
Abstract
Background
Major depressive disorder (MDD) is a common mental illness that affects millions of people worldwide and imposes a heavy burden on individuals, families and society. Previous studies on MDD predominantly focused on neurons and employed bulk homogenates of brain tissues. This paper aims to decipher the relationship between oligodendrocyte lineage (OL) development and MDD at the single-cell resolution level.
Methods
Here, we present the use of a guided regularized random forest (GRRF) algorithm to explore single-nucleus RNA sequencing profiles (GSE144136) of the OL at four developmental stages, which contains dorsolateral prefrontal cortex of 17 healthy controls (HC) and 17 MDD cases, generated by Nagy C et al. We prioritized and ordered differentially expressed genes (DEGs) based on Nagy et al., which could predominantly discriminate cells in the four developmental stages and two adjacent developmental stages of the OL. We further screened top-ranked genes that distinguished between HC and MDD in four developmental stages. Moreover, we estimated the performance of the GRRF model via the area under the curve value. Additionally, we validated the pivotal candidate gene Malat1 in animal models.
Results
We found that, among the four developmental stages, the onset development of OL (OPC2) possesses the best predictive power for distinguishing HC and MDD, and long noncoding RNA MALAT1 has top-ranked importance value in candidate genes of four developmental stages. In addition, results of fluorescence in situ hybridization assay showed that Malat1 plays a critical role in the occurrence of depression.
Conclusions
Our work elucidates the mechanism of MDD from the perspective of OL development at the single-cell resolution level and provides novel insight into the occurrence of depression.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD) is a heterogeneous disease with multiple causes that affects an estimated 350 million people of all ages worldwide [1]. Research related to neuropsychiatric disorders such as MDD has dominantly focused on neurons [2]. Glia make up roughly half of the total cells in the central nervous system (CNS). However, glia have been considered static bystanders in the formation and function of the CNS [3]. Glia and neurons were first described during the same period, but due to the limitations of research methods, the study of glial cells remained at the morphological level for a long time [4]. A deep insight of glial cells could contribute to understanding of mental illness.
During the past two decades, oligodendrocyte progenitor cells (OPCs) have become known as the fourth member of the glia family, in addition to astrocytes, microglia, and oligodendrocytes [5]. It is universally acknowledged that the human brain comprises 3–10% OPCs, 25% oligodendrocytes, 20% astrocytes, and 5–15% microglia [6,7,8]. OPCs are widely distributed in the adult brain and are the most proliferative cell type in the adult CNS [9]. They differentiate and mature into oligodendrocytes during development as well as throughout adulthood. Differentiation of OPCs into oligodendrocytes follows a complex, multistep, tightly regulated process [9]. OPCs and oligodendrocytes are uniformly referred to as the oligodendrocyte lineage (OL). There is growing evidence that the OL is not just a “passive supporter” of neurons, and the cells comprising the OL are now recognized as metabolic exchangers of neurons, a cellular interface of blood vessels and responders to gut-derived metabolites or changes in the social environment [10]. Both preclinical [11,12,13,14] and clinical [15, 16] studies have shown that the OL plays an important role in the pathogenesis of depression. Therefore, studying the development of the OL is of great importance.
Studies have suggested that patients with depression suffer damage to multiple brain regions, including the hippocampus, prefrontal cortex, amygdala and hypothalamus [17, 18]. In addition to the complexity of the brain regions involved, each brain region comprises multiple types of cells with different neural circuits [19]. Each cell type acts differently and has intricate interactions with others. Given the complicated structure and multitudinous cell types of the brain, there is a high probability of the low abundance of some cell types and transcripts [20, 21]. More in-depth and accurate research approaches will pave the way for studying an elusive mental illness such as MDD [22, 23].
The rapid development of sequencing technology has occurred during the twenty-first century. The invention of single-cell RNA sequencing (scRNA-seq) technology has enabled a breakthrough in research involving the molecular mechanism profiling of many inherently complex diseases [24]. In conventional bulk RNA-sequencing (RNA-seq) analysis, the final signal is actually the average of signals from numerous cells from different regions and/or different cell types. As a result, much of the cell type-specific information is usually overlooked. For example, when a transcript has low abundance, it is very difficult to judge whether it is highly expressed in a rare cell type or expressed at low levels in most of the dominant cell types. Unlike bulk RNA-seq, scRNA-seq, a revolutionary tool, enables the analysis of transcriptomes at single-cell resolution. Since the first publication of an scRNA-seq study, this technique has been widely used in biomedical research in many contexts, including the study of tumour heterogeneity [25, 26], the identification of new cell types [27, 28], the study of tissue development and cell differentiation [29, 30], studies of gene regulatory network (GRN) [31], and investigations of differences in allelic gene expression [32].
Although single-cell sequencing technology conveniently addresses research heterogeneity, subsequent data analysis is also a problem that cannot be ignored. Due to the temporal and spatial specificity of gene expression in cell types, many genes are not expressed in a certain type of cell at a certain time, which leads to node sparsity issues when creating tree-based predictive models. When instances are recursively split in the tree, the number of instances decreases, and measures calculated from nodes with a spot of instances cannot effectively distinguish between features with different predictive information. The extraction of significant features contributes to implement the efficient model, improve the accuracy and reduce the training time. An ensemble machine learning algorithm, random forest (RF), comprises decision trees, which is quick and robust to the noise of target data [33].
Guided regularized random forest (GRRF) is an enhanced method for RF analysis, which is computationally efficient and compact feature subsets [34]. GRRF obtains a subset of relevant and non-redundant features by the regularization of the information gain in the random forest nodes [35]. The importance scores in GRRF are from a preliminary RF, and each feature in the RF is assigned a penalty coefficient. GRRF model couple with prior or statistical information to well select feature. Previous study demonstrated that integration of statistical feature extraction and GRRF feature selection can enhance the detection accuracy compared to conventional detection methods [33]. GRRF is constantly applied to analyse highly heterogeneous data, GRN, integrated multiple data and single-cell sequencing data [36,37,38,39].
In this study, we attempt to illustrate the relationship between OL development and depression at the single-cell level using GRRF methods. We addressed the following three problems, which are listed in order of increasing complexity. (1) Which genes distinguish the four stages of development? (2) Which genes promote the transformation of two adjacent developmental stages? (3) Which of the four developmental stages has better predictive power of MDD? Our work links the development of the OL with the occurrence of depression, providing a new perspective for the study of the pathogenesis of depression.
