
Abstract
Juvenile Idiopathic Inflammatory Myopathies (IIM) are a group of rare diseases that are heterogeneous in terms of pathology that can include proximal muscle weakness, associated skin changes and systemic involvement. Despite options for treatment, many patients continue to suffer resistant disease and lasting side-effects. Advances in the understanding of the immunopathology and genetics underlying IIM may specify new therapeutic targets, particularly where conventional treatment has not achieved a clinical response. An upregulated type I interferon signature is strongly associated with disease and could be a prime target for developing more specific therapeutics. There are multiple components of the IFN pathway that could be targeted for blockade therapy.
Downstream of the cytokine receptor complexes are the Janus kinase-signal transducers and activators of transcription (JAK-STAT) pathway, which consists of JAK1–3, TYK2, and STAT1–6. Therapeutic inhibitors have been developed to target components of this pathway. Promising results have been observed in case studies reporting the use of the JAK inhibitors, Baricitinib, Tofacitinib and Ruxolitinib in the treatment of refractory Juvenile Dermatomyositis (JDM). There is still the question of safety and efficacy for the use of JAK inhibitors in JDM that need to be addressed by clinical trials. Here we review the future for the use of JAK inhibitors as a treatment for JDM.
Similar content being viewed by others

Introduction
Idiopathic Inflammatory Myopathies (IIM) are a group of rare immune-mediated diseases that are heterogeneous in terms of pathology, clinical phenotypes and age of onset (Table 1). JDM is very rare with an annual incidence of three cases per million children [2, 23, 24] and median age of onset 6.3 years old (IQR; 3.8–9.6) [1]. Children typically present with symmetrical proximal and axial muscle weakness and characteristic skin changes including Gottron’s papules and heliotrope rash. Long-term complications include lung fibrosis, lipodystrophy and calcinosis [25,26,27,28,29] . In most JDM cohorts, 60–70% of children with JDM are positive for an autoantibody [30,31,32,33]. A number of myositis specific antibodies (MSA) have been described associated with a variety of phenotypes in JDM [10].
The need for new treatments
The mainstay treatments for IIM are prednisolone and methotrexate, and even those patients who respond well to these drugs can have prolonged disease [34, 35]. Other immunotherapy treatments used include mycophenolate mofetil, cyclophosphamide, intravenous immunoglobulin (IvIG), azathioprine, cyclosporine and tacrolimus [36,37,38]. Biological targets include blockade of tumour necrosis factor alpha (TNFα) and B cells (anti-CD20). As potential treatments for JDM, efficacy was reported in a case series of the use of adalimumab and infliximab (TNFα blockades), and also in an International study of B cell depletion by rituximab (anti-CD20) [39, 40]. However, there is a need for more targeted treatments and methods to identify patients who will require these.
Several more recent emerging biologic therapies for the treatment of IIM have been reported including; belimumab, abatacept, bimagrumab, spiponimod, apremilast, gevokizumab, eculizumab and basiliximab (Table 2) [41,42,43,44,45,46,47,48]. Sifalimumab, is a fully human immunoglobulin G1 κ anti-IFNα monoclonal antibody that binds to and neutralizes the majority of IFN-α subtypes, is an important candidate therapeutic due to the wealth of evidence of the strong IFN signature identified in myositis [11, 12, 50,51,52,53,54,55]. A phase 1b clinical trial of sifalimumab in adult patients with dermatomyositis (DM) and polymyositis (PM), used outcome measures of IFN gene signature suppression against disease improvement. Initial results suggested that targeting the IFN pathway with sifalimumab showed more neutralisation of IFN gene expression in patients that had greater improvement of disease, but blockade of the type I IFN receptor (IFNAR) may offer superior clinical benefit [49]. Beyond the therapeutics highlighted in Table 2 there are potential new therapies for the treatment of IIM including JAK inhibitors to target the IFN pathway.
Interferon: mechanisms in autoimmune disease
While the interferon family are a group of molecules central to the anti-viral responses, many autoimmune diseases also have an aberrant interferon response. Gene activation is the main mechanism for the interferon anti-viral response, but interferons are also integral to intra-cellular signalling in the immune system (Additional file 1: Supplementary Fig. 1 [56]). Many autoimmune diseases have been found to have an up-regulated IFN type I signature, including systemic lupus erythematous (SLE), rheumatoid arthritis (RA) and myositis [11, 50, 51, 57,58,59]. The IFN type I comprise of thirteen types including IFN-α, IFN-β, IFN-κ, IFN-ω and IFN-ν; these bind to a common receptor, IFN-α receptor (IFNAR), but the differences in induction of cellular responses is poorly defined [60]. There are three proposed mechanisms. The first is that plasmacytoid dendritic cells (pDCs) are activated by endogenous IFN inducers to produce IFN-α [61]. The second is that genes associated with autoimmune disease risk, lie within the IFN type I signalling pathway that in turn effect the production and response of IFN-α. IFN-regulatory factor (IRF) 5 was identified as a SLE risk gene as it has increased expression and is activated in SLE patients [62,63,64]. Other autoimmune diseases have specific risk genes that associate with the IFN signature [65]. The third mechanism proposes that regulation and control of plasmacytoid dendritic cells (pDC) and the expression of interferon regulatory genes (IRG) is not functioning correctly [61]. A decrease in reactive oxygen species (ROS) production from monocytes can lead to enhanced autoimmunity. In addition there is a predominant STAT1 signature in ROS deficient disease [66]. The relative contribution of these three mechanisms may differ between autoimmune disease, severity and patient.
Role of interferons in myositis
The most abundant IFN type I are IFN-α and IFN-β. The IFNs bind to the IFN-α receptor (IFNAR) and activate the Janus kinase (JAK)-signal transducer and transcription (STAT) pathway that in turn lead to the transcription of IFN-stimulated genes (ISGs) [67, 68]. The over production of IFN in the blood and muscle is an abnormality in the pathogenesis of dermatomyositis [13, 14, 69]. The release of IFN type I leads to immune cell activation and vasculopathy. A major source of IFN type I is from plasmacytoid dendritic cells (pDC) after activation by either self-DNA or viral nucleic acid [70, 71]. Plasmacytoid dendritic cells (pDC) have been identified in JDM muscle, but IFN type I is difficult to detect in serum due to limits of sensitivity of existing assays until recently [11, 72]. The Simoa assay developed by Rodero et al. can detect IFN-α at differential levels and determine cellular sources measured from lysed cell-subsets [57]. Using this assay IFN-α levels were significantly increased in sera from a JDM cohort compared to a healthy cohort [57, 73].
