
Abstract
Background
Juvenile Dermatomyositis (JDM) is the most common chronic idiopathic inflammatory myopathy in children. The diagnosis is clinical. Baseline laboratory and complementary studies trace the phenotype of these patients. The objective of this study was to describe epidemiological, clinical and laboratory characteristics at diagnosis of JDM patients included in the Spanish JDM registry, as well as to identify prognostic factors on these patients.
Methods
We retrospectively reviewed clinical features, laboratory tests, and complementary studies at diagnosis of JDM patients included on the Spanish JDM registry. These data were analyzed to assess whether there was a relationship with the development of complications and time to disease inactivity.
Results
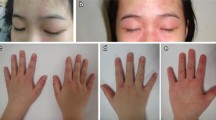
One hundred and sixteen patients from 17 Spanish paediatric rheumatology centres were included, 76 girls (65%). Median age at diagnosis was 7.3 years (Interquartile range (IQR) 4.5–10.2). All patients had pathognomonic skin lesions at the beginning of the disease. Muscle weakness was present in 86.2%. Median Childhood Muscle Assessment Scale was 34 (IQR 22–47). Twelve patients (34%) had dysphagia and 3,5% dysphonia. Anti-p155 was the most frequently detected myositis specific antibody, followed by anti-MDA5. Twenty-nine patients developed calcinosis and 4 presented with macrophage activation syndrome. 70% reached inactivity in a median time of 8.9 months (IQR 4.5–34.8). 41% relapsed after a median time of 14.4 months (IQR 8.6–22.8) of inactivity. Shorter time to treatment was associated with better prognosis (Hazard ratio (HR) = 0.95 per month of evolution, p = 0.02). Heliotrope rash at diagnosis correlates with higher risk of development complications.
Conclusions
We describe heliotrope rash as a risk factor for developing complications in our cohort of JDM patients, an easy-to-evaluate clinical sign that could help us to identify the group of patients we should monitor closely for this complication.
Similar content being viewed by others

Background
Juvenile Dermatomyositis (JDM) is the most common chronic idiopathic inflammatory myopathy in children (85%). It’s a systemic vasculopathy characterised by muscle and skin involvement. Diagnosis is clinical, with the identification of pathognomonic cutaneous rashes (Gottron’s papules, heliotrope rash), usually accompanied by proximal muscle weakness. Baseline laboratory and complementary studies trace the phenotype of these patients [1]. The goal of treatment is disease remission, reducing the development of complications, such as calcinosis. First line therapy includes systemic corticosteroids and subcutaneous methotrexate, adding intravenous immunoglobulins in selected patients. Although mortality remains below 4%, morbidity continues to be high (70–80%), predominantly on cutaneous, endocrine, muscular, and skeletal domains [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17].
The objectives of this study were to describe epidemiological, clinical and laboratory findings at time of diagnosis of JDM patients included in the Spanish JDM registry, as well as to identify prognostic factors in these patients.
Methods
Study population and inclusion criteria
This is a retrospective descriptive observational and multicentre study of the JDM Spanish registry. Sant Joan de Déu Hospital (Barcelona) was the coordinator centre. Registration data were entered from January 2013 to January 2021. This study was approved by the Ethics committee with the code CEIC PIC-74-13. Inclusion criteria were diagnosis of JDM according to the Bohan and Peter criteria and/or expert diagnosis supported by Magnetic resonance image (MRI), electomyogram (EMG) or muscle biopsy evidence of myositis. Informed consent was obtained via the signature of the patient or their parent/legal guardian. Overlapping syndrome were not included in the registry.
Data collection
Data collection was carried out retrospectively, reviewing data from the medical records until the last visit made at the centres of origin. Of the patients diagnosed before 2013, only those who still had a visit to the pediatric rheumatology unit between the period from 2013 to 2021 were included. We registered demographic (age, sex, concomitant medical history of immune-mediated pathology and family history of immune-mediated pathology, previous vaccines and infections to the onset of symptoms, age at disease onset, age at diagnosis, time to diagnosis since onset symptoms); clinical features at diagnosis like skin manifestations: Gottron’s papules, erythematous lesions, heliotrope rash, vasculitis lesions, subcutaneous oedema, atrophy, livedo reticularis, periungueal erythema, skin ulcers, shawl rash, poikiloderma, oral ulcers; weakness: pelvic girdle weakness, scapular girdle weakness and axial weakness; other symptoms: arthritis, constitutional symptoms (fever, weight loss, asthenia), gastrointestinal symptoms (dysphagia, abdominal pain, perforation bowel), pulmonary symptoms (dysphonia, interstitial lung disease (ILD), cardiovascular involvement and Raynaud’s phenomenon).