Materials and methods
Single-nucleus RNA-Seq data collection
-
(1)
The GSE144136 dataset was downloaded from the Gene Expression Omnibus database. GSE144136 contains postmortem dorsolateral prefrontal cortex (BA9) tissue from 17 healthy controls (HC) and 17 MDD cases who died by suicide [13]. All subjects were male. The sample collection process lasted nearly 15 years, and single-nucleus RNA sequencing (snRNA-seq) was performed on the frozen samples.
-
(2)
A total of 26 distinct cell types were identified in GSE144136, among which are five cell types belonging to the OL, namely, OPC1, OPC2, oligodendrocytes1, oligodendrocytes2, and oligodendrocytes3 (Hereafter, oligodendrocytes are designated as “Oligos”). Four cell types were selected: OPC1, OPC2, Oligos1, and Oligos3. After quality control filtering, there were merely few Oligos2 cells, which was inappropriate for constructing prediction model. Therefore, this cell type was excluded from this study. The numbers of the four cell clusters in the HC and MDD groups are displayed in Table 1. We downloaded the differentially expressed genes (DEGs) in these four cell clusters from the supplementary materials in reference [13] for subsequent analysis.
-
(3)
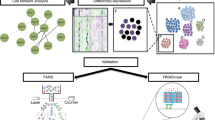
The reconstruction of the OL developmental trajectory indicated that OPC2 cells were the youngest cell type, followed by OPC1, Oligos3 and Oligos1 [13]. The subsequent analysis was based on the developmental trajectory (OPC2 → OPC1 → Oligos3 → Oligos1). In this study, we defined this developmental trajectory as four developmental stages: Stage 1 (OPC2), Stage 2 (OPC1), Stage 3 (Oligos3) and Stage 4 (Oligos1). The workflow of this study is shown in Fig. 1.

Data analysis flow chart. OPC, oligodendrocyte progenitor cell; Oligos, oligodendrocytes; HC, healthy control; MDD, major depressive disorder
Identification of genes distinguishing the four developmental stages of OL
The GSE144136 dataset in “SRA” format was downloaded and converted into “fastq” format using FASTQ-dump in SRAToolkit. The “sampling” and “RRF” R packages were used as stratified sampling methods and for GRRF analysis, respectively. These analytic packages were used in R software (version 3.6.3, https://www.r-project.org). In the GRRF classifier, DEGs in four cell clusters based on Nagy et al. were validated by a fivefold cross validation method. The top-ranked genes of the four cell clusters were further subjected to partial least squares discriminant analysis (PLS-DA) to screen candidate genes, which obviously separated the four cell clusters. PLS-DA was analysed by the “MetaboAnalystR” R package [40]. PLS-DA enables the analysis of variable importance in projection (VIP) values, which emphasizes the importance of each variable during prediction.
Identified genes distinguishing two adjacent developmental stages (stage 1 & stage 2, stage 2 & stage 3, stage 3 & stage 4)
Normally, developmental stages can be identified by stage-specific marker genes. To more precisely delineate developmental stages and enrich stage-specific marker genes, we detected genes that distinguish adjacent developmental stages via GRRF. Simultaneously, the discriminative ability of each classifier was measured by receiver operating characteristic (ROC) curves, and the area under the ROC curve (AUC) was calculated using the R package “ROCR”. We screened genes with top-ranked Gini scores, which were able to distinguish Stage 1 & Stage 2, Stage 2 & Stage 3, Stage 3 & Stage 4 and defined these genes as Group1, Group2, Group3.
Identification genes distinguishing HC and MDD cases in four developmental stages
Each of the four developmental stages of the OL may play a role in the occurrence and development of depression, but to what extent each contributes is unknown. To assess the four developmental stages, we employed GRRF to establish predictive models based on DEGs between HC and MDD cases, and ROC curves were rendered on four cell types using the R package “ROCR”.
Matching of candidate genes with published MDD-related databases and pathways
To improve the correlation between candidate genes and depression, we established a candidate gene list of depression-related genes through three approaches to facilitate the selection of candidate genes.
Approach 1: selecting genes associated with depression from the ingenuity pathways.
analysis (IPA) database (https://www.bilibili.com/video/av540856330/).
Approach 2: selecting genes associated with depression from the publicly available.
Database PsyGeNET http://www.psygenet.org/web/PsyGeNET/menu;jsessionid = 1t7yjgsorwmwz1ta1fv2k1yoib).
Approach 3: selecting genes from pathways closely related to depression.
(1) PATHWAY: hsa04726 Serotonergic synapse-Homo sapiens (human).
(https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04726).
(2) PATHWAY: hsa04722 Neurotrophin signaling pathway-Homo sapiens (human).
(https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04722).
(3) PATHWAY: hsa04080 Neuroactive ligand-receptor interaction-Homo sapiens (human).
(https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04080).
(4) PATHWAY: hsa04020 Calcium signaling pathway-Homo sapiens (human).
(https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04020).
(5) PATHWAY: hsa04915 Estrogen signaling pathway-Homo sapiens (human).
(https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04915).
(6) PATHWAY: hsa04010 MAPK signaling pathway-Homo sapiens (human).
(https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04010).
(7) PATHWAY: hsa04014 Ras signaling pathway-Homo sapiens (human).
(https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04014).
(8) PATHWAY: hsa04630 JAK-STAT signaling pathway-Homo sapiens (human).
(https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04630).
(9) PATHWAY: hsa04750 Inflammatory mediator regulation of TRP channels-Homo sapiens (human) (https://www.kegg.jp/dbget-bin/www_bget?pathway+hsa04750).
The selected depression-related genes from the IPA database, PsyGeNET and MDD-related pathways are listed in Additional file 1: Tables S1-S3, and the union of the two databases and pathways (Hereafter, the union of the two databases and pathways is designated as Union) is listed in Additional file 1: Table S4.
Animals
The animal study was carried out using male C57BL/6 mice (n = 20) weighing 16–18 g purchased from the Company of Experimental Animals of Hunan Slack King (Hunan, China). Before the experiment, the mice were acclimated to the laboratory environment for one week. All mice were maintained under standard laboratory conditions with a 12-h light/dark cycle (lights on at 08:00), 22 ± 2 °C, and relative humidity 45%-55% and had free access to food and water. All animal care and experimental procedures were in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) Guidelines and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Chronic unpredictable mild stress (CUMS) model
Mice were randomly divided into two groups: the CUMS group (n = 10) and the Control group (n = 10). The CUMS model was based on a previous study and was slightly modified [41]. The CUMS group was exposed to nine kinds of mild stressors for 4 weeks: food deprivation for 24 h, water deprivation for 24 h, damp sawdust for 24 h, 45° tilted cages for 24 h, swimming in ice water for 5 min, swimming in hot water at 45 °C for 5 min, tail clamping for 5 min, day and night reversal and pushing and squeezing. The CUMS group received one type of stress per day, all of which were applied randomly, and the same type of stress was not applied for two consecutive days.