Due to the difficulties in measuring IFN directly, gene expression is often used as a marker of the activation of the IFN type I pathway. An IFN score was developed to encompass a selection of the IFN response genes, IFI27, IFI44L, IFIT1, ISG15, RSAD2 and SIGLEC1, these are measured by quantitative reverse transcription polymerase chain reaction (qPCR) [74]. Other studies have also measured expression of additional genes including ISG15 ubiquitin-like modifier (G1P2), and interferon regulatory factor 7 (IRF7) [51, 71]. Variations of this score have been used to correlate with disease in multiple studies [53, 75, 76]. A signature of 43 genes was elevated in myositis compared to controls [14]. A positive correlation has been shown between an IFN score (6 genes) compared to serum IFN-α levels (n = 24, Rs = 0.620, p = 0.0004) taken from JDM patients [57]. The type II IFN signature also correlates to disease activity in JDM and other chemokines [77]. This suggests that as a whole the IFN family are upregulated in the context of JDM and adult DM. The clinical trial of sifalimumab in DM/PM showed suppression of the IFN gene signature in blood and muscle tissue of the IIM patient cohort. Patients with 15% or greater improvement from baseline manual muscle testing scores (MMT8) showed greater neutralisation of the interferon gene signature than patients with less than 15% improvement [49]. This trial highlights the potential for the therapeutic targeting of interferon in DM and JDM.
Another indirect measure is the IFN-driven protein signature which may include measurement of levels of monocyte chemoattractant protein 1 (MCP-1), monocyte chemoattractant protein 2 (MCP-2), interferon gamma-induced protein 10 (IP-10), tumour necrosis factor receptor II (TNFRII), galectin 9 and chemokine (C-XC motif) ligand 9 (CXCL9). These proteins, measured in serum, significantly correlated with disease activity in JDM [22, 78,79,80]. Chemokines and cytokines have shown to correlate with the IFN signature in peripheral blood mononuclear cells. A study in JDM showed an expansion of peripheral blood naïve immature B cells, skewed to an inflammatory profile, in early disease, that correlated with an IFN type I score taken from RNA-seq analysis of B cells and downstream IFN proteins [81].
Circulating endothelial cells (CEC) have been detected in peripheral blood and associated with vascular injury [82]. An in vitro study has shown that IFN type I treatment of HUVECs impaired endothelial cell function, with significant reduction of tubule formation when HUVECs were cultured with IFN type I + VEGF and anti-IP10 [83]. A recent study in JDM, identified higher CEC in both active and definite inactive disease (JDM n = 90; median 96(IQR 40–192) cell/ml compared to controls n = 79; median 12(IQR 8–24) cells/ml, p < 0.0001). They also showed a strong correlation with other markers of vascular injury including endothelial microparticles and galectin-9 [9]. Another study showed that CEC correlated with extra muscular disease activity but not muscular damage [84]. In JDM, CEC may prove to be a useful biomarker for underlying disease pathology.
Key sites of inflammation in JDM are the muscle and skin. Both muscle and skin tissue biopsy material can provide valuable insights to our understanding of an individual JDM patients disease [85,86,87,88]. These tissue samples are the key to understanding the pathophysiology of disease at the tissue site. IFN type I and other cytokines have been detected within the inflamed muscle [89]. The IFN proteins (IFN-α,-β,-γ) themselves have been detected in muscle, but also the IFN-stimulated proteins ISG15, MxA and class I MHC [90,91,92]. Higher levels of ISG15 were quantified in JDM muscle tissue compared to non-JDM [93]. Markers of disease activity and muscle damage have been shown to correlate with the expression of MxA in the muscle tissue [94, 95]. Research has been carried out to identify the direct effects that IFN type I has on muscle tissue types. Muscle atrophy and loss of myogenin has been detected on muscle myotubes, reduced junctions and capillary growth on endothelium [96]. A recent study has shown that these effects have been blocked in vitro by the JAK inhibitor Ruxolitinib [97]. These findings build a picture of the interferonopathy at both the tissue site and the peripheral blood.
The JAK-STAT pathway – a therapeutic target
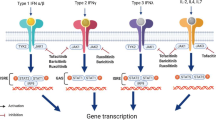
When IFN binds to its respective receptor, IFN-R, on the cell surface membrane, this in turn activates the signalling cascade inducing the Janus kinase-signal transducers and activators of transcription (JAK-STAT) pathway [98, 99] (Fig. 1). The JAK-STAT pathway consists of JAK1–3, TYK2, and STAT1–6, of these JAK1 and TYK2 are directly activated by IFN type 1 proteins. This signalling cascade triggers the receptor-associated JAK to phosphorylate the receptor and other JAKs [100]. If specific tyrosine motifs are phosphorylated in the cytokine receptor, then a docking-site for STATs is opened enabling further phosphorylation of STATs. When STATs are phosphorylated they dimerize through their Src homology domain-2 (SH2) domains, this allows them to translocate to the nucleus and activate specific genes [101]. An individual receptor is made up of several subunits, each is associated with a specific JAK. Therefore, each receptor chain can have more or less specificity to an individual JAK. The JAK-STAT pathway could offer a potential target for the blockade of the transcription of IFN genes [100].

JAK-STAT pathway with JAK inhibitor targets. The activation of the JAK-STAT pathway after IFN type 1 has engaged with the associated receptor, IFNR. This induces the transcription of proteins. Tofacitinib inhibits JAK1/2/3. Ruxolitinib and Baricitinib inhibit JAK 1/2, inhibtion prevents STAT phosphorylation, dimeraziation and transolcation into the nucleuse. This in turn stops the transcription of pro-inflammatory proteins
JAK inhibition
JAKs are constructed from four domains made of seven homologous regions (JH1–7) (Fig. 2). To date JAK inhibitors (JAK-inhibitors) have generally targeted the JH1 domain. JH1 is the active catalytic phosphotransferase domain and competes with adenosine triphosphate at the catalytic site [102]. JH2 is a pseudokinase domain that supresses ligand-independent kinase activity, the mode of action is direct interaction with JH1 and activation of ligand-induced JAK [103]. Deucravacitinib is an example of a JAK inhibitor that targets the JH2 psuedokinase domain [104, 105]. JH3/4 have a primary role in stabilising the structure of the enzyme. JH5–7 associate JAKs with their cognate receptors [106]. There have been multiple JAK-inhibitors that have been or are in development. These can be defined in two categories; first-generation or next-generation JAK-inhibitors [100]. The first-generation exert pan-inhibition on all four of the JAKs, these include; tofacitinib, ruxolitinib, baricitinib, and oclacitinib [107]. The next-generation of JAK-inhibitors are more specific in their target blockade, these include; fedratinib, momelotinib, and pacritinib [108]. This specificity should help with disease targeted treatment and reduce associated side-effects.

JAK domains and homologous regions. JAKs are constructed from four domains made of seven homologous regions (JH1–7)
Inhibition of TYK2 is an example of a more specific next-generation Jakinib. TYK2 has been associated with several autoimmune conditions including; RA, JIA, SLE, type 1 diabetes and MS [109,110,111,112,113,114]. A GWAS analysis of IIM in Caucasian individuals identified that a non-synonymous SNP rs2304256 in TYK2 was associated with DM, IIM but not PM (Bonferroni correction p = 0.17, [115]. In a study of a Chinese Han population, analysis of TYK2 SNPS associated with DM and PM excluded TYK2 rs2304256 as it deviated from the Hardy-Weinberg equilibrium (HWE) in healthy controls [116]. This SNP is in the protein FERM (4.1 protein, erzin, radixin, moesin) domain, mediating interaction with JAK and microtubule interacting proetin1, this is thought to be increased in DM. Examples of TYK2 inhibitors trialled in psoriasis include; brepocitinib, BMS-986165 and PF-06826647 [117]. TYK2 is just one proposed target for inhibition in IIM.
