
Abstract
Chronic kidney disease (CKD) affects more than 10% population worldwide and becomes a huge burden to the world. Recent studies have revealed multifold interactions between CKD and gut microbiome and their pathophysiological implications. The gut microbiome disturbed by CKD results in the imbalanced composition and quantity of gut microbiota and subsequent changes in its metabolites and functions. Studies have shown that both the dysbiotic gut microbiota and its metabolites have negative impacts on the immune system and aggravate diseases in different ways. Herein, we give an overview of the currently known mechanisms of CKD progression and the alterations of the immune system. Particularly, we summarize the effects of uremic toxins on the immune system and review the roles of gut microbiota in promoting the development of different kidney diseases. Finally, we discuss the current sequencing technologies and novel therapies targeting the gut microbiome.
Similar content being viewed by others
Introduction
Chronic kidney disease (CKD) is one of the most important chronic diseases worldwide, affecting the well-being of many people. Currently, the main therapies for CKD are renin–angiotensin aldosterone system inhibitors with drugs for symptomatic treatment and dietary restriction. However, these interventions have achieved limited benefits. Recently, an increasing body of data has implicated that both the progression of CKD and its complications are related to the disturbed gut microbiome in patients with CKD.
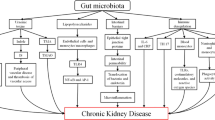
Gut microbiome is considered a “living organism” that coevolves with the host. Compared with the human body, it exceeds the number of cells in the human body by a factor of 10 [1]. The gut microbial genes are 100-fold greater than human genes, indicating the diversity of microbial species [2]. In healthy individuals, microbial communities as well as their metabolites perform multiple beneficial functions through bacterial-host and bacterial-bacterial interactions. The flora composition can be altered by many environmental factors such as dietary habits, drugs and disease conditions [3]. The kidney diseases, such as IgA nephropathy (IgAN), diabetic nephropathy (DN) and end-stage renal disease (ESRD) with hemodialysis (HD) or peritoneal dialysis (PD), can discompose intestinal microbial ecologies, referred to as dysbiosis [4]. Extensive efforts have been invested toward unveiling the changes. For example, Vaziri et al. discovered that 175 operational taxonomic units (OTUs) were different between CKD and healthy rats, and the richness of OTUs was decreased in CKD group [5]. At the family level, both Lactobacillaceae and Prevotellaceae were notably reduced, resulting in a decreased production of short chain fatty acids (SCFAs). This is attributed to the enrichment of phosphotransbutyrylase and butyrate kinase in these two taxa. Whereas the urease- and uricase-containing and indole- and p-cresol-forming intestinal microbiota increased, such as Enterobacteriaceae, Pseudomonadaceae, and Clostridiaceae [5, 6]. In return, gut dysbiosis is also involved in the onset and progression of CKD and complications. The potential mechanisms are associated with immune dysfunction, translocation of bacteria via impaired intestinal barriers and increased production of gut-derived uremic toxins [7, 8]. Among them, the protein-bound uremic toxins (PBUTs), such as indoxyl sulfate (IS), p-cresol sulfate (pCS) and indole-3-acetic acid (IAA), are defined as potential immune-modulatory candidates. These metabolites cause the dysfunction of immune cells and induce the production of pro-inflammatory cytokines and chemokines. The permeability of the gut barrier increases in CKD, leading to the translocation of inflammatory factors and uremic toxins into circulation. This contributes to vascular damage, glomerular sclerosis, kidney fibrosis and tubular damage, promoting inflammation and resulting in adverse outcomes and complications of CKD, such as cardiovascular disease (CVD) [9,10,11]. Thus, the pathological interactions between kidney disease and dysbiosis could generate vicious cycles accelerating kidney disease progression (Fig. 1).

The vicious cycle between chronic kidney disease and gut dysbiosis
Recent years witness the emergence of new methods, such as next-generation sequencing (NGS) and third-generation sequencing (TGS). In addition, researchers prefer to integrate multi-omics analysis to unravel the intricate interactions between the gut microbiome and CKD. These methods have improved our understanding of the gut flora ecosystem, which fuels the search for novel diagnostic methods and treatments for CKD.
In this review, we will focus on the effects of the gut microbiome on CKD progression and immune dysfunction. Additionally, advanced detection techniques and potential therapeutic avenues for CKD will also be discussed.