Body weight and behavioural tests
The body weights of mice in both the Control group and the CUMS group were measured before and after CUMS. Three behavioural tests were performed, including the sucrose preference test (SPT), forced swimming test (FST) and open field test (OFT). Anhedonia, an important clinical symptom of depression, was measured by SPT, and SPT was carried out as described in a previous study [42]. The FST is based on the assumption that when an animal is in a container filled with water and initially tries to escape but ends up staying still, the length of time it stays motionless reflects the degree of behavioural despair [43]; this test was implemented as described previously [44]. The OFT is a method to evaluate the autonomous and inquiry behaviour of rodents in new environments. The experimental apparatus consisted of an open field reaction box (50 cm*50 cm*50 cm) and an automatic video tracking system (Ethovision XT 11.5). Each mouse was placed in the centre of the box, and the frequency of rearing and distance travelled in the box were recorded during a 5-min session in a quiet environment.
Sample collection
After all behavioural tests and data analyses, five mice in the Control group and CUMS group were anaesthetized with sodium pentobarbital (60 mg/kg, i.p.) and internally infused with 4% paraformaldehyde. Whole brain tissues were collected, postfixed with 4% paraformaldehyde for 24 h, embedded in paraffin and sectioned at 5 μm thickness for the following fluorescence in situ hybridization (FISH) assay.
FISH assay and statistical analysis
The probes used in the FISH assay were synthesized by GenePharma (Shanghai, China), and the sequences and modifications are listed in Table 2. The FISH assay was performed according to the protocol provided by the RNA FISH Kit (GenePharma, #F2220). Images were collected using a fluorescence microscope (Olympus BX51, Japan), and the fluorescence intensity was analysed by ImageJ (National Institutes of Health, Bethesda, MD). GraphPad Prism (version 5.0, GraphPad, USA) was employed for statistical analysis and graphing. The data are presented as the mean ± standard error of the mean (SEM). A two-tailed unpaired t test was used for comparisons between two groups. Statistical significance was indicated by a p value < 0.05.
Results
Identification of genes distinguishing the four developmental stages of the OL
To identify genes distinguishing the four developmental stages of the OL, we selected four cell types, OPC2 (Stage 1), OPC1 (Stage 2), Oligos3 (Stage 3), and Oligos1 (Stage 4), for subsequent analyses. By employing the GRRF algorithm and fivefold cross-validation method, classifiers based on DEGs were constructed. Gene symbols and Gini scores of the top 30 candidates are shown in Fig. 2A. We further screened 100 top-ranked genes based on Gini scores to detect candidate genes using PLS-DA. The PLS-DA score plots (Figs. 2B, C) indicated obvious separation between the four cell types, with the first component accounting for 31.4%. The VIP scores of the top 5 components of the top 48 candidates were greater than 1 (highlighted in bold in Additional file 1: Table S5), suggesting that the candidate genes possess high classification ability. Figure 2D shows the top 30 gene symbols and VIP scores with the direction of gene expression among the four cell types shown in the right panel. The heatmap (Fig. 2E) exhibits the changes in the top 100 genes among the four cell types. Genes with the top 100 Gini scores in the GRRF classifier are listed in Additional file 1: Table S6. Overall, these results demonstrate that the top 100 genes are capable of distinguishing between the four developmental stages. The screened genes might be important supplements for developmental stage-specific markers.

The top100 genes from GRRF model to distinguish four cell types. A Gene symbol and Gini scores of top 30 candidates. B PLS-DA score plots of four cell types based on top100 genes. C PLS-DA score plots of the top 5 components. D VIP scores of top 30 genes, with direction of gene expression shown on right panel. E Heatmap of top 100 genes among four cell types
Identification of genes distinguishing two adjacent developmental stages
When cells transition from one developmental stage to another, certain characteristics are observed, in addition to the regulation and expression of some key genes [10]. To identify these key genes, the GRRF method was employed to construct predictive models to distinguish every two adjacent developmental stages (Stage 1 & Stage 2, Stage 2 & Stage 3, Stage 3 & Stage 4). The ROC curves (Fig. 3A–C) showed the performance of the GRRF method and a high accuracy, with the AUC equalling 0.998, 0.998 and 0.997 when distinguishing Stage 1 & Stage 2, Stage 2 & Stage 3, Stage 3 & Stage 4, respectively. Figure 3D–E show the gene symbols and Gini scores of the top 30 genes corresponding to two adjacent developmental stages. Group 1, Group 2 and Group 3 of the top 100 genes were listed in Additional file 1: Tables S7–S9. The genes identified are likely to be key factors that propel cells to the next developmental stage, as well as potential complementary molecular markers that distinguish each stage of development.

Constructing GRRF model for distinguishing two adjacent stages. ROC plot in A Stage1/ Stage2, B Stage2/ Stage3, C Stage2/ Stage3. Top 30 genes according to ranking Gini scores in D Stage1/ Stage2, E Stage2/ Stage3, F Stage2/ Stage3
To explore the biological relevance of the top 100 genes in the three groups (Group 1, Group 2, and Group 3), we performed gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses and obtained the following results. Group 1 was found to be involved in cell adhesion molecules, axon guidance and microRNAs in cancer, while Group 2 and Group 3 were found to be involved in cell adhesion molecules, glutamatergic synapses, neuroactive ligand-receptor interactions and mineral absorption, endocytosis, and GABAergic synapses, respectively. The KEGG pathways in which each group were found to be involved in are listed in Table 3, and GO analysis of the top 100 genes in the three groups is listed in Additional file 1: Tables S10–S12. We found that both Group 1 and Group 2 are involved in the cell adhesion molecule signalling pathway, while Group 2 and Group 3 are involved in different signalling pathways. Further analysis revealed that the intersection of Group 1 and Group 2 was 36, and the intersection of Group 2 and Group 3 was 17 (Fig. 4). This indicates that the conversion between Stage 1, Stage 2 and Stage 3 is relatively similar, while the conversion between Stage 3 and Stage 4 is quite different from the previous transitions.

