. Cochrane Database Syst Rev. 2014;11, CD008721.
Fernando Arevalo J. Intravitreal bevacizumab as anti-vascular endothelial growth factor in the management of complications of proliferative diabetic retinopathy. Med Hypothesis Discov Innov Ophthalmol. 2013;2:20–4.
Dugel PU. Ranibizumab treatment of patients with ocular diseases. Int Ophthalmol Clin. 2006;46:131–40.
Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, et al. Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology. 2012;119:789–801.
Rosenfeld PJ, Fung AE, Puliafito CA. Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for macular edema from central retinal vein occlusion. Ophthalmic Surg Lasers Imaging. 2005;36:336–9.
Arevalo JF, Wu L, Sanchez JG, Maia M, Saravia MJ, Fernandez CF, et al. Intravitreal bevacizumab (avastin) for proliferative diabetic retinopathy: 6-months follow-up. Eye (Lond). 2009;23:117–23.
Do DV, Schmidt-Erfurth U, Gonzalez VH, Gordon CM, Tolentino M, Berliner AJ, et al. The DA VINCI study: phase 2 primary results of VEGF trap-eye in patients with diabetic macular edema. Ophthalmology. 2011;118:1819–26.
Das A, Stroud S, Mehta A, Rangasamy S. New treatments for diabetic retinopathy. Diabetes Obes Metab. 2014.
Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–8.
Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22:6401–7.
Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP, et al. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. FASEB J. 2001;15:1218–20.
Shikari H, Silva PS, Sun JK. Complications of intravitreal injections in patients with diabetes. Semin Ophthalmol. 2014;29:276–89.
Falavarjani KG, Modarres M, Hashemi M, Parvaresh MM, Naseripour M, Zare-Moghaddam A, et al. Incidence of acute endophthalmitis after intravitreal bevacizumab injection in a single clinical center. Retina. 2013;33:971–4.
Hayman SR, Leung N, Grande JP, Garovic VD. VEGF inhibition, hypertension, and renal toxicity. Curr Oncol Rep. 2012;14:285–94.
Osaadon P, Fagan XJ, Lifshitz T, Levy J. A review of anti-VEGF agents for proliferative diabetic retinopathy. Eye (Lond). 2014;28:510–20.
Kaminski NE. Regulation of the cAMP cascade, gene expression and immune function by cannabinoid receptors. J Neuroimmunol. 1998;83:124–32.
Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (−)delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A. 1998;95:8268–73.
Rajesh M, Mukhopadhyay P, Batkai S, Hasko G, Liaudet L, Drel VR, et al. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am J Physiol Heart Circ Physiol. 2007;293:H610–619.
Weiss L, Zeira M, Reich S, Har-Noy M, Mechoulam R, Slavin S, et al. Cannabidiol lowers incidence of diabetes in non-obese diabetic mice. Autoimmunity. 2006;39:143–51.
El-Remessy AB, Al-Shabrawey M, Khalifa Y, Tsai NT, Caldwell RB, Liou GI. Neuroprotective and blood-retinal barrier-preserving effects of cannabidiol in experimental diabetes. Am J Pathol. 2006;168:235–44.
Sharma S, Anjaneyulu M, Kulkarni SK, Chopra K. Resveratrol, a polyphenolic phytoalexin, attenuates diabetic nephropathy in rats. Pharmacology. 2006;76:69–75.
Kumar A, Kaundal RK, Iyer S, Sharma SS. Effects of resveratrol on nerve functions, oxidative stress and DNA fragmentation in experimental diabetic neuropathy. Life Sci. 2007;80:1236–44.
Holian O, Wahid S, Atten MJ, Attar BM. Inhibition of gastric cancer cell proliferation by resveratrol: role of nitric oxide. Am J Physiol Gastrointest Liver Physiol. 2002;282:G809–816.
Ghadiri Soufi F, Arbabi-Aval E, Rezaei Kanavi M, Ahmadieh H. Anti-inflammatory properties of resveratrol in the retinas of type 2 diabetic rats. Clin Exp Pharmacol Physiol. 2015;42:63–8.
Kim SH, Park JH, Kim YJ, Park KH. The neuroprotective effect of resveratrol on retinal ganglion cells after optic nerve transection. Mol Vis. 2013;19:1667–76.
Soheilian M, Karimi S, Ramezani A, Peyman GA. Pilot study of intravitreal injection of diclofenac for treatment of macular edema of various etiologies. Retina. 2010;30:509–15.
Miyake K, Ibaraki N. Prostaglandins and cystoid macular edema. Surv Ophthalmol. 2002;47 Suppl 1:S203–218.
Soheilian M, Karimi S, Ramezani A, Montahai T, Yaseri M, Soheilian R, et al. Intravitreal diclofenac versus intravitreal bevacizumab in naive diabetic macular edema: a randomized double-masked clinical trial. Int Ophthalmol. 2015;35:421–8.
Acknowledgements
We are grateful to Dr. Ji Ming Wang, National Cancer Institute, National Institutes of Health, for his helpful critique of the manuscript. This project was supported in part by the grants from the National Natural Science Foundation of China (31471122).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YY studied the literature and drafted the manuscript. HC and SBS reviewed and edited the article. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yu, Y., Chen, H. & Su, S.B. Neuroinflammatory responses in diabetic retinopathy. J Neuroinflammation 12, 141 (2015). https://doi.org/10.1186/s12974-015-0368-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-015-0368-7