JAK inhibitor use in IIM – clinical trials
For the potential treatment of autoimmune conditions, multiple JAK inhibitors have been developed, trialled or approved [104, 107]. In adults the metabolism, pharmacokinetics and efficacy of JAK-inhibitors are highlighted in Additional file 1: Supplementary Table 1. Clinical trials are ongoing to determine the safety and efficacy of the use of multiple JAK-inhibitors as a therapy for treatment-resistant adult IIM. In addition to their small open-label, proof-of concept study of tofacitinib in 10 treatment-resistant DM patients (6 were anit-TIF1-γ positive), Paik et al. are carrying out a larger randomised controlled trial, with results pending (NCT03002649). Initial results from 10 participants showed they all met the primary outcome DOI at 12 weeks, 5 of 10 (50%) had moderate improvement and 5 of 10 (50%) had minimal improvement according to the 2016 ACR/EULAR myositis response criteria. The secondary outcome showed a significant change in CDASI disease activity score (mean average 28 ± 15.4 (baseline) vs. 9.5 ± 8.5 (12 weeks), p = 0.0005). There was also a trend towards a reduction of CXCL9/10 in serum and STAT1 signalling in 3 of 9 skin biopsies [118]. Another case report of 3 patients with refractory DM and calcinosis treated with tofacitinib showed an improvement of their calcinosis after 12 weeks (3 months) on treatment [119]. There is little know about the pathology of calcinosis but if JAK-inhibitors are effective then the JAK-STAT pathway may play a role in the underlying mechanism. Chen et al. are conducting a single centre, open-label clinical study of the use of tofacitinib in amyopathic dermatomyositis-associated ILD (Chinese Clinical Trial Registry number, ChiCTR-1,800,016,629). Initial results showed that 26 week (6-month) survival after onset of ILD was significantly higher in the prospective group (18 of 18, 100%) compared to the historical controls (25 of 32, 78%, p = 0.04), more conclusive results are pending [120]. Another ongoing study of the use of Baricitinib in adult IIM, is the MYOJAK study, a phase II, multicentre, randomised treatment delayed-start trial to receive active treatment (Baricitinib) or delayed-start after 13 weeks (NCT04208464). These trials are currently only including adult IIM patients of which have different clinical features to that of juvenile disease. Children have distinct developmental and physiological differences to adults, as such it is important to test the pharmokinectics and formulation of any given drug in the appropriate age populations.
Evidence for the use of JAK inhibitors in JDM
There have been several reports and case series which support the need to pursue testing JAK-inhibitors for the future therapeutic use in juvenile DM (Table 3). The potential for Ruxolitinib was shown in a report of compassionate treatment for a case of severe vasculopathic refractory JDM. The thirteen year old patient presented with severe disease and was admitted to ICU after 3 weeks of diagnosis with multi-symptom, systemic disease. Over a period of 78 weeks (18 months) the patient was poorly controlled with combination therapy, and developed lower limb oedema and diffuse fascia calcinosis. The IFN type I signature was investigated, which showed IFN-α serum levels and IFN score were increased compared to controls, this was also the case with constitutive phosphorylation of STAT1/3 in T-cells and monocytes. From these results the patient was taken off MMF, rituximab infusions were stopped, and switched to Ruxolitinib (10 mg BD) with Prednisolone. After 2 table there was a noted improvement in disease activity scores and no reported adverse events. During the 52 weeks (12 months) of Ruxolitnib treatment the IFN measures did not normalise, but there was decreased STAT 1 phosphorylation in T cells [121].
Positive results were seen in a compassionate case of the use of the Jakinib, Baricitinib for an eleven year old male with a seven year history of refractory JDM positive for anti-TIF1-γ and anti-Ro52 autoantibodies. When Baricitinib therapy was started clear improvement of disease was recorded. The IFN biomarkers, IFN type I signature and STAT1 phosphorylation in T cells and monocytes, decreased to comparative levels seen in controls. Also observed was a marked decrease of CEC. To note this was a singular, very severe case, however for the first time in seven years prednisolone could be tapered down, progression of calcinosis was halted and the disease improved as a whole [122]. Further prospective studies need to be carried out to investigate the safety and efficacy of Baricitnib for the use in the treatment of JDM.
A report of 2 patients with anti-MDA5 AB+ JDM with uncontrolled disease were treated with tofacitinib. Disease activity scores decreased within 26 weeks (6 months) following the start of tofacitinib therapy; IFN score, STAT1 phosphorylation of T-cells and monocytes decreased. This report shows evidence that tofacitinib improves JDM at an immunopathogenic level [123]. Another recent report of 3 cases of refractory JDM showed that 26 weeks (6 months) of treatment with tofacitinib was tolerated and the patients responded well to the treatment. Comparing 0–26 weeks (0–6 months) on treatment there were significant improvements in physician global VAS (p < 0.001), manual muscle testing-8 (MMT) (p = 0.002), child myositis assessment scale (CMAS) (p = 0.006), C-HAQ (p < 0.001) and DAS (p = 0.002). This set of case reports showed that tofacitinib treatment improved signs and symptoms of JDM and could be a promising treatment option [124].
A recent retrospective study included nine refractory and one new-onset JDM patients treated with ruxolitinib (n = 7) or baricitinib (n = 3). At 26 weeks (6 months) of follow up five of the ten patients (three Ruxolitnib and two Baricitinib) had reached clinically inactive disease (CID). In these patients the mean daily dose of steroids decreased from 1.1 mg/kg (range 0.35–2) to 0.1 (range, 0–0.3, p = 0.008). Serum IFN-α levels normalised 26 weeks (6 months) after the start of treatment in all patients [125].
A larger case series of refractory JDM patients, 8/25(35%) treatment was ineffective and 17/25 (68%) glucocorticoid dependant, were treated with tofacitinib 7/25(28%) or ruxolitinib 18/25 (72%). All 25 patients were followed up for a median of 30 weeks (7 months) (range = 3–21 months). 24/25 (96%) of patients had improvement of their rash of which 16/24 (66.7%) the rash completely resolved. The cutaneous assessment tool binary method score significantly decreased (7.0(3.0–10.0) to 0.0(0.0–1.0) p < 0.001). As a measure of muscle activity 7/25 (28%) of patients showed an improvement of CMAS score (from 18.6 ± 15.0 to 35.7 ± 6.3, p = 0.018). As of follow-up in August 2019 7/25 (28%) of patients had discontinued glucocorticoids. This case series has shown promise for the use of both drugs especially to improve skin disease [126].