Venn diagram of top100 genes distinguishing two adjacent stages (Stage1&Stage2, Stage2&Stage3, Stage3&Stage4)
Identification of candidate genes in the four developmental stages for distinguishing HC and MDD
Each of the four developmental stages of the OL may play a role in the occurrence of depression. To determine which of the four developmental stages is most closely associated with the onset of depression, we adopted the GRRF algorithm to construct disease predictive models based on the DEGs between HC and MDD cases. To screen genes distinguishing MDD cases from HC at each developmental stage, we selected the top 100 genes (Additional file 1: Tables S13-16) according to the Gini scores at each developmental stage. Figure 5A–D show that OPC2 exhibited the best predictive ability (AUC = 0.870), followed by Oligos1 (AUC = 0.682), OPC1 (AUC = 0.648) and Oligos3 (AUC = 0.644). Figure 5E–H show the gene symbols and importance scores of the top 30 genes in the four cell types. Surprisingly, the numbers of cells in the OPC2 (HC = 312, MDD = 164) and Oligos1 (HC = 166, MDD = 70) categories were much smaller than those in the OPC1 (HC = 793 MDD = 888) and Oligos3 (HC = 1989, MDD = 1632) categories; however, the former two possess significantly better predictive power than the latter two.

Constructing GRRF model from DEGs in four cell types distinguishing HC and MDD. ROC plot in A OPC2, B OPC1, C Oligos3, D Oligos1. Top 30 genes according to ranking Gini scores in E OPC2, F OPC1, G Oligos3, H Oligos1
According to the original sequencing results, the number of cells in the OPC2 and Oligos1 categories was very small in the MDD group and HC group, accounting for less than 10%, without considering the technical systematic error. The number of cells in OPC1 and Oligos3 accounted for approximately 30% and 60%, respectively. We speculate that the conversion process from Stage 1 to Stage 2 was relatively fast, but the process from Stage 3 to Stage 4 was slow, resulting in more than half of the cells remaining in Stage 3. The results indicate that among the effects of the OL on depression, a few cells could play an important role in the occurrence of depression. The findings also highlight the importance of single-cell sequencing and provide a new perspective for studying diseases.
Identification of MALAT1 and DLG2 by comparison with published MDD-related databases and pathways
To further compare our candidate genes with previously reported findings in MDD, we used the IPA database, the publicly available Database PsyGeNET and pathways closely related to depression. We retrieved the top 100 genes associated with the four developmental stages in the Union, and the number of overlapping genes of the four developmental stages was 25, 35, 40 and 21. The overlapping of the top 100 genes at each stage and in candidate gene list are listed in Table 4. Further analysis of the top 100 genes in the predictive model of four developmental stages revealed that two genes, namely, MALAT1 and DLG2, appeared in all four stages (Fig. 6A). Interestingly, MALAT1 and DLG2 appeared simultaneously in the overlapping of the top 100 genes and Union at four stages (Fig. 6B).

Venn diagram of top 100 genes distinguishing between HC and MDD from four cell types A Without retrieving in the list of candidate genes. B With retrieving in the list of candidate genes
MALAT1 ranked second in Stage 1 and first in the other three stages. MALAT1 has been widely studied in cancer, but little is known about its role in depression. DLG2 is among the top 30 genes in Stage 2, Stage 3 and Stage 4. DLG2 encodes the postsynaptic scaffolding protein DLG2 (also referred to as PSD93), which interacts with NMDA receptors, potassium channels and cytoskeletal regulators. Genetic variation in the DLG2 locus has been associated with a variety of psychiatric disorders. In view of the high ranking of MALAT1, we hypothesized that MALAT1 may play an important role in the development of depression and selected it as a follow-up verification object.
CUMS-susceptible mice showed lower body weight and obvious depressive-like behaviour
Due to the difficulty of obtaining the human prefrontal cortex, we established a mouse CUMS model, which is recognized as reliable, practical, and widely employed to study the mechanism of depression. After 4 weeks of CUMS, mice in the CUMS group (n = 6) showed lower body weight than mice in the Control group [t(12) = 4.233, **P < 0.01, Fig. 7A] and obvious depression-like behaviour, including decreased sucrose preference rate [t(12) = 3.979, **P < 0.01, Fig. 7B]; total distance moved [t(12) = 4.639, ***P < 0.001, Fig. 7D], time in centre [t(12) = 4.305, **P < 0.01, Fig. 7E], frequency of rearing [t(12) = 4.14, **P < 0.01, Fig. 7F] in OFT; and increased immobility time in the FST [t(12) = − 4.571, ***P < 0.01, Fig. 7C]. The results indicated that mice with depression-like behaviour were successfully selected.

CUMS-susceptible mice showed lower body weight and obvious depression-like behavior. Effects of CUMS on body weight (A) and sucrose preference rate in the SPT (B); immobility time in the FST (C); total distance moved (D), time in the center (E), and rearing frequency (F) in the OFT. *P < 0.01, **P < 0.05, ***P < 0.001 vs. the Control group
Validation of Malat1 expression in oligodendrocytes at different developmental stages in the PFC
Our analysis shows that the expression of MALAT1 was increased in oligodendrocytes at different developmental stages in the PFC. To validate this prediction, we conducted FISH assay with PFC slices from both the Control group and the CUMS group. Pdgfα and Mbp were selected as markers of immature oligodendrocytes and mature oligodendrocytes, respectively. FISH assay showed that Malat1 expression was higher in both immature [t(8) = − 4.210, **P < 0.01, Fig. 8A and C] and mature [t(8) = − 4.877, **P < 0.01, Fig. 8B and D] oligodendrocytes in the CUMS group than in the Control group. The experimental results were consistent with the prediction, indicating that Malat1 in oligodendrocytes at different developmental stages in the PFC is associated with the occurrence of depression; however, the specific mechanism needs to be further studied.

Malat1 up-regulated in both immature and mature oligodendrocytes. Representative images of FISH assays of Malat1 in immature (A) and mature (B) oligodendrocytes in the PFC, respectively. Increased Malat1 signal intensity in immature (C) and mature (D) oligodendrocytes in the PFC, respectively. Nuclei were stained with DAPI; Malat1 was labeled with Cy3, Pdgfα and Mbp were labeled with FAM. Scale bars represent 50 μm. The data were expressed as means ± SEM (n = 5). **P < 0.05 vs. the Control group
Discussion
This study investigated the pathogenesis of depression from the perspective of oligodendrocyte development. The GRRF method was adopted to analyse the four developmental stages of the OL, and the following results were obtained. First, we screened the top-ranked genes distinguishing the four developmental stages based on DEGs, as well as those between two adjacent developmental stages. Then, we analysed the power of the four developmental stages to predict the occurrence of depression and found that the analysis of genes associated with the onset of OL development (Stage 1) possessed better predictive power than other developmental stages. Through comparison with the top-ranked candidate genes and subsequent FISH assay, we revealed that lncRNA Malat1 might be closely related to the occurrence of depression.