Laboratory tests collected were acute phase reactants: erythrocyte sedimentation rate (ESR) and C reactive protein (CRP); muscle enzymes: creatine phosphokinase (CK), aspartate aminotransferase (GOT), alanine aminotransferase (GPT), aldolase and lactate dehydrogenase (LDH), and myositis specific antibodies (MSA) and myositis associated antibodies (SMA) by immunoblot/blot-line. The reagent kit we used is Euroline Myositis Profile 16 from Euroimmun (Lübeck, Germany). Laboratory parameters were adjusted based on age-defined upper limits of normal. We used the Childhood Muscle Assessment Scale (CMAS) to assess muscle strength when applicable, we considered a normal score ≥ 48/52 points. Medical test evaluated: MRI and whole body MRI (WBMRI), EMG, muscle biopsy, video fluoroscopy/barium studies, echocardiography and electrocardiogram (ECG), pulmonary function tests and nailfold capillaroscopy.
We considered disease inactivity based on a modification of the Paediatric Rheumatology International Trials Organisation (PRINTO) criteria: absence of skin disease at the time of assessment, and at least 3 of the following 4 criteria: (1) creatine kinase (CK) ≤ 150 units/liter, (2) Childhood Myositis Assessment Scale (CMAS) score ≥ 48/52, (3) Manual Muscle Testing 8 (MMT-8) score ≥ 78/80, and (4) physician global assessment ≤ 0.2 (of a possible 10).
We registered time to inactivity disease and complications. Complications were calcinosis, macrophage activation syndrome (MAS), others included infections, osteoporosis and lipodystrophy. We analysed each clinical, laboratory and medical tests at diagnosis to detect prognostic factors that predispose to the development of complications and the time to reach inactivity. The times of disease inactivity and relapses were calculated retrospectively according to the status of the patients on the date of inclusion in the registry. We considered shorter time to reach remission and absence of complications as optimal treatment objectives.
Statistical data analysis
We performed statistical analysis with SPSS 19.0 statistical software (Armonk, NY: IBM Corp). Descriptive statistics of the study variables with absolute frequency and percentage in the case of qualitative variables and median and interquartile range (IQR) for continuous variables. A verification of the study groups was carried out using the X test or Fisher’s exact test when necessary for qualitative variables. The comparison of the quantitative variables according to the study groups was carried out using the Student t-test for independent samples (or Welh test) or the MannWhitney U test (depending on whether or not they followed a normal distribution). The level of statistical significance was set at p < 0.05.
Survival analysis for each categorical variable (skin manifestations; type of weakness; other symptoms; laboratory tests; myositis specific antibodies and myositis associated antibodies; medical test evaluated) of interest was obtained by analysing the time until inactivity and complications, estimating the survival curve for each value of the variable using the Kaplan-Meier method and comparing them using the log-rank test. If the variable was numerical, we used Cox Proportional Hazards Regression.
Results
Demographic and clinical features
One hundred and sixteen patients from 17 Spanish paediatric rheumatology centres were included, 76 girls (65%). Of the 116 patients, 70 were diagnosed before 2013. Median age at diagnosis was 7.3 years (Interquartile range (IQR) 4.5–10.5). All patients had pathognomonic skin lesions at the beginning of the disease. Muscle weakness was present in 86.2% (108/116). Median Childhood Muscle Assessment Scale was 34 (IQR 22–47). 37% (43/116) of patients were associated with other symptoms at the beginning of the disease. 34% (12/116) were referred for dysphagia and 3.5% (4/116) dysphonia. Table 1 summarises demographic and clinical features. Median time to start treatment since the beginning of the disease was 2.5 moths (IQR 1.2–6.3), with systemic steroids as the most frequently used drug (98.3%), followed by methotrexate (76.7%) and hydroxychloroquine (53.4%).