Recently data has been published from a compassionate use study (NCT01724580) for the treatment of JDM with Baricitinib. Four JDM patients with chronically active disease were assessed at regular intervals over a 24 week period. There was significant improvement in clinical scores from 4 weeks (Physicians Global Assessment, Pt Global activity and CDASI activity score) and down-regulation of IRG score (28 genes) and serum IP-10. In CD4+ and CD8+ T Cells there were lower levels IFN-α stimulated pSTAT1 and interleukin-2 (IL-2) stimulated pSTAT5 IC50s. In CD4+ T cells and CD19+ B cells there were lower levels of IL-10- stimulated pSTAT3 IC50s [127].
Overall, these reports provide more supportive evidence for the use of JAK-inhibitors in JDM, but these are limited case studies with the use of several distinct JAK-inhibitors. Along with specific clinical trials of the use of JAK-inhibitors in the treatment of JDM, there is a need for standardised outcome measures for both clinical and pathological disease improvement.
The future of JAK inhibitors
Clinical trials currently only include adult IIM patients. Successful results from these trials and validation of the case studies in JDM should be translatable to trials and treatment in juvenile disease. There are multiple JAK-inhibitors that are being trialled as potential new therapeutics for adult IIM, but these differ in their JAK targets and pharmokinetics. JAK-inhibitors provide one step further towards more targeted treatment beyond IFN blockade. It is vital to continue to investigate the exact pathogenic mechanism of the JAK/STAT pathway in IIM. If a more specific target can be found then a refined Jakinib can be developed for clinical trial in juvenile disease.
Concluding remarks
There is a wealth of information and evidence for the potential use of JAK-inhibitors as a therapy for JDM. There is a desperate need for therapeutics that target defined pathogenic pathways in JDM. The IFN pathway is a clear point of target. JAK-inhibitors appear to be promising, but there is still the question of safety and efficacy for the use in JDM. The choice of agent will need careful consideration before choice of trials of first generation pan-JAK-inhibitors or next-generation JAK specific inhibitors. An international collaborative approach, or novel trial design for disease trials, may be required in order to achieve clear evidence of efficacy.
Availability of data and materials
Not applicable.
Abbreviations
- JAK-STAT:
-
Janus kinase-signal transducers and activators of transcription
- JDM:
-
Juvenile dermatomyositis
- IIM:
-
Idiopathic Inflammatory Myopathies
- JDCBS:
-
UK Juvenile Dermatomyositis cohort and biobank study
- MSA:
-
Myositis specific autoantibodies
- TNFα:
-
Tumour necrosis factor alpha
- DM:
-
Dermatomyositis
- PM:
-
Polymyositis
- IFNAR:
-
Type I IFN receptor
- SLE:
-
Systemic lupus erythematous
- IFN type I:
-
Interferon type I
- pDCs:
-
Plasmacytoid dendritic cells
- IFR:
-
IFN-regulatory factor
- IRG:
-
Interferon regulatory genes
- PCR:
-
Polymerase chain reaction
- G1P2:
-
ISG15 ubiquitin-like modifier
- IRF7:
-
Interferon regulatory factor 7
- MMT8:
-
Manual muscle testing scores
- IL-6:
-
Interleukin 6
- MCP1:
-
Monocyte chemoattractant protein 1
- MCP2:
-
Monocyte chemoattractant protein 2
- IP-10:
-
Interferon gamma-induced protein 10
- TNFRII:
-
Tumour necrosis factor receptor II
- CEC:
-
Circulating endothelial cells
- SH2:
-
Src homology domain-2
- HWE:
-
Hardy-Weinberg equilibrium
- TIS:
-
Total improvement score
- MMT:
-
Manual muscle testing-8
- CMAS:
-
Child myositis assessment scale
- RA:
-
Rheumatoid arthritis
- PsA:
-
Psoriatic arthritis
- Ps:
-
Psoriasis
- AS:
-
Ankylosing spondylitis
- AA:
-
Alopecia areata
- AD:
-
Atopic dermatitis
- CD:
-
Crohn’s disease
- UC:
-
Ulcerative colitis
- Vit:
-
Vitiligo
- HPS:
-
Hemophagocytic syndrome
- GCA:
-
Giant cell arteritis
- NIU:
-
Non-infectious uveitis
- CLE:
-
Cutaneous lupus erythematosus
- LN:
-
Lupus nephritis
- DLE:
-
Discoid lupus erythematosus
- dSc:
-
Diffuse scleroderma
References
Martin N, Krol P, Smith S, Murray K, Pilkington CA, Davidson JE, et al. A national registry for juvenile dermatomyositis and other paediatric idiopathic inflammatory myopathies: 10 years’ experience; the juvenile dermatomyositis national (UK and Ireland) cohort biomarker study and repository for idiopathic inflammatory Myopat. Rheumatology. 2011;50(1):137–45. https://doi.org/10.1093/rheumatology/keq261.
Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatol. 2015;54(1):50–63.
Ramanan AV, Feldman BM. Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin N Am. 2002;28(4):833–57. https://doi.org/10.1016/S0889-857X(02)00024-8.
Fisler RE, Liang MG, Fuhlbrigge RC, Yalcindag A, Sundel RP. Aggressive management of juvenile dermatomyositis results in improved outcome and decreased incidence of calcinosis. J Am Acad Dermatol. 2002;47(4):505–11. https://doi.org/10.1067/mjd.2002.122196.
Huber AM, Lang B, LeBlanc CMA, Birdi N, Bolaria RK, Malleson P, et al. Medium- and long-term functional outcomes in a multicenter cohort of children with juvenile dermatomyositis. Arthritis Rheum. 2000;43(3):541–9. https://doi.org/10.1002/1529-0131(200003)43:3<541::AID-ANR9>3.0.CO;2-T.
Huemer C, Kitson H, Malleson PN, Sanderson S, Huemer M, Cabral DA, et al. Lipodystrophy in patients with juvenile dermatomyositis - evaluation of clinical and metabolic abnormalities. J Rheumatol. 2001;28(3):610–5.
Morinishi Y, Oh-Ishi T, Kabuki T, Joh K. Juvenile dermatomyositis: clinical characteristics and the relatively high risk of interstitial lung disease. Mod Rheumatol. 2007;17(5):413–7. https://doi.org/10.3109/s10165-007-0610-y.
Cantez S, Gross GJ, MacLusky I, Feldman BM. Cardiac findings in children with juvenile Dermatomyositis at disease presentation. Pediatr Rheumatol Online J. 2017;15(1):54.
Papadopoulou C, Hong Y, Krol P, Al Obaidi M, Pilkington C, Wedderburn LR, et al. The vasculopathy of juvenile dermatomyositis: endothelial injury, hypercoagulability, and increased arterial stiffness. Arthritis Rheum. 2021;73(7):1253–66. https://doi.org/10.1002/art.41639.
Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. 2016;280(1):8–23. https://doi.org/10.1111/joim.12451.