ScRNA-seq technology has indicated that in brain tissues, gene expression patterns are cell-type specific in both dominant cell groups such as neurons and glial cells and in subtypes of neuronal cells or glial cells [45]. Generally, OL cells can be divided into OPCs and mature oligodendrocytes according to the degree of development. PTGDS, PDGFRA, PCDH15, OLIG2, and OLG1 are common markers of OPCs, and PLP1, MAG, MOG, MOBP, and MBP are frequently-used markers of mature oligodendrocytes [10]. However, the division of these markers is not very rigorous; for instance, PDGFRA and PCDH15 are only expressed in immature oligodendrocytes, while OLIG2 and SOX10 are expressed in both immature and mature oligodendrocytes [13]. The top-ranked genes identified included PCDH15, PLP1, MAG, MBP and other routine markers, and among the top 100 candidates, PCDH15 ranked first, indicating the high feasibility of our prediction method.
By reviewing the single-cell gene expression profile in GSE144136, we found that three genes, including PCDH15, DSCAM and PTPRZ1, are only expressed in OPCs but not in neurons and are all associated with cell adhesion. DSCAM is located on human chromosome 21 and belongs to the immunoglobulin superfamily of cell adhesion molecules. It is unquestionable that DSCAM plays a role in regulating cell recognition, neural circuit formation, and the delamination of neurons in the developing midbrain [46, 47]. Furthermore, Amano found an association between increased DSCAM expression and bipolar disorder in a genetic screen of patients with bipolar disorder. PTPRZ, a protein tyrosine phosphatase, is mainly expressed in astrocytes, oligodendrocyte precursor cells (OPCs), and immature and mature oligodendrocytes of the developing and adult nervous system [48]. This receptor binds to the cell adhesion molecules on the surface of OPCs and participates in the proliferation and differentiation of OPCs [49]. Studies have shown that PTPRZ affects the balance between OPC proliferation and maturation by forming a complex with CNTN1 on the surface of OPCs, inhibiting their proliferation and promoting their transformation into mature oligodendrocytes [50]. Our analysis showed that the screened genes could be used as potential developmental markers of OL.
After the 1950s, with the development of intracellular recording methods and histochemistry, our understanding of glial cells became more comprehensive. At present, OL cells are generally classified as mature or immature. Our analysis divided the development of the OL into four stages and identified the key genes that distinguish each of the two adjacent stages. Quaking (qki), encoding a conserved RNA-binding protein QKI, ranked first in Group 1. Reportedly, QKI plays a specific role in myelin defects in the aetiology of psychiatric disorders and is critical to the myelination decision of the OL in MDD suicide victims [51]. Studies have shown that Qki participates in the regulation of myelin lipid homeostasis, and deletion of Qki in oligodendrocytes did not affect oligodendrocyte survival but resulted in rapid demyelination in adult mice within one week and progressive neurological dysfunction [52]. Group 2, distinguishing OPC1 (Stage 2) and Oligos3 (Stage 3), promotes the transformation of immature oligodendrocytes into mature oligodendrocytes. Our results show that PCDH15, PLP1 and MBP were all within the top 10 of Group 2, consistent with previous studies wherein PCDH15 was found to be highly expressed in late OPCs and was a marker of immature oligodendrocytes [53], and PLP1 and MBP are markers of mature oligodendrocytes [54].
Oligodendrocyte development is also accompanied by changes in the expression of neurotransmitter receptors on the cell surface. KEGG pathway analysis showed that Group 2 was involved in the glutamatergic synapse pathway, while Group 3 participated in the GABAergic pathway. Bergles first reported that OPCs accepted the introduction of excitatory glutamatergic through AMPA receptors [55]. Subsequently, Lin found that OPCs expressed GABA receptors in response to GABAergic input [56]. Ablation of OPCs in the prefrontal cortex of adult mice altered AMPA receptor membrane trafficking, impaired excitatory glutamatergic neurotransmission and extracellular glutamate uptake, and ultimately led to depressive-like behaviour in mice [57]. OPCs can form synaptic complexes with hippocampal interneurons. Photostimulation of OPCs stimulates GABA release and affects hippocampal excitatory-inhibitory balance, resulting in anxiety-like behaviours in mice [58].
For a long time, OL cells were regarded as a source of myelinating cells. Recent research has shown that OL cells have other roles, such as regulating the function of neurons and astrocytes, ultimately affecting behaviour, responding to central nervous system damage and acting as innate immune cells [59]. Recent studies have shown that the loss of OPCs in the prefrontal cortex alters glutamate energy signalling and promotes depression-like behaviour in mice [57]. Nagy et al. also found that OPCs played a significant role in the occurrence of depression in humans [13]. Recently, an interesting discovery showed that the immune system utilizes OPCs to maintain its immune response in the demyelination state [60, 61]. Both OPCs and oligodendrocytes act as antigen-presenting cells and activate CD8+ T cells in humans and mice [62, 63]. Neuroinflammation is a well-known molecular mechanism of depression, and we hypothesize that OPCs may be involved in neuroinflammation leading to depression.
The OPC2 subcellular type, the initiation stage of the OL, had the best predictive power, and we infer that there may be two reasons. On the one hand, OPCs account for approximately 5% of the total number of mature brains and retain the ability to self-proliferate throughout life [9]. On the other hand, although the primary function of OPCs is to proliferate and differentiate into mature oligodendrocytes, OPCs can also differentiate into astrocytes [64] and neurons [65] at certain developmental stages and brain regions. Depression is associated with neurogenesis and neuroplasticity, and the continued proliferation and transformation of OPCs into neurons or glia may play a compensatory role.
The LncRNA MALAT1, also known as NEAT2, was first discovered by Ji [66]. MALAT1 is dominantly found in nuclear speckles and was highly conserved during mammalian evolution. It is widely expressed in normal mammalian tissues and has elevated levels in most malignant tissues [67]. Recently, an increasing number of studies have also linked MALAT1 to neurological disorders such as schizophrenia (SZ), Alzheimer’s disease (AD) and neuropathic pain. The level of MALAT1 in peripheral blood decreased in patients with SZ [68]. MALAT1 positively regulates the expression of CDK5R1 and affects the occurrence of AD [69], and regulation of the miR-129-5p/HMGB1 axis causes the occurrence of neuropathic pain in a chronic constriction injury model in rats [70]. Currently, there are few reports on the relationship between MALAT1 and depression [71,72,73]. Our results provide a new target for the study of long noncoding RNAs in depression.