Mechanisms of CKD progression
CKD is a common chronic disease with a high rate of morbidity and mortality, affecting approximately 843.6 million population worldwide [12]. Kidney Disease: Improving Global Outcomes (KDIGO) defines CKD as abnormalities of kidney structure or function, present for > 3 months, with implications for health [13]. Based on the glomerular filtration rate (GFR), CKD is classified into 5 categories. From 2017 to March 2020, the prevalence of CKD stages 1–4 was 14.7%, and the crude incidence rate of CKD stage 5, namely ESRD, will increase 11–18% from 2015 to 2030, which is a huge medical burden for the whole world [14, 15].
The mechanisms of CKD progression are enormously complex and only partly understood. Several factors have been implicated in contributing to the pathophysiological and pathological alterations observed in individuals with CKD, including but not limited to hyperglycemia, hypertension, infections, high-protein diets, and others. From a pathophysiological point of view, hyperglycemia, hypertension and immune dysfunction can directly cause cellular damage. In response to these insults, the reactive oxygen species (ROS), pro-inflammatory and pro-fibrosis factors increase in circulation, and the immune complexes deposit in the glomerulus. This leads to damage to the glomerulus and decreased GFR. The declined GFR activates the renin-angiotensin system (RAS), shown by the increasing levels of angiotensin-II to elevate the glomerular hydrostatic pressure and filtration, ultimately compromising the glomerular barrier [16]. Apart from controlling vascular pressure, angiotensin-II also induces factors that regulate inflammation and fibrosis [16]. These pathophysiological changes further result in glomerular sclerosis, tubulointerstitial fibrosis, and proliferation of mesangial cells. The pathological remodeling of kidney infrastructure is closely associated with clinical manifestations such as albuminuria and hematuria. Furthermore, uremic toxins are retained and actively participate in the processes of CKD progression stated above. The resulting disturbances in the humoral environment can impair other systems, causing CVD, bone diseases, abnormal immunity, etc. [17].
The immune dysfunction of CKD has been well reviewed elsewhere [18]. These immune abnormalities can further exert negative effects on patients with CKD. Toll-like receptor (TLR) 2 and TLR4 play a significant role in the inflammation pathway [19]. Patients with CKD exhibit the upregulation of Toll-like receptor (TLR) 2 and TLR4 on neutrophils and monocytes partly due to the elevated production of pro-inflammatory cytokines and ROS [20]. These inflammatory factors can also cause tissue impairment and are related to low GFR [21]. There is an increase in the number of peripheral polymorphonuclear leukocytes (PMNLs) in patients with CKD, which is demonstrated to be negatively correlated with GFR [22]. Besides, the proportion of CD14+CD16+ monocytes and CD4+CD28− T cells increases [23, 24]. Both subtypes exhibit notable proinflammation ability and are related to CVD in patients with ESRD [25, 26]. Moreover, the anti-bactericidal activity of neutrophils decreased, leading to increased susceptibility to infection. For instance, the ability of neutrophils to generate extracellular NET formation (NETosis) is severely impaired, favoring the invasion of pathogens [27]. In addition to causing complications, immune dysfunction also contributes to poor vaccine response in patients with ESRD [28]. It may be related to the decreased stimulation ability of dendritic cells (DCs) and impaired antigen-specific T cell differentiation [29].
Effects of gut-derived metabolites on CKD
Uremic toxins can be divided into three categories: small water-soluble, protein-bound and middle molecules. Among them, several metabolites, such as IS, pCS and IAA, are in the limelight. All three are PBUTs. Unfortunately, they can hardly be removed by dialysis, because the molecule size of PBUTs exceeds the upper limit of dialysis membranes [30]. Moreover, HD cannot replace tubular secretion, which is the main mechanism of PBUTs excretion. Similar to HD, PD cannot compensate for the reduced renal function, and the restriction coefficient of its membrane for macromolecules increases over time [31]. PD even has a lower clearance of PBUTs than HD [32]. Besides, studies showed that hemodiafiltration (HDF) has no notable effects on removing PBUTs in the long term, even though its efficacy is higher than HD [33, 34]. Therefore, PBUTs accumulate in patients with CKD as the kidney function decreases, especially in advanced CKD stages.