Baechler EC, Bilgic H, Reed AM. Type I interferon pathway in adult and juvenile dermatomyositis. Arthritis Res Ther. 2011;13(6):249. https://doi.org/10.1186/ar3531.
Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57(5):664–78.
Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener PA, Pinkus JL, et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56(11):3784–92.
Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. 2007;13(1–2):59–68.
McDouall RM, Dunn MJ, Dubowitz V. Nature of the mononuclear infiltrate and the mechanism of muscle damage in juvenile dermatomyositis and Duchenne muscular dystrophy. J Neurol Sci. 1990;99(2–3):199–217. https://doi.org/10.1016/0022-510X(90)90156-H.
Vercoulen Y, Bellutti Enders F, Meerding J, Plantinga M, Elst EF, Varsani H, et al. Increased presence of FOXP3+ regulatory T cells in inflamed muscle of patients with active juvenile dermatomyositis compared to peripheral blood. PLoS One. 2014;9(8):e105353. https://doi.org/10.1371/journal.pone.0105353.
López De Padilla CM, Vallejo AN, McNallan KT, Vehe R, Smith SA, Dietz AB, et al. Plasmacytoid dendritic cells in inflamed muscle of patients with juvenile dermatomyositis. Arthritis Rheum. 2007;56(5):1658–68. https://doi.org/10.1002/art.22558.
Ekholm L, Michelle Kahlenberg J, Helmers SB, Tjärnlund A, Yalavarthi S, Zhao W, et al. Dysfunction of endothelial progenitor cells is associated with the type I IFN pathway in patients with polymyositis and dermatomyositis. Rheumatol (United Kingdom). 2016;55(11):1987–92.
Xu D, Kacha-Ochana A, Morgan GA, Huang CC, Pachman LM. Endothelial progenitor cell number is not decreased in 34 children with juvenile dermatomyositis: a pilot study. Pediatr Rheumatol. 2017;15(1):3–7. https://doi.org/10.1186/s12969-017-0171-3.
Yokota M, Suzuki K, Tokoyoda K, Meguro K, Hosokawa J, Tanaka S, et al. Roles of mast cells in the pathogenesis of inflammatory myopathy. Arthritis Res Ther. 2014;16(2).
Briones MR, Morgan GA, Amoruso MC, Rahmani B, Ryan ME, Pachman LM. Decreased CD3-CD16+CD56+ natural killer cell counts in children with orbital myositis: a clue to disease activity. RMD Open. 2017;3(1):1–5. https://doi.org/10.1136/rmdopen-2016-000385.
Wienke J, Deakin CT, Wedderburn LR, van Wijk F, van Royen-Kerkhof A. Systemic and Tissue Inflammation in Juvenile Dermatomyositis: From Pathogenesis to the Quest for Monitoring Tools. Front Immunol. 2018;9:2951.
Pachman LM, Lipton R, Ramsey-Goldman R, Shamiyeh E, Abbott K, Mendez EP, et al. History of infection before the onset of juvenile dermatomyositis: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Registry. Arthritis Rheum. 2005;53(2):166–72.
Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Med. 2013;92(1):25–41.
McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)--clinical characteristics of children recruited within the first 5 yr. Rheumatol. 2006;45(10):1255–60.
Guseinova D, Consolaro A, Trail L, Ferrari C, Pistorio A, Ruperto N, et al. Comparison of clinical features and drug therapies among European and Latin American patients with juvenile dermatomyositis. Clin Exp Rheumatol. 2011;29(1):117–24.
Ravelli A, Trail L, Ferrari C, Ruperto N, Pistorio A, Pilkington C, et al. Long-term outcome and prognostic factors of juvenile dermatomyositis: a multinational, multicenter study of 490 patients. Arthritis Care Res. 2010;62(1):63–72.
Sato JO, Sallum AM, Ferriani VP, Marini R, Sacchetti SB, Okuda EM, et al. A Brazilian registry of juvenile dermatomyositis: onset features and classification of 189 cases. Clin Exp Rheumatol. 2009;27(6):1031–8.
Christen-Zaech S, Seshadri R, Sundberg J, Paller AS, Pachman LM. Persistent association of nailfold capillaroscopy changes and skin involvement over thirty-six months with duration of untreated disease in patients with juvenile dermatomyositis. Arthritis Rheum. 2008;58(2):571–6.
Tansley SL, Betteridge ZE, Shaddick G, Gunawardena H, Arnold K, Wedderburn LR, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatol. 2014;53(12):2204–8.
Tansley SL, Betteridge ZE, Gunawardena H, Jacques TS, Owens CM, Pilkington C, et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther. 2014;16(4):R138. https://doi.org/10.1186/ar4600.
Gunawardena H, Wedderburn LR, North J, Betteridge Z, Dunphy J, Chinoy H, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatol. 2008;47(3):324–8. https://doi.org/10.1093/rheumatology/kem359.
Rider LG, Shah M, Mamyrova G, Huber AM, Rice MM, Targoff IN, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Med. 2013;92(4):223–43.
Vencovský J, Alexanderson H, Lundberg IE. Idiopathic inflammatory myopathies. Rheum Dis Clin N Am. 2019;45(4):569–81. https://doi.org/10.1016/j.rdc.2019.07.006.
Deakin CT, Yasin SA, Simou S, Arnold KA, Tansley SL, Betteridge ZE, et al. Muscle biopsy in combination with myositis-specific autoantibodies aids prediction of outcomes in juvenile dermatomyositis. Arthritis Rheum. 2016;11(68):2806–16. https://doi.org/10.1002/art.39753.
Deakin CT, Campanilho-Marques R, Simou S, Moraitis E, Wedderburn LR, Pullenayegum E, et al. Efficacy and Safety of Cyclophosphamide Treatment in Severe Juvenile Dermatomyositis Shown by Marginal Structural Modeling. Arthritis Rheum. 2018;70(5):785–93.
Albayda J, Christopher-Stine L. Novel approaches in the treatment of myositis and myopathies. Ther Adv Musculoskelet Dis. 2012;4(5):369–77. https://doi.org/10.1177/1759720X12447705.
Dagher R, Desjonquères M, Duquesne A, Quartier P, Bader-Meunier B, Fischbach M, et al. Mycophenolate mofetil in juvenile dermatomyositis: a case series. Rheumatol Int. 2012;32(3):711–6. https://doi.org/10.1007/s00296-010-1653-5.
Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65(2):314–24.
Campanilho-marques R, Deakin CT, Simou S, Papadopoulou C, Wedderburn LR, Pilkington CA, et al. Retrospective analysis of infliximab and adalimumab treatment in a large cohort of juvenile dermatomyositis patients. 2020;5(1):1–9. https://doi.org/10.1186/s13075-020-02164-5.
Amato AA, Sivakumar K, Goyal N, David WS, Salajegheh M, Praestgaard J, et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology. 2014;83(24):2239–46. https://doi.org/10.1212/WNL.0000000000001070.