Unfortunately, there are four main limitations to this study. First, since the sample collection lasted 15 years, single nuclei could only be isolated from tissues, not single cells, and transcripts within the cytoplasm were lost; thus, the analysis could not be performed at the overall transcript level. Second, only the relationship between the development of the OL and the occurrence of depression was analysed, and the role of other cell types was ignored. In addition, brain samples from patients with depression are limited and extremely difficult to collect, and therefore, we verified the candidate gene Malat1 in animal models. It is more logical to perform this analysis in human samples. Finally, this manuscript is a reanalysis of data obtained in a previous work of Nagy Corina’s group [13], and is not a replication or confirmation. We conducted a correlation analysis of single-nucleus transcriptomics data of oligodendrocyte lineage in female samples (HC = 18, MDD = 20), also generated by Nagy Corina’s group [74]. The results showed that, 1) the pseudotime trajectory of oligodendrocyte lineage in female samples was similar to that in male samples, and 2) Malat1 also distinguished HC and MDD at different developmental stages of oligodendrocyte in female samples (Additional file 1: Table S17, S18 and Figure S1-S4).
Conclusion
This study investigated the mechanism of MDD from the perspective of OL development at the single-cell level. We adopted a GRRF algorithm to screen critical candidate genes in single-cell sequencing data processing. The results demonstrate that the initiation developmental stage of the OL was a better predictor of the occurrence of depression than other developmental stages, and that the lncRNA Malat1 might be closely related to the occurrence of depression. Our work highlights the importance of single-cell sequencing in mental disorders and provides a novel direction for research on the occurrence of MDD.
Availability of data and materials
All the necessary data are included within the article. Further data will be shared by request.
Abbreviations
- AD:
-
Alzheimer’s disease
- AUC:
-
Area under the curve
- CUMS:
-
Chronic unpredictable mild stress
- CNS:
-
Central nervous system
- GRN:
-
Gene regulatory network
- GRRF:
-
Guided regularized random forest
- RF:
-
Random forest
- FISH:
-
Fluorescence in situ hybridization
- FST:
-
Forced swimming test
- HC:
-
Healthy controls
- IPA:
-
Ingenuity pathways analysis
- MDD:
-
Major depressive disorder
- OFT:
-
Open field test
- OL:
-
Oligodendrocyte lineage
- OPCs:
-
Oligodendrocyte progenitor cells
- PLS-DA:
-
Partial least squares discriminant analysis
- RNA-seq:
-
RNA-sequencing
- ROC:
-
Receiver operating characteristic
- SPT:
-
Sucrose preference test
- SZ:
-
Schizophrenia
- scRNA-seq:
-
Single-cell RNA sequencing
References
GBD. Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet Psychiatry. 2022;9:137–50.
Hirbec H, Déglon N, Foo LC, Goshen I, Grutzendler J, Hangen E, Kreisel T, Linck N, Muffat J, Regio S, et al. Emerging technologies to study glial cells. Glia. 2020;68:1692–728.
Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science. 2018;362:181–5.
No-authers: Recherches Anatomiques et Physiologiques sur la Structure Intime des Animaux et des Végétaux, et sur leur Motilité. Edinb Med Surg J. 1829, 31:369–394.
Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–71.
Dimou L, Götz M. Glial cells as progenitors and stem cells: new roles in the healthy and diseased brain. Physiol Rev. 2014;94:709–37.
Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23:1018–27.
von Bartheld CS, Bahney J, Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol. 2016;524:3865–95.
Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–88.
Elbaz B, Popko B. Molecular control of oligodendrocyte development. Trends Neurosci. 2019;42:263–77.
Makinodan M, Rosen KM, Ito S, Corfas G. A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science. 2012;337:1357–60.
Liu J, Dietz K, DeLoyht JM, Pedre X, Kelkar D, Kaur J, Vialou V, Lobo MK, Dietz DM, Nestler EJ, et al. Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat Neurosci. 2012;15:1621–3.
Nagy C, Maitra M, Tanti A, Suderman M, Théroux JF, Davoli MA, Perlman K, Yerko V, Wang YC, Tripathy SJ, et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat Neurosci. 2020;23:771–81.
Zhou B, Zhu Z, Ransom BR, Tong X. Oligodendrocyte lineage cells and depression. Mol Psychiatry. 2021;26:103–17.
Lutz PE, Tanti A, Gasecka A, Barnett-Burns S, Kim JJ, Zhou Y, Chen GG, Wakid M, Shaw M, Almeida D, et al. Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: convergent epigenetic, transcriptional, and morphological evidence. Am J Psychiatry. 2017;174:1185–94.
Tanti A, Kim JJ, Wakid M, Davoli MA, Turecki G, Mechawar N. Child abuse associates with an imbalance of oligodendrocyte-lineage cells in ventromedial prefrontal white matter. Mol Psychiatry. 2018;23:2018–28.
Peng W, Jia Z, Huang X, Lui S, Kuang W, Sweeney JA, Gong Q. Brain structural abnormalities in emotional regulation and sensory processing regions associated with anxious depression. Prog Neuropsychopharmacol Biol Psychiatry. 2019;94: 109676.
Zhang FF, Peng W, Sweeney JA, Jia ZY, Gong QY. Brain structure alterations in depression: psychoradiological evidence. CNS Neurosci Ther. 2018;24:994–1003.
Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, Zhang K. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 2018;36:70–80.
La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, Zeisel A, Borm LE, Stott SRW, Toledo EM, Villaescusa JC, et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell. 2016;167:566-580.e519.
Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–90.
Dutta R, Trapp BD. Gene expression profiling in multiple sclerosis brain. Neurobiol Dis. 2012;45:108–14.
Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53.
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–82.
Ding S, Chen X, Shen K. Single-cell RNA sequencing in breast cancer: understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020;40:329–44.
Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, Yu EA, Schenk EL, Tan W, Zee A, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182:1232-1251.e1222.
Grün D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, Clevers H, van Oudenaarden A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–5.
Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 2016;24:593–607.
Li L, Dong J, Yan L, Yong J, Liu X, Hu Y, Fan X, Wu X, Guo H, Wang X, et al. Single-cell RNA-Seq analysis maps development of human germline cells and gonadal niche interactions. Cell Stem Cell. 2017;20:858-873.e854.