Laboratory parameters and diagnostic features
Laboratory parameters
Forty-two and 52.9% of patients had erythrocyte sedimentation rate (ESR) and C reactive protein (CRP) increased. According to muscular enzymes, aldolase was the most frequently raised (78%), followed by alanine aminotransferase (GPT) 75%, aspartate aminotransferase (GOT) 66%, lactate dehydrogenase (LDH) 56.5%, and creatine phosphokinase (CK) 52%.
We identified a myositis specific antibody (MSA) in 27.3% of patients (21/77), with anti-p155 as the most frequently detected (12.8%), followed by anti-MDA5 (11.1%). Table 2 describes laboratory parameters.
Diagnostic features
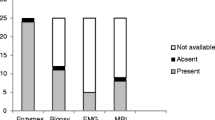
Magnetic resonance imaging was obtained in 73 patients at diagnosis. As expected, pelvic girdle muscles were the muscular group most frequently involved (79%). Whole-body MRI (WBMRI) was even more sensitive, detecting muscle inflammation in 94% of the patients studied.
Only 17 patients (14.7%) had video fluoroscopy/Barium studies, 16 of them asymptomatic from a gastrointestinal standpoint. We detected swallowing dysfunction in 9/16 (56.3%) asymptomatic patients.
At diagnosis twenty-seven of 74 patients (35.1%), had pulmonary function tests (PFTs) if feasible due to age. Four patients (15.4%) presented abnormal results (3 patients with restrictive pattern, 1 obstructive pattern). None of these four patients had interstitial lung disease (ILD) related with JDM on high resolution CT. According to MSA, we performed PFTs in two of the four MDA5 + patients. Both were normal.
Muscle biopsy at diagnosis was performed in 66 patients (56.9%). Ninety-six per cent (63/65) of patients had abnormal microscopic findings. The two patients with a muscle biopsy reported as normal, had classic JDM symptoms, including muscle involvement documented by physical examination and/or other complementary tests. Table 3 describes diagnostic features.
Disease course
Sixty-one patients (52.6%) achieved inactivity until the last follow-up visit registered. The median time to achieve inactivity was 8.9 months (IQR 4.5–34.8). Seventy patients were diagnosed before opening the registry. Laboratory parameters normalized 1.4 months before reaching clinical inactivity, with skin disease as the latest to be controlled (median 8.0 months (IQR 3.5–30.0). Twenty-nine (25%) developed calcinosis and four patients (3.4%) presented MAS. Forty-one per cent (37/90) relapsed after 14.4 months of inactivity (IQR 8.6–22.8). The number of each relapse was not recorded, only the first. The denominator 90 refers to the total number of patients in whom the relapse variable could be analyzed; in the remaining 26, the data was not collected.
Prognostic factors
Clinical, laboratory, and medical tests at diagnosis were analysed to detect prognostic factors. The longer the time to start treatment, the lower the probability of reaching inactivity disease (Hazard ratio (HR) 0.95 per month of treatment delay, p = 0.02). The presence of heliotrope rash at diagnosis was associated with a higher risk of development of complications (Fisher test 0,009; OR 0.35, IC 95% 0,14-0.83) with a close statistical significance for the development of calcinosis (Fisher test 0,07; OR 0.33, IC 95% 0,10-1.06). We cannot calculate the survival curve for the time until the onset of calcinosis because the date of the calcinosis event was not recorded. We did not detect any variable that could help us to predict MAS.
Discussion
To date, this is the largest multicentre cohort of JDM patients in our country. Patient characteristics are similar to previously reported series, with a female predominance (65%) 18,19,20, 24,25,26,27,28,29,30,31,32,33, but an older mean age at diagnosis compared with the CARRA cohort 19, 20. Cutaneous disease was the first organ involved, this could explain the shorter time to diagnosis compared with the British cohort 18, 21. 37% of patients complained about other symptoms at diagnosis apart from cutaneous and muscular disease, with arthritis as the most frequently described symptom (14.7%). We would like to remark this data because according to our experience, in some cases there is a misdiagnosis with juvenile idiopathic arthritis, mainly in JDM anti-MDA5 + patients, with the consequent delay in completing the basal study and starting the adequate immunosuppression for JDM.