Ioannis M, Foivos P, Dimitrios K. A review on the treatment of sporadic inclusion body myositis with Bimagrumab and Alemtuzumab. Int J Neurosci. 2019;129(3):297–302. https://doi.org/10.1080/00207454.2018.1527329.
Hatemi G, Melikoglu M, Tunc R, Korkmaz C, Ozturk BT, Mat C, et al. Apremilast for Behçet’s syndrome - a phase 2, placebo-controlled study. N Engl J Med. 2015;372(16):1510–8. https://doi.org/10.1056/NEJMoa1408684.
Anyanwu CO, Chansky PB, Feng R, Carr K, Okawa J, Werth VP. The systemic management of cutaneous dermatomyositis: results of a stepwise strategy. Int J Women’s Dermatology. 2017;3(4):189–94. https://doi.org/10.1016/j.ijwd.2017.05.001.
Bitar C, Maghfour J, Ho-Pham H, Stumpf B, Boh E. Apremilast as a potential treatment for moderate to severe dermatomyositis: a retrospective study of 3 patients. JAAD Case Reports. 2019;5(2):191–4. https://doi.org/10.1016/j.jdcr.2018.11.019.
Patwardhan A, Spencer CH. Biologics in refractory myositis: experience in juvenile vs. adult myositis; part II: emerging biologic and other therapies on the horizon. Pediatr Rheumatol Online J. 2019;17(1):56.
Faguer S, Belliere J, Ribes D. Complement C5-blocking Agent in Refractory Dermatomyositis. J Rheumatol Canada. 2018;45(12):1710–1. https://doi.org/10.3899/jrheum.180060.
Zou J, Li T, Huang X, Chen S, Guo Q, Bao C. Basiliximab may improve the survival rate of rapidly progressive interstitial pneumonia in patients with clinically amyopathic dermatomyositis with anti-MDA5 antibody. Ann Rheum Dis. 2014;73(8):1591–3. https://doi.org/10.1136/annrheumdis-2014-205278.
Higgs BW, Zhu W, Morehouse C, White WI, Brohawn P, Guo X, et al. A phase 1b clinical trial evaluating sifalimumab, an anti-IFN-alpha monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann Rheum Dis. 2014;73(1):256–62.
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100(5):2610–5.
Bilgic H, Ytterberg SR, Amin S, McNallan KT, Wilson JC, Koeuth T, et al. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. 2009;60(11):3436–46.
Gallay L, Mouchiroud G, Chazaud B. Interferon-signature in idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2019;31(6):634–42. https://doi.org/10.1097/BOR.0000000000000653.
Greenberg SA, Higgs BW, Morehouse C, Walsh RJ, Won Kong S, Brohawn P, et al. Relationship between disease activity and type 1 interferon- and other cytokine-inducible gene expression in blood in dermatomyositis and polymyositis. Genes Immun. 2012;13(3):207–13. https://doi.org/10.1038/gene.2011.61.
Kim H, Gunter-Rahman F, McGrath JA, Lee E, De Jesus AA, Targoff IN, et al. Expression of interferon-regulated genes in juvenile dermatomyositis versus Mendelian autoinflammatory interferonopathies. Arthritis Res Ther. 2020;22(1):1–12. https://doi.org/10.1186/s13075-020-02160-9.
De Jesus AA, Canna SW. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases Graphical abstract Find the latest version. 2019;130(4):1669–82.
Ronnblom L, Elkon KB. Cytokines as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6(6):339–47.
Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med. 2017;214(5):1547–55. https://doi.org/10.1084/jem.20161451.
Muskardin TLW, Niewold TB. Type i interferon in rheumatic diseases. Nat Rev Rheumatol. 2018;14(4):214–28. https://doi.org/10.1038/nrrheum.2018.31.
Cooles FAH, Anderson AE, Lendrem DW, Norris J, Pratt AG, Hilkens CMU, et al. The interferon gene signature is increased in patients with early treatment-naive rheumatoid arthritis and predicts a poorer response to initial therapy. J Allergy Clin Immunol. 2018;141(1):445–448.e4.
Crow MK. Autoimmunity: interferon alpha or beta: which is the culprit in autoimmune disease? Nat Rev Rheumatol. 2016;12(8):439–40. https://doi.org/10.1038/nrrheum.2016.117.
Ronnblom L, Eloranta ML. The interferon signature in autoimmune diseases. Curr Opin Rheumatol. 2013;25(2):248–53. https://doi.org/10.1097/BOR.0b013e32835c7e32.
Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76(3):528–37. https://doi.org/10.1086/428480.
Feng D, Barnes BJ. Bioinformatics analysis of the factors controlling type I IFN gene expression in autoimmune disease and virus-induced immunity. Front Immunol. 2013;4:291. https://doi.org/10.3389/fimmu.2013.00291.
Jiang SH, Athanasopoulos V, Ellyard JI, Chuah A, Cappello J, Cook A, et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat Commun. 2019;10(1):1–12. https://doi.org/10.1038/s41467-019-10242-9.
Stone RC, Feng D, Deng J, Singh S, Yang L, Fitzgerald-Bocarsly P, et al. Interferon regulatory factor 5 activation in monocytes of systemic lupus erythematosus patients is triggered by circulating autoantigens independent of type I interferons. Arthritis Rheum. 2012;64(3):788–98. https://doi.org/10.1002/art.33395.
Kelkka T, Kienhöfer D, Hoffmann M, Linja M, Wing K, Sareila O, et al. Reactive oxygen species deficiency induces autoimmunity with type 1 interferon signature. Antioxid Redox Signal. 2014;21(16):2231–45. https://doi.org/10.1089/ars.2013.5828.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49.
George R, Stark1,* and James E. Darnell Jr.2 *. No Title. J Immuni. 2012;36(4):503–14.
Greenberg SA, Bradshaw EM, Pinkus JL, Pinkus GS, Burleson T, Due B, et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. 2005;65(11):1782–7.
Mauri C, Menon M. The many faces of type I interferon in systemic lupus erythematosus. J Clin Invest. 2015;125(7):2562–4. https://doi.org/10.1172/JCI82574.
Menon M, Blair PA, Isenberg DA, Mauri C. A regulatory feedback between Plasmacytoid dendritic cells and regulatory B cells is aberrant in systemic lupus erythematosus. Immunity. 2016;44(3):683–97. https://doi.org/10.1016/j.immuni.2016.02.012.
Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Elevated serum interferon-alpha activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum. 2009;60(6):1815–24. https://doi.org/10.1002/art.24555.
Gitiaux C, Bondet V, Bekaddour N, Nusbaum P, Hubas A, Melki I, et al. Inhibition of IFNα secretion in cells from patients with juvenile dermatomyositis under TBK1 inhibitor treatment revealed by single-molecular assay technology. Rheumatol (United Kingdom). 2020;59(5):1171–4.
Rice GI, Melki I, Frémond ML, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol. 2017;37(2):123–32.
Kim H, De Jesus AA, Brooks SR, Liu Y, Huang Y, Vantries R, et al. Development of a validated interferon score using NanoString technology. J Interf Cytokine Res. 2018;38(4):171–85. https://doi.org/10.1089/jir.2017.0127.