Rizvi AH, Camara PG, Kandror EK, Roberts TJ, Schieren I, Maniatis T, Rabadan R. Single-cell topological RNA-seq analysis reveals insights into cellular differentiation and development. Nat Biotechnol. 2017;35:551–60.
Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, Amit I. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-Seq. Cell. 2016;167:1883-1896.e1815.
Reinius B, Mold JE, Ramsköld D, Deng Q, Johnsson P, Michaëlsson J, Frisén J, Sandberg R. Analysis of allelic expression patterns in clonal somatic cells by single-cell RNA-seq. Nat Genet. 2016;48:1430–5.
Thakur D, Biswas S. Guided regularized random forest feature selection for smartphone based human activity recognition. J Ambient Intell Humaniz Comput. 2023;14:9767–79.
Deng H, Runger GJPR: Gene selection with guided regularized random forest. 2013, 46:3483-3489.
Izquierdo-Verdiguier, Emma; Zurita-Milla, Raul: Use of Guided Regularized Random Forest for Biophysical Parameter Retrieval. In IEEE IGARSS 2018 - 2018 IEEE International Geoscience and Remote Sensing Symposium. pp. 5776–5779; 2018: 5776–5779.
Zhang Y, Chen Q, Gao D, Zou Q: GRRFNet: Guided Regularized Random Forest-based Gene Regulatory Network Inference Using Data Integration. In 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). pp. 132–139; 2020:132–139.
Sylvester EVA, Bentzen P, Bradbury IR, Clément M, Pearce J, Horne J, Beiko RG. Applications of random forest feature selection for fine-scale genetic population assignment. Evol Appl. 2018;11:153–65.
Guan X, Runger G, Liu L. Dynamic incorporation of prior knowledge from multiple domains in biomarker discovery. BMC Bioinformatics. 2020;21:77.
Lee J, Geng S, Li S, Li L. Single cell RNA-Seq and machine learning reveal novel subpopulations in low-grade inflammatory monocytes with unique regulatory circuits. Front Immunol. 2021;12: 627036.
Pang Z, Chong J, Li S, Xia J. MetaboAnalystR 3.0: toward an optimized workflow for global metabolomics. Metabolites. 2020;10:186.
Cai X, Kallarackal AJ, Kvarta MD, Goluskin S, Gaylor K, Bailey AM, Lee HK, Huganir RL, Thompson SM. Local potentiation of excitatory synapses by serotonin and its alteration in rodent models of depression. Nat Neurosci. 2013;16:464–72.
Si L, Xiao L, Xie Y, Xu H, Yuan G, Xu W, Wang G. Social isolation after chronic unpredictable mild stress perpetuates depressive-like behaviors, memory deficits and social withdrawal via inhibiting ERK/KEAP1/NRF2 signaling. J Affect Disord. 2023;324:576–88.
Yankelevitch-Yahav R, Franko M, Huly A, Doron R. The forced swim test as a model of depressive-like behavior. J Vis Exp. 2015;97:52587.
Bian H, Wang G, Huang J, Liang L, Zheng Y, Wei Y, Wang H, Xiao L, Wang H. Dihydrolipoic acid protects against lipopolysaccharide-induced behavioral deficits and neuroinflammation via regulation of Nrf2/HO-1/NLRP3 signaling in rat. J Neuroinflammation. 2020;17:166.
Ofengeim D, Giagtzoglou N, Huh D, Zou C, Yuan J. Single-cell RNA sequencing: unraveling the brain one cell at a time. Trends Mol Med. 2017;23:563–76.
Hattori D, Millard SS, Wojtowicz WM, Zipursky SL. Dscam-mediated cell recognition regulates neural circuit formation. Annu Rev Cell Dev Biol. 2008;24:597–620.
Arimura N, Okada M, Taya S, Dewa KI, Tsuzuki A, Uetake H, Miyashita S, Hashizume K, Shimaoka K, Egusa S, et al. DSCAM regulates delamination of neurons in the developing midbrain. Sci Adv. 2020. https://doi.org/10.1126/sciadv.aba1693.
Harroch S, Palmeri M, Rosenbluth J, Custer A, Okigaki M, Shrager P, Blum M, Buxbaum JD, Schlessinger J. No obvious abnormality in mice deficient in receptor protein tyrosine phosphatase beta. Mol Cell Biol. 2000;20:7706–15.
Peles E, Schlessinger J, Grumet M. Multi-ligand interactions with receptor-like protein tyrosine phosphatase beta: implications for intercellular signaling. Trends Biochem Sci. 1998;23:121–4.
Lamprianou S, Chatzopoulou E, Thomas JL, Bouyain S, Harroch S. A complex between contactin-1 and the protein tyrosine phosphatase PTPRZ controls the development of oligodendrocyte precursor cells. Proc Natl Acad Sci USA. 2011;108:17498–503.
Klempan TA, Ernst C, Deleva V, Labonte B, Turecki G. Characterization of QKI gene expression, genetics, and epigenetics in suicide victims with major depressive disorder. Biol Psychiatry. 2009;66:824–31.
Zhou X, He C, Ren J, Dai C, Stevens SR, Wang Q, Zamler D, Shingu T, Yuan L, Chandregowda CR, et al. Mature myelin maintenance requires Qki to coactivate PPARβ-RXRα-mediated lipid metabolism. J Clin Invest. 2020;130:2220–36.
Huang W, Bhaduri A, Velmeshev D, Wang S, Wang L, Rottkamp CA, Alvarez-Buylla A, Rowitch DH, Kriegstein AR. Origins and proliferative states of human oligodendrocyte precursor cells. Cell. 2020;182:594-608.e511.
Kuhn S, Gritti L, Crooks D, Dombrowski Y. Oligodendrocytes in Development, Myelin generation and beyond. Cells. 2019;8:1424.
Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–91.
Lin SC, Bergles DE. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat Neurosci. 2004;7:24–32.
Birey F, Kloc M, Chavali M, Hussein I, Wilson M, Christoffel DJ, Chen T, Frohman MA, Robinson JK, Russo SJ, et al. Genetic and stress-induced loss of NG2 glia triggers emergence of depressive-like behaviors through reduced secretion of FGF2. Neuron. 2019;104:825–6.
Zhang X, Liu Y, Hong X, Li X, Meshul CK, Moore C, Yang Y, Han Y, Li WG, Qi X, et al. NG2 glia-derived GABA release tunes inhibitory synapses and contributes to stress-induced anxiety. Nat Commun. 2021;12:5740.