According to laboratory tests, CK was normal in 48% of patients, with aldolase as the most frequently increased (78%) despite being the less frequently ordered. EMG and MR increased the probability of detecting muscle involvement (87.40 and 86.3% respectively). Sensitivity increased to 95% when we assessed WBMRI. All but one patient who had a muscle biopsy performed had pathological results, supporting the importance of taking biopsy in case of diagnostic uncertainty [10].
A MSA was identified in 27.3% of patients, a lower percentage compared to other series [18,19,20,21,22], explained because we are in front of a retrospective study and not all the MSA were performed in each patient. The most frequently MSA detected was anti-p155 (12.8%), followed by anti-MDA5 (11.1%) and anti-NXP2 (8.7%), similar to previously published [2,3,4].
At the time of enrolment to the registry, there was no consensus about performing high-resolution CT pulmonary scan at diagnosis as part of the baseline study except in case of pathological PFTs. We detected ILD in one of the four MDA5 + patients when she altered PFTs after 3.6 years of JDM diagnosis.
Despite just 34% (12/116) referred for dysphagia, swallowing disturbances were presented in 56.3% (9/16) asymptomatic patients. This result emphasizes the importance of performing a complete evaluation of dysphagia, not limited to anamnesis, especially considering the implication of swallowing impairment for patient management.
Calcinosis was present in 10.3% of patients (12/116) at diagnosis, with an increase to 25% if we consider all the patients with calcinosis at the time of being included in the registry (29 patients, median of follow up of 59 months since diagnosis (IQR 16.25–109). Only four patients were anti-NXP2+, therefore we could not evaluate if there was a higher risk of calcinosis on these patients in our series. Heliotrope rash at diagnosis was related with a higher risk of development of complications, Moreover, it was close to statistical significance for the development of calcinosis. To the best of our knowledge, this is the first time that a clinical variable, as easy to assess as heliotrope rash, can be related to complications like calcinosis and it raises the question of whether in patients with heliotrope we should perform a low-radiation total body CT to detect calcinosis at JDM diagnosis. In others publications like of Nozawa et al. described nailfold capillary changes as predictors of calcinosis in JDM [23].
Unfortunately, we did not identify any prognostic factor to recognize patients with higher risk of developing MAS. Despite MAS being a rare JDM complication, 3.4% of our cohort presented it in the three-month period after diagnosis, and due its severity it must be considered and actively searched. Moreover, awareness of this complication is important because elevation of AST and/or ALT could be interpreted as muscular origin causing a delay on MAS diagnosis.
41% of patients relapsed after a median time of 14.4 months of inactivity underscoring the need for continued regular follow-up. We would also like to highlight the importance of new biomarkers, such as plasma interferon signature or circulating endothelial cells that allow us to detect a relapse prior to clinical or analytical findings [2,3,4]. We have no data of these biomarkers in our cohort but could be interesting to study in future studies.
We find some limitations in our study, most of them due to being a retrospective study. Another important limitation was that of the patients diagnosed before 2013, only those who still had a visit to the pediatric rheumatology unit between the period from 2013 to 2021 were included. Furthermore, another limitation is the low percentage of patients in whom SMA were performed. Recent publications, of Papadopoulou et al. and McCann et al. have described how having an SMA modifies a patient´s prognosis, as it is the case of anti-MDA5 patients with a higher risk of ILD.