Ekholm L, Vosslamber S, Tjärnlund A, de Jong TD, Betteridge Z, McHugh N, et al. Autoantibody specificities and type I interferon pathway activation in idiopathic inflammatory myopathies. Scand J Immunol. 2016;84(2):100–9. https://doi.org/10.1111/sji.12449.
Moneta GM, Pires Marafon D, Marasco E, Rosina S, Verardo M, Fiorillo C, et al. Muscle expression of type I and type II interferons is increased in juvenile dermatomyositis and related to clinical and histologic features. Arthritis Rheum. 2019;71(6):1011–21. https://doi.org/10.1002/art.40800.
Nistala K, Varsani H, Wittkowski H, Vogl T, Krol P, Shah V, et al. Myeloid related protein induces muscle derived inflammatory mediators in juvenile dermatomyositis. Arthritis Res Ther. 2013;15(5):R131. https://doi.org/10.1186/ar4311.
Bellutti Enders F, van Wijk F, Scholman R, Hofer M, Prakken BJ, van Royen-Kerkhof A, et al. Correlation of CXCL10, tumor necrosis factor receptor type II, and galectin 9 with disease activity in juvenile dermatomyositis. Arthritis Rheum. 2014;66(8):2281–9.
Van Den Hoogen LL, Van Roon JAG, Mertens JS, Wienke J, Lopes AP, De Jager W, et al. Galectin-9 is an easy to measure biomarker for the interferon signature in systemic lupus erythematosus and antiphospholipid syndrome. Ann Rheum Dis. 2018;77(12):1810–4. https://doi.org/10.1136/annrheumdis-2018-213497.
Piper CJM, Wilkinson MGL, Deakin CT, Otto GW, Dowle S, Duurland CL, et al. CD19(+)CD24(hi)CD38(hi) B Cells Are Expanded in Juvenile Dermatomyositis and Exhibit a Pro-Inflammatory Phenotype After Activation Through Toll-Like Receptor 7 and Interferon-alpha. Front Immunol. 2018;9:1372.
Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105(1):71–7. https://doi.org/10.1172/JCI8071.
Jia H, Thelwell C, Dilger P, Bird C, Daniels S, Wadhwa M. Endothelial cell functions impaired by interferon in vitro: insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb Res. 2018;163(December 2017):105–16. https://doi.org/10.1016/j.thromres.2018.01.039.
Kishi T, Chipman J, Evereklian M, Nghiem K, Stetler-Stevenson M, Rick ME, et al. Endothelial activation markers as disease activity and damage measures in juvenile dermatomyositis. J Rheumatol. 2020;47(7):1011–8.
Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344–7.
Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292(8):403–7.
Wedderburn LR, Varsani H, Li CK, Newton KR, Amato AA, Banwell B, et al. International consensus on a proposed score system for muscle biopsy evaluation in patients with juvenile dermatomyositis: a tool for potential use in clinical trials. Arthritis Rheum. 2007;57(7):1192–201.
Wedderburn LR, Rider LG. Juvenile dermatomyositis: new developments in pathogenesis, assessment and treatment. Best Pr Res Clin Rheumatol. 2009;23(5):665–78. https://doi.org/10.1016/j.berh.2009.07.007.
Lundberg I, Ulfgren AK, Nyberg P, Andersson U, Klareskog L. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 1997;40(5):865–74.
Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, et al. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci U S A. 2000;97(16):9209–14.
Li CK, Varsani H, Holton JL, Gao B, Woo P, Wedderburn LR, et al. MHC Class I overexpression on muscles in early juvenile dermatomyositis. J Rheumatol. 2004;31(3):605–9.
Tournadre A, Lenief V, Eljaafari A, Miossec P. Immature muscle precursors are a source of interferon-β in myositis: role of toll-like receptor 3 activation and contribution to HLA class i up-regulation. Arthritis Rheum. 2012;64(2):533–41. https://doi.org/10.1002/art.33350.
Salajegheh M, Kong SW, Pinkus JL, Walsh RJ, Liao A, Nazareno R, et al. Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann Neurol. 2010;67(1):53–63. https://doi.org/10.1002/ana.21805.
Uruha A, Nishikawa A, Tsuburaya RS, Hamanaka K, Kuwana M, Watanabe Y, et al. Sarcoplasmic MxA expression: a valuable marker of dermatomyositis. Neurology. 2017;88(5):493–500. https://doi.org/10.1212/WNL.0000000000003568.
Soponkanaporn S, Deakin CT, Schutz PW, Marshall LR, Yasin SA, Johnson CM, et al. Expression of myxovirus-resistance protein a: a possible marker of muscle disease activity and autoantibody specificities in juvenile dermatomyositis. Neuropathol Appl Neurobiol. 2019;45(4):410–20. https://doi.org/10.1111/nan.12498.
Allenbach Y, Leroux G, Suárez-Calvet X, Preusse C, Gallardo E, Hervier B, et al. Dermatomyositis with or without anti-melanoma differentiation-associated gene 5 antibodies common interferon signature but distinct NOS2 expression. Am J Pathol. 2016;186(3):691–700. https://doi.org/10.1016/j.ajpath.2015.11.010.
Ladislau L, Suárez-Calvet X, Toquet S, Landon-Cardinal O, Amelin D, Depp M, et al. JAK inhibitor improves type i interferon induced damage: proof of concept in dermatomyositis. Brain. 2018;141(6):1609–21. https://doi.org/10.1093/brain/awy105.
Coskun M, Salem M, Pedersen J, Nielsen OH. Involvement of JAK/STAT signaling in the pathogenesis of inflammatory bowel disease. Pharmacol Res. 2013;76:1–8. https://doi.org/10.1016/j.phrs.2013.06.007.
Gadina M, Le MT, Schwartz DM, Silvennoinen O, Nakayamada S, Yamaoka K, et al. Janus kinases to jakinibs: From basic insights to clinical practice. Rheumatol (United Kingdom). 2019;58:i4–16.
Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK–STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77(5):521–46. https://doi.org/10.1007/s40265-017-0701-9.
Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25(5):745–55. https://doi.org/10.1016/j.immuni.2006.09.009.
Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57(12):5023–38. https://doi.org/10.1021/jm401490p.
Hammarén HM, Ungureanu D, Grisouard J, Skoda RC, Hubbard SR, Silvennoinen O. ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc Natl Acad Sci U S A. 2015;112(15):4642–7. https://doi.org/10.1073/pnas.1423201112.
Spinelli FR, Meylan F, O’Shea JJ, Gadina M. JAK inhibitors: ten years after. Eur J Immunol. 2021;51(7):1615–27. https://doi.org/10.1002/eji.202048922.
Liu C, Lin J, Langevine C, Smith D, Li J, Tokarski JS, et al. Discovery of BMS-986202: a clinical Tyk2 inhibitor that binds to Tyk2 JH2. J Med Chem. 2021;64(1):677–94.