Moreover, aside from toxins, gut microbiota can also produce metabolites with renoprotective properties such as SCFAs. The impacts of PBUTs and renoprotective metabolites are outlined in Table 1.
Effects of PBUTs on immune cells
These accumulated toxins have multiple biological effects, of which the harmful effects on immune cells have gained more interest.
Indoxyl sulfate
Indoxyl sulfate (IS) is metabolized from tryptophan. Tryptophan first is metabolized into indole by gut bacteria such as Escherichia coli, Proteus vulgaris, Paracolobactrum coliforme, Achromobacter liquefaciens, and Micrococcus aerogenes and then is hydroxylated and sulfated to circulating IS in liver [35].
The effects of IS on immune cells have been shown to contribute to kidney and vascular damage in patients with CKD. The tryptophan derivates are the ligands of aryl hydrocarbon receptor (AhR), which is expressed on kinds of immune cells and provides immune function [36]. Through the AhR pathway, IS induces human monocytes to produce interleukin (IL)-1β and tumor necrosis factor-α (TNF-α). IL-1β is involved in the onset and progression of tubulointerstitial fibrosis [37]. The increasing level of TNF-α can elicit the production of CX3CL1 in vascular endothelial cells, which will recruit CD4+CD28− T cells that express CX3CR1. The activated CD4+CD28− T cells cause endothelial cells to undergo apoptosis [38]. In macrophages, IS induces TNF-α production involving crosstalk among the AhR, NF-kB, and the suppressor of cytokine signaling (SOCS)2 [39]. In addition, IL-6, one of the pro-inflammatory factors, is proven to be notably related to the free IS level [40]. IS also induces adhesion and extravasation of leukocytes in a murine experiment, causing vascular impairment [41].
Indole-3-acetic acid
Indole-3-acetic acid (IAA) also originates from tryptophan and belongs to the family of indole, which is metabolized by bacterial tryptophan monooxygenases and indole-3-acetamide hydrolases [42]. As a tryptophan derivate, IAA can also activate AhR. In addition, IAA has multiple effects on neutrophils, which may contribute to the development of infection. The production of \({\text{O}}_{2}^{-}\) and H2O2 activated by IAA leads to death and significant ultrastructural changes in cultured neutrophils [43]. IAA damages the DNA of neutrophils in a dose-dependent manner, resulting in the death of neutrophils [44]. Similar to IS, IAA can impair the vascular. In cultured human endothelial cells, IAA upregulates the expression and activity of COX-2 by activating an AhR/p38 MAPK/NF-κB signaling pathway and increases ROS production in endothelial cells, which induces endothelial inflammation and oxidative stress [45]. A study has demonstrated that serum IAA has predictive value for mortality and cardiovascular events in CKD [45].
p-Cresol sulfate
p-Cresol is the product of tyrosine, most of which is sulfated to p-Cresol sulfate (pCS) in colon [46].
pCS exhibits both pro-inflammatory and immunosuppressive effects at different stages of CKD, which involves the progression of CKD and its complications. Schepers et al. showed the pro-inflammatory effect of pCS for the first time, which functions by increasing the percentage of free radical-producing leucocytes [47]. Similarly, Azevedo et al. described the elevated ability of NO production and phagocytosis in macrophages in the early stage of CKD. However, in advanced stages, the high concentration of pCS could cause immunosuppression [48]. Studies showed that pCS could inhibit the production of IFN-γ by decreasing Th1 cells and the production of IL-12 in macrophages, thus suppressing immune responses [49, 50]. In adenine-induced renal dysfunction mice, pCS could suppress the phosphorylation of STAT5, which is a vital process in the proliferation and survival of CD43+ B-cell progenitors. Therefore, pCS inhibited the proliferation of CD43+ B-cell progenitors [51]. The resulting reduction of B cells is associated with a decreased response to T-cell-independent vaccines in patients with ESRD [52]. The immunosuppression mechanisms may contribute to infection including septicemia, which is one of the leading causes of death in ESRD patients.