Conclusions
In conclusion, we present data from the JDM Spanish registry and identify heliotrope rash as a risk factor in the development of complications in JDM patients. This easy-to-evaluate clinical sign could help us to identify a subgroup of patients with higher risk whom we should monitor more closely. We need larger registries, preferably prospective, that can confirm these findings.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- JDM:
-
Juvenile Dermatomyositis
- CMAS:
-
Childhood Muscle Assessment Scale
- MAS:
-
Macrophage activation syndrome
- IQR:
-
Interquartile range
- N:
-
Sample
- ILD:
-
Interstitial lung disease
- ESR:
-
Erythrocyte sedimentation rate
- CRP:
-
C reactive protein
- GPT:
-
Alanine aminotransferase
- GOT:
-
Aspartate aminotransferase
- LDH:
-
Lactate dehydrogenase
- CK:
-
Creatine phosphokinase
- MSA:
-
Myositis specific antibody
- SMA:
-
Myositis associated antibodies
- MRI:
-
Magnetic resonance image
- WBMRI:
-
Whole-body MRI
- PFTs:
-
Pulmonary function tests
- CT:
-
Computed axial tomography
- EMG:
-
Electromyogram
- ECG:
-
Electrocardiogram
- HR:
-
Hazard ratio
References
Clarissa A, Pilkington BM, Feldman W. Sontichai. Juvenile Dermatomyositis and Other inflammatory muscle diseases. Petty,Laxer, Lindsley, Wedderburn, Mellins, Fuhlbrigge. Pediatric Rheumatology. Eighth edition. Philadelphia. Elsevier, 2021. Chapter 26, pg 360–376.
Papadopoulou C, Chew C, Wilkinson MGL, McCann L, Wedderburn LR. Juvenile idiopathic inflammatory myositis: an update on pathophysiology and clinical care. Volume 19. Nature Reviews Rheumatology. Nature Research; 2023. pp. 343–62.
McCann LJ, Livermore P, Wilkinson MGL, Wedderburn LR. Juvenile dermatomyositis. Where are we now? Clin Exp Rheumatol. 2022;40(2):394–403.
Rider LG, Nistala K. The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. Journal of Internal Medicine. Volume 280. Blackwell Publishing Ltd; 2016. pp. 24–38.
McLellan K, Papadopoulou C. Update on biomarkers of Vasculopathy in Juvenile and Adult Myositis. Curr Rheumatol Rep. 2022;24(7):227–37.
Gitiaux C, Bondet V, Bekaddour N, Nusbaum P, Hubas A, Melki I, et al. Inhibition of IFNα secretion in cells from patients with juvenile dermatomyositis under TBK1 inhibitor treatment revealed by single-molecular assay technology. Volume 59. Oxford University Press; 2020. pp. 1171–4. Rheumatology (United Kingdom).
Melki I, Devilliers H, Gitiaux C, Bondet V, Duffy D, Charuel JL, Miyara M, Bokov P, Kheniche A, Kwon T, Authier FJ, Allenbach Y, Belot A, Bodemer C, Bourrat E, Dumaine C, Fabien N, Faye A, Frémond ML, Hadchouel A, Bader-Meunier B. Anti-MDA5 juvenile idiopathic inflammatory myopathy: a specific subgroup defined by differentially enhanced interferon-α signalling. Rheumatology (Oxford). 2020;59(8):1927–37.
Giancane G, Lavarello C, Pistorio A, Oliveira SK, Zulian F, Cuttica R, et al. The PRINTO evidence-based proposal for glucocorticoids tapering/discontinuation in new onset juvenile dermatomyositis patients. Pediatr Rheumatol. 2019;17(1):1–11.
Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76(2):329–40.
Spencer CH, Rouster-Stevens K, Gewanter H, Syverson G, Modica R, Schmidt K et al. Biologic therapies for refractory juvenile dermatomyositis: five years of experience of the Childhood Arthritis and Rheumatology Research Alliance in North America. Pediatr Rheumatol. 2017;15(1).
Kim S, Kahn P, Robinson AB, Lang B, Shulman A, Oberle EJ et al. Childhood Arthritis and Rheumatology Research Alliance consensus clinical treatment plans for juvenile dermatomyositis with skin predominant disease. Pediatr Rheumatol. 2017;15(1).
Huber AM, Kim S, Reed AM, Carrasco R, Feldman BM, Hong SD, et al. Childhood arthritis and rheumatology research alliance consensus clinical treatment plans for juvenile dermatomyositis with persistent skin rash. J Rheumatol. 2017;44(1):110–6.
Orandi AB, Baszis KW, Dharnidharka VR, Huber AM, Hoeltzel MF. Assessment, classification and treatment of calcinosis as a complication of juvenile dermatomyositis: a survey of pediatric rheumatologists by the childhood arthritis and rheumatology research alliance (CARRA). Pediatr Rheumatol. 2017;15(1).