Fernandez-Castaneda A, Gaultier A. Adult oligodendrocyte progenitor cells—multifaceted regulators of the CNS in health and disease. Brain Behav Immun. 2016;57:1–7.
Harrington EP, Bergles DE, Calabresi PA. Immune cell modulation of oligodendrocyte lineage cells. Neurosci Lett. 2020;715: 134601.
Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J, Strasburger H, Herbst L, Alexis M, Karnell J, et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun. 2019;10:3887.
Jäkel S, Agirre E, Mendanha Falcão A, van Bruggen D, Lee KW, Knuesel I, Malhotra D, Ffrench-Constant C, Williams A, Castelo-Branco G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 2019;566:543–7.
Falcão AM, van Bruggen D, Marques S, Meijer M, Jäkel S, Agirre E, Samudyata, Floriddia EM, Vanichkina DP, Ffrench-Constant C, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018;24:1837–44.
Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–57.
Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–401.
Ji P, Diederichs S, Wang W, Böing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–41.
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–38.
Li J, Zhu L, Guan F, Yan Z, Liu D, Han W, Chen T. Relationship between schizophrenia and changes in the expression of the long non-coding RNAs Meg3, Miat, Neat1 and Neat2. J Psychiatr Res. 2018;106:22–30.
Spreafico M, Grillo B, Rusconi F, Battaglioli E, Venturin M. Multiple layers of CDK5R1 regulation in Alzheimer’s disease implicate long non-coding RNAs. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19072022.
Ma X, Wang H, Song T, Wang W, Zhang Z. lncRNA MALAT1 contributes to neuropathic pain development through regulating miR-129-5p/HMGB1 axis in a rat model of chronic constriction injury. Int J Neurosci. 2020;130:1215–24.
Xue X, Pan J, Zhang H, Lu Y, Mao Q, Ma K. Baihe Dihuang (Lilium Henryi Baker and Rehmannia Glutinosa) decoction attenuates somatostatin interneurons deficits in prefrontal cortex of depression via miRNA-144-3p mediated GABA synthesis and release. J Ethnopharmacol. 2022;292: 115218.
Bian Q, Chen J, Wu J, Ding F, Li X, Ma Q, Zhang L, Zou X, Chen J. Bioinformatics analysis of a TF-miRNA-lncRNA regulatory network in major depressive disorder. Psychiatry Res. 2021;299: 113842.
Seki T, Yamagata H, Uchida S, Chen C, Kobayashi A, Kobayashi M, Harada K, Matsuo K, Watanabe Y, Nakagawa S. Altered expression of long noncoding RNAs in patients with major depressive disorder. J Psychiatr Res. 2019;117:92–9.
Maitra M, Mitsuhashi H, Rahimian R, Chawla A, Yang J, Fiori LM, Davoli MA, Perlman K, Aouabed Z, Mash DC, et al. Cell type specific transcriptomic differences in depression show similar patterns between males and females but implicate distinct cell types and genes. Nat Commun. 2023;14(1):2912.
Acknowledgements
Not applicable.
Funding
This research was supported by Fundamental Research Funds for the Central Universities (Grant number 2042022kf1101), the National Natural Science Foundation of China (Grant numbers 82071523, 62076229), and the Medical Science Advancement Program of Wuhan University (Grant number TFLC2018001).
Author information
Authors and Affiliations
Contributions
GW and LX designed the study. YX and LC conducted the data analysis and drafted the manuscript. YZ, LS, HG and HX were responsible for data screening and collection. LW and TL were responsible for the layout of the charts and proofreading of the manuscripts.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures involving animals were carried out per the Regulations of Experimental Animal Administration issued by the State Committee of Science and Technology of the People’s Republic of China, and approved by the ethics committee in Renmin Hospital of Wuhan University (IACUC Issue No. WDRM20200704).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1:Oligodendrocyte lineage cell clusters of 38 female HC and MDD visualized by UMAP. (A): Colors indicate a female subject (HC or MDD). (B): Colors indicate cell types. Each dot represents one nucleus. HC: healthy control; MDD: major depressive disorder; UMAP: uniform manifold approximation and projection. Figure S2:Pseudotime trajectory (Monocle analysis) of the Oligodendrocyte lineage.(A): Cells are colored based according to the cell type; (B): Cells are colored based according to the predicted pseudotime. Figure S3:Constructing GRRF model for distinguishing two adjacent developmental stages. ROC plot in (A) OPC1/OPC2, (B) OPC2/Oligos1, (C) Oligos1/Oligos2, (D) Oligos2/Oligos3. Top 30 genes and Gini scores in (E) OPC1/OPC2, (F) OPC2/Oligos1,(E) Oligos1/Oligos2, (F) Oligos2/Oligos3. Figure S4:Constructing GRRF model from DEGs in four cell types distinguishing HC and MDD. ROC plot in (A) OPC1, (B) OPC2, (C) Oligos1, (D) Oligos2, (E) Oligos3. Top 30 genes and Gini scores in (F) OPC1, (G) OPC2, (H) Oligos1, (I) Oligos2, (J) Oligos3. Table S1: Depression-associated genes in IPA. Table S2: Depression-associated genes in PsyGeNET. Table S3: Depression-associated genes in MDD-related pathways. Table S4: The union of supplementary1&2&3 (Union). Table S5: VIP scores of the top 5 components of the top 100 genes distinguishing four development stages. Table S6: Genes with top 100 Gini scores in four stages. Table S7: Top 100 genes distingush between Stage1&Stage2. Table S8: Top 100 genes distingush between Stage2&Stage3. Table S9: Top 100 genes distingush between Stage3&Stage4. Table S10: GO analysis of top 100 genes in Stage1&Stage2 (Group1). Table S11: GO analysis of top 100 genes in Stage2&Stage3 (Group2). Table S12: GO analysis of top 100 genes in Stage3&Stage4 (Group3). Table S13: Gini scores of top 100 genes of HC and MDD in Stage1. Table S14: Gini scores of top 100 genes of HC and MDD in Stage2. Table S15: Gini scores of top 100 genes of HC and MDD in Stage3. Table S16: Gini scores of top 100 genes of HC and MDD in Stage4. Table S17: Number of DEGs for 5 cell types. Table S18: Number of DEGs for developmental stage of different cell types.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xie, Y., Chen, L., Wang, L. et al. Single-nucleus transcriptomic analysis reveals the relationship between gene expression in oligodendrocyte lineage and major depressive disorder. J Transl Med 22, 109 (2024). https://doi.org/10.1186/s12967-023-04727-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04727-x