In addition to the reduced clearance, increased protein fermentation of gut flora also contributes to the elevated level of PBUTs. In CKD patients, the colon transit times are prolonged [53]. Moreover, the protein digestion of the upper gut is impaired, leading to the protein increase in colon [53]. A recent study linked gut flora in patients with CKD to uremic toxins levels. In this study, the abundance of Escherichia_Shigella is positively correlated with serum levels of IS; Alistipes is positively associated with total levels of IS and pCS [54].
Effects of PBUTs on CKD progression
PBUTs are involved in fibrosis and inflammation in CKD. A recent study revealed that IS induces epithelial-to-mesenchymal transition (EMT) in human proximal tubular cells (HK-2), providing a potential mechanistic link to renal fibrosis [55]. Poveda et al. have reported that pCS causes cell death in HK-2 cells. The findings also showed that pCS increases expression of the TWEAK receptor Fn14 and cooperates with inflammatory cytokine TWEAK, collectively contributing to kidney damage [56]. Watanabe et al. explored the effects of pCS on renal tubular cells. They found that pCS led to the elevation of Nox4 and p22phox expression, resulting in the increase of ROS production through Nox4 and p22phox NADPH oxidase. Thereafter, ROS mediates the increase of pro-inflammatory and pro-fibrotic factors, ultimately contributing to damage in tubular cells [11]. PBUTs have been found to elicit comparable deleterious effects on cellular function. IS and pCS have been shown to inhibit the expression of Klotho in renal tubules of mice by DNA methylation, contributing to renal tubule senescence [57]. Moreover, both IS and pCS can elicit an increase in Tgfb1 expression in proximal renal tubular cells, which is related to immunomodulation and fibrosis [58]. The activation of RAAS is widely acknowledged as a critical mechanism driving the progression of CKD [16]. Both IS and pCS have been demonstrated to increase levels of renin, angiotensinogen and the expression of AT1 receptor, contributing to kidney injury [10]. Furthermore, it has been observed that uremic toxins can also affect metabolism in human renal proximal tubule cells. A study has revealed the involvement of both IAA and IS in reducing the activity of the mitochondrial electron transport chain, leading to decreased NAD + levels and subsequent attenuation of UDP-glucuronosyltransferases (UGT)-mediated metabolism [59].
Reno-protective metabolites
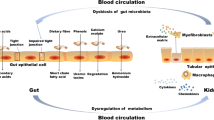
In healthy individuals, gut microbiota ferments carbohydrates into SCFAs e.g., acetate, propionate and butyrate. Unfortunately, these microbiota-producing SCFAs are decreased in CKD. Notably, SCFAs have been attributed with a protective role in CKD, exerting anti-inflammatory effects, enhancement of gut barrier integrity, and amelioration of insulin sensitivity. Li et al. found that butyrate improves renal fibrosis, renal lesion and tubular injury through the GPR43 pathway. Moreover, it inhibits lipopolysaccharide-induced overexpression of proinflammatory Il1b and Il6 in both kidney tubular epithelium cell and renal podocyte. The researchers observed reduced infiltration of macrophage in the kidney upon butyrate treatment [60]. In addition, Gonzalez et al. demonstrated the improvement of 5ʹ adenosine monophosphate-activated protein kinase (AMPK) phosphorylation by butyrate, resulting in enhanced gut barrier and reduced leakage of gut-derived uremic toxins. Butyrate mediates the secretion of glucagon-like peptide-1 (GPL-1), ameliorating insulin sensitivity [61]. Furthermore, acetate mitigates the inflammation in CKD by inhibiting the histone acetylation (HDAC) activity of T cells, correcting the oxidant-antioxidant imbalance in T cell [62]. As for propionate, researchers found that it alleviates the increased serum Cr (Scr) and BUN as well as inflammation via the GPR43 or GPR41 pathway [63, 64].