Mamyrova G, Rider LG, Ehrlich A, Jones O, Pachman LM, Nickeson R, et al. Environmental factors associated with disease flare in juvenile and adult dermatomyositis. Rheumatol (United Kingdom). 2017;56(8):1342–7.
Hinze CH, Speth F, Oommen PT, Haas JP. Current management of juvenile dermatomyositis in Germany and Austria: an online survey of pediatric rheumatologists and pediatric neurologists. Pediatr Rheumatol. 2018;16(1):1–8.
Leclair V, Lundberg IE. New Myositis classification Criteria—what we have learned since Bohan and Peter. Curr Rheumatol Rep. 2018;20(4).
Wienke J, Deakin CT, Wedderburn LR, van Wijk F, van Royen-Kerkhof A. Systemic and tissue inflammation in Juvenile Dermatomyositis: from pathogenesis to the Quest for Monitoring Tools. Front Immunol. 2018;9(December):2951.
Martin N, Krol P, Smith S, Murray K, Pilkington CA, Davidson JE, et al. A national registry for juvenile dermatomyositis and other paediatric idiopathic inflammatory myopathies: 10 years’ experience; the Juvenile Dermatomyositis National (UK and Ireland) Cohort Biomarker Study and Repository for idiopathic inflammatory myopat. Rheumatology. 2011;50(1):137–45.
Neely J, Ardalan K, Huber A, Kim S, Abel N, Abulaban K et al. Baseline characteristics of children with juvenile dermatomyositis enrolled in the first year of the new Childhood Arthritis and Rheumatology Research Alliance registry. Pediatr Rheumatol. 2022;20(1).
Robinson AB, Hoeltzel MF, Wahezi DM, Becker ML, Kessler EA, Schmeling H, et al. Clinical characteristics of children with juvenile dermatomyositis: the childhood arthritis and rheumatology research alliance registry. Arthritis Care Res. 2014;66(3):404–10.
McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland) - clinical characteristics of children recruited within the first 5 year. Rheumatology. 2006;45(10):1255–60.
Guseinova D, Consolaro A, Trail L, Ferrari C, Pistorio A, Ruperto N, et al. Paediatric rheumatology comparison of clinical features and drug therapies among European and latin American patients with juvenile dermatomyositis. Volume 29. Clinical and Experimental Rheumatology; 2011.
Tomo Nozawa A, Bell-Peter J-A, Marcuz K, Whitney O, Vinik R, Shupak. Saunya Dover and Brian M. Feldman. Early abnormal Nailfold Capillary Changes are Predictive of Calcinosis Development in Juvenile Dermatomyositis. J Rheumatol November. 2022;49(11):1250–5.
Okong’o LO, Esser M, Wilmshurst J, Scott C. Characteristics and outcome of children with juvenile dermatomyositis in Cape Town: a cross-sectional study. Pediatr Rheumatol. 2016;14(1):1–8.
Sharma A, Gupta A, Rawat A, Suri D, Singh S. Long-term outcome in children with juvenile dermatomyositis: a single-center study from north India. Int J Rheum Dis. 2020;23(3):392–6.
Al-Mayouf SM, AlMutiari N, Muzaffer M, shehata R, Al-Wahadneh A, Abdwani R, et al. Phenotypic characteristics and outcome of juvenile dermatomyositis in arab children. Rheumatol Int. 2017;37(9):1513–7.
Dressler F, Frosch M, Mönkemöller K, Thon A, Weißbarth-Riedel E, Horneff G. Results of the German ESPED-recording of new patients with Juvenile Dermatomyositis (JDM). Klin Padiatr. 2011;223(5):280–2.
Peloro TM, Miller OF, Hahn TF, Newman ED. Juvenile dermatomyositis: a retrospective review of a 30-year experience. J Am Acad Dermatol. 2001;45(1):28–34.
Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord. 2000;10(1):1–9.
Kobayashi S, Higuchi K, Tamaki H, Wada Y, Wada N, Kubo M, et al. Characteristics of juvenile dermatomyositis in Japan. Pediatr Int. 1997;39(2):257–62.