Babon JJ, Liau NPD, Kershaw NJ. JAK1 takes a FERM hold of type II cytokine receptors. Structure. 2016;24(6):840–2. https://doi.org/10.1016/j.str.2016.05.007.
Fragoulis GE, Mcinnes IB, Siebert S, JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatol (United Kingdom). 2019;58:i43–54.
Patel AA, Odenike O. The next generation of JAK inhibitors: an update on Fedratinib, Momelotonib, and Pacritinib. Curr Hematol Malig Rep. 2020;15(6):409–18. https://doi.org/10.1007/s11899-020-00596-z.
Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. 2012;44(12):1336–40.
Hinks A, Marion MC, Cobb J, Comeau ME, Sudman M, Ainsworth HC, et al. Brief Report: The Genetic Profile of Rheumatoid Factor-Positive Polyarticular Juvenile Idiopathic Arthritis Resembles That of Adult Rheumatoid Arthritis. Arthritis Rheum. 2018;70(6):957–62.
Suarez-Gestal M, Calaza M, Endreffy E, Pullmann R, Ordi-Ros J, Domenico Sebastiani G, et al. Replication of recently identified systemic lupus erythematosus genetic associations: a case-control study. Arthritis Res Ther. 2009;11(3):1–9.
Wallace C, Smyth DJ, Maisuria-armer M, Walker NM, Todd JA. UKPMC funders group alters susceptibility to type 1 diabetes. Diabetes Res. 2010;42(1):1–13.
Mero IL, Lorentzen ÅR, Ban M, Smestad C, Celius EG, Aarseth JH, et al. A rare variant of the TYK2 gene is confirmed to be associated with multiple sclerosis. Eur J Hum Genet. 2010;18(4):502–4. https://doi.org/10.1038/ejhg.2009.195.
Papp K, Gordon K, Thaçi D, Morita A, Gooderham M, Foley P, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379(14):1313–21. https://doi.org/10.1056/NEJMoa1806382.
Jani M, Massey J, Wedderburn LR, Vencovský J, Danko K, Lundberg IE, et al. Genotyping of immune-related genetic variants identifies TYK2 as a novel associated locus for idiopathic inflammatory myopathies. Ann Rheum Dis. 2014;73(9):1750–2. https://doi.org/10.1136/annrheumdis-2014-205440.
Li L, Chen S, Wang Q, Wu C, Wen X, Yang F, et al. GLIS3 and TYK2 single nucleotide polymorphisms are not associated with dermatomyositis/polymyositis in Chinese Han population. Genet Test Mol Biomarkers. 2017;21(9):565–70. https://doi.org/10.1089/gtmb.2017.0059.
Nogueira M, Puig L, Torres T. JAK inhibitors for treatment of psoriasis: focus on selective TYK2 inhibitors. Drugs. 2020;80(4):341–52. https://doi.org/10.1007/s40265-020-01261-8.
Paik JJ, Casciola-Rosen L, Shin JY, Albayda J, Tiniakou E, Leung DG, et al. Study of Tofacitinib in refractory dermatomyositis: an open-label pilot study of ten patients. Arthritis Rheum. 2021;73(5):858–65.
Shneyderman M, Ahlawat S, Christopher-Stine L, J. Paik J. Calcinosis in refractory dermatomyositis improves with tofacitinib monotherapy: a case series. Rheumatology. 2021;0:1–2.
Zhiwei C, Xiaodong Wang MD, M.D. Shuang Ye MD. Tofacitinib in Amyopathic dermatomyositis–associated interstitial lung disease. N Engl J Med. 2019;381(3):291–3. https://doi.org/10.1056/NEJMc1900045.
Aeschlimann FA, Frémond ML, Duffy D, Rice GI, Charuel JL, Bondet V, et al. A child with severe juvenile dermatomyositis treated with ruxolitinib. Brain. 2018;141(11):e80.
Papadopoulou C, Hong Y, Omoyinmi E, Brogan PA, Eleftheriou D. Janus kinase 1/2 inhibition with baricitinib in the treatment of juvenile dermatomyositis. Brain. 2019;142(3):E8.
Sabbagh S, De Jesus AA, Hwang SJ, Kuehn HS, Kim H, Jung L, et al. Treatment of anti-MDA5 autoantibody-positive juvenile dermatomyositis using tofacitinib. Brain. 2019;142(11):E59. https://doi.org/10.1093/brain/awz293.
Yu Z, Wang L, Quan M, Zhang T, Song H. Successful management with Janus kinase inhibitor tofacitinib in refractory juvenile dermatomyositis: a pilot study and literature review. Rheumatology. 2020;60(4):1–8. https://doi.org/10.1093/rheumatology/keaa558.
Le Voyer T, Gitiaux C, Authier F-J, Bodemer C, Melki I, Quartier P, et al. JAK inhibitors are effective in a subset of patients with juvenile dermatomyositis: a monocentric retrospective study. Rheumatology. 2021:1–8.
Ding Y, Huang B, Wang Y, Hou J, Chi Y, Zhou Z, et al. Janus kinase inhibitor significantly improved rash and muscle strength in juvenile dermatomyositis. Ann Rheum Dis. 2021;80(4):543–5. https://doi.org/10.1136/annrheumdis-2020-218582.
Kim H, Dill S, O’Brien M, Vian L, Li X, Manukyan M, et al. Janus kinase (JAK) inhibition with baricitinib in refractory juvenile dermatomyositis. Ann Rheum Dis. 2021;80(3):406–8. https://doi.org/10.1136/annrheumdis-2020-218690.
Acknowledgments
MGLlW would like to thank all co-authors for their contributions to this review.
Funding
All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. MGLlW was supported by fellowships from Cure JM (Early Career Investigator Award GOSH042019), NIHR Great Ormond Street Hospital (GOSH) Biomedical Research Centre (BRC) (NIHR GOSH BRC 18IR33). CP was funded by the Remission Charity. CTD, DE and LRW were supported by Arthritis Research UK, now Versus Arthritis (grant 20164 and 21593). DE and LRW also acknowledge support from Great Ormond Street Hospital Children’s Charity. LRW was also supported by a NIHR Senior Investigator Award. LRW and CTD supported by NIHR Great Ormond Street Hospital (GOSH) Biomedical Research Centre (BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR.
Author information
Authors and Affiliations
Contributions
MGLlW (wrote and prepared manuscript), CTD (review and edit); CP (review and edit); DE (review and edit); LRW (review and edit). The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
None required.
Consent for publication
Authors consent.
Competing interests
No conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure 1
The role of type I IFN and the interaction with other cytokines in the immune system.
Additional file 2: Supplementary Table 1
Metabolism, pharmacokinetics and efficacy of JAK-inhibitors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ll Wilkinson, M.G., Deakin, C.T., Papadopoulou, C. et al. JAK inhibitors: a potential treatment for JDM in the context of the role of interferon-driven pathology. Pediatr Rheumatol 19, 146 (2021). https://doi.org/10.1186/s12969-021-00637-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-021-00637-8