Mendez EP, Lipton R, Ramsey-Goldman R, Roettcher P, Bowyer S, Dyer A, et al. US incidence of juvenile dermatomyositis, 1995–1998: results from the National Institute of Arthritis and Musculoskeletal and skin diseases Registry. Arthritis Care Res. 2003;49(3):300–5.
Constantin T, Ponyi A, Orbán I, Molnár K, Dérfalvi B, Dicso F, et al. National registry of patients with juvenile idiopathic inflammatory myopathies in Hungary - Clinical characteristics and disease course of 44 patients with juvenile dermatomyositis. Autoimmunity. 2006;39(3):223–32.
Balci MA, Donmez S, Saritas F, Bas V, Pamuk ON. The epidemiology of dermatomyositis in northwestern Thrace region in Turkey: epidemiology of dermatomyositis in Turkey. Rheumatol Int. 2017;37(9):1519–25.
Acknowledgements
The authors would like to thank all the participants/families and hospital sites that recruited patients from the JDM Spanish registry. In addition, we are grateful to the juvenile dermatomyositis working group of Spanish Society of Paediatric Rheumatology (SERPE) as well as National Juvenile Dermatomyositis Association (ANADEJU).
Funding
SERPE scholarship for research in pediatric rheumatology from Spanish Society of Paediatric Rheumatology (SERPE).
Author information
Authors and Affiliations
Contributions
1. SCA. Sant Joan de Déu Hospital, Barcelona. Spain. Database creation, data analysis and interpretation, article writing.
1. JMM. Sant Joan de Déu Hospital, Barcelona. Spain. Article review.
2. EQM. Vall d’Hebron University Hospital, Barcelona. Spain. Data entry and article review.
2. ML. Vall d’Hebron University Hospital, Barcelona. Spain. Data entry and article review.
3. DC. Niño Jesús Hospital, Madrid. Spain. Data entry and article review.
4. AB. Ramón y Cajal University Hospital, Madrid. Spain. Data entry and article review.
5. CU. La Paz University Children’s Hospital. Madrid. Spain. Data entry and article review.
6. JI. University Hospital 12 de octubre, Madrid. Associate Professor of Pediatrics Complutense University of Madrid. Spain. Data entry and article review.
7. JCN. Gregorio Marañón General University Hospital, Madrid. Spain. Data entry and article review.
8. LR. Valdecilla Hospital, Santander. Spain. Data entry.
9. EN. Pediatric Unit. Maternal and Child Hospital, Regional University Hospital of Malaga. Spain. Data entry and article review.
10. JSM. MD, PhD. Parc Taulí University Hospital, Sabadell. University Autónoma, Barcelona. Spain. Data entry and article review.
11. MJL. Pediatric. Universtiy Hospital Macarena, Sevilla. Spain. Data entry.
12. RR. Rheumatology Department. University Hospital Reina Sofía, Córdoba. Spain. Data entry.
13. MC. Virgen del Rocío University Hospital, Sevilla. Spain. Data entry and article review.
14. MM. Germans Trias i Pujol University Hospital, Badalona. Spain. Data entry and article review.
15. MM. Miguel Servet University Hospital, Zaragoza. Spain. Data entry.
16. PA. Pediatra. Virgen de la Arrixaca University Clinical Hospital, Murcia. Spain. Data entry and article review.
1. JA. Sant Joan de Déu Hospital, Barcelona. Associate Professor University of Barcelona (UB). Spain. Article review.
1. EI. Sant Joan de Déu Hospital, Barcelona. Spain. Database creation, data analysis and interpretation, article writing.
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The local ethical committee of Sant Joan de Déu Hospital approved it with the code CEIC PIC-74-13. All patients signed the informed consent for participation in the study, use and analysis of the data.
Consent for publication
All patients or their legal representatives signed the informed consent prepared by the juvenile dermatomyositis working group of Spanish Society of Paediatric Rheumatology (SERPE) to participate in the study, use and analysis of the data.
Competing interests
The authors have declared no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Carriquí-Arenas, S., Mosquera, J.M., Quesada-Masachs, E. et al. Clinical characteristics and prognostic factor in juvenile dermatomyositis: data of the Spanish registry. Pediatr Rheumatol 22, 66 (2024). https://doi.org/10.1186/s12969-024-00999-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-024-00999-9