Roles of the microbiome in CKD
The impacts of the gut microbiome on CKD
IgA nephropathy
IgA nephropathy (IgAN) is the most prevalent disease among primary glomerulonephritis with 25 cases per 100,000 persons worldwide [65]. The pathogenesis of IgAN has been described as a multi-hit mechanism, the most important of which is the synthesis of galactose-deficient IgA1 (Gd-IgA1) [66]. Evidence has emerged suggesting the involvement of the gut microbiome in this pathogenetic process. Several studies have shown a reduction in cell densities and diversity of intestinal microbiota in IgAN patients when compared to healthy individuals [67, 68]. Additionally, research has indicated that Sutterellaceae and Enterobacteriaceae increase, both of which are LPS-producing bacteria [68]. In addition to inducing systemic inflammation, LPS significantly inhibits the expression of core I β3-Gal-T-specific molecular chaperone (Cosmc) mRNA and leads to methylation of the Cosmc by activating toll-like receptor 4 (TLR4) [69]. Cosmc is a critical molecular chaperone of IgA1 glycosylation [70]. Therefore, the decrease of Cosmc can cause the increase of circulating Gd-IgA1. The accumulated Gd-IgA1 can stimulate B cells to produce IgG, resulting in the sequential deposition of IgG-Gd-IgA1 immune complexes in kidney [71]. The complexes induce mesangial cell proliferation and the production of extracellular matrix components [66]. Moreover, in BAFF-overexpressing transgenic mice (BAFF-Tg mice), the presence of commensal microbiota is indispensable for IgA production and collaborates with high levels of BAFF to participate in the pathogenesis of IgAN [72]. Besides, the uremic toxins produced by imbalanced gut flora induce the release of pro-inflammatory cytokines and cause subsequent systemic inflammation, further contributing to the progression of IgAN [73].
Furthermore, a recent investigation has elucidated the correlation between intestinal microbiota and clinical manifestation of IgAN, shown by increased Escherichia-Shigella and reduced Bifidobacterium in patients with severe haematuria and proteinuria [74]. In another study, antibiotic administration leads to decreased formation of glomerular deposition and prevents proteinuria in a humanized mouse model of IgAN [75]. Therefore, targeting the gut microbiome may be a promising strategy to prevent the initiation and progression of IgAN.
Diabetic nephropathy
Approximately 40% of patients with diabetes that is poorly controlled develop diabetic nephropathy (DN), which has been the leading cause of CKD over the past years [76, 77]. DN is characterized by decreased glomerular filtration rate and increased albuminuria and seriously affects patients’ quality of life.
Current views hold that the activated RAS plays a pivotal role in renal dysfunction of DN [78]. The release of renin and Ang II can cause extracellular matrix accumulation and injury of renal cells [79]. The important metabolites of gut microbiota, SCFAs, are reported to participate in RAS regulation [80]. Acetate can activate olfactory receptor 78 (Olfr78) that is expressed on renal juxtaglomerular afferent arteriole, leading to the release of renin and subsequent increase in blood pressure [81]. In addition, acetate activates G protein-coupled receptor 43 (GPR43) to cause a disorder of cholesterol homeostasis, contributing to the lipid accumulation in the tubulointerstitium of DN patients [82]. The activation of GPR43 also mediates insulin resistance and thereby causes impairment in podocyte [83]. Podocyte injury is one of the vital mechanisms of albuminuria [84]. Moreover, insulin resistance (IR) also contributes to the progression of DN. IR leads to high insulin levels, which activates the production of plasminogen activator inhibitor 1 (PAI-1), thereby causing mesangial matrix expansion and fibrosis in kidney [85]. Administration of Bifidobacteria and Lactobacillus is observed to improve insulin resistance in type 2 diabetes mellitus (T2DM) [86].
Besides, hyperglycemia can impair the gut barrier, resulting in the increased translocation of gut microbiota and its products [87]. The increases of these metabolites in circulation exert negative effects on patients. For example, the upregulation of pro-inflammatory cytokines and inflammation can be caused by elevated LPS levels [88]. Moreover, LPS is associated with the occurrence of IR [88]. A recent study observed the translocation of gut microbiota and demonstrated its damage to kidney in mitochondrial antiviral signaling protein (MAVS) knockout diabetic mice [89]. MAVS is important in protecting the gut barrier [90]. Furthermore, phenyl sulfate (PS) is one of the gut microbiota-derived PBUTs, which increases in circulation of patients with DN [91]. Koichi et al. demonstrated that PS exerts multiple negative effects, including podocyte injury, mitochondrial dysfunction of the podocyte, and fibrosis in kidney [91]. This study also suggests that PS levels may serve as a predictive biomarker for the progression of albuminuria for incipient patients with DN [91]. In addition to kidney injury, PBUTs also contribute to the complication of DN. IS induces the proliferation of vascular smooth muscle cells, indicating its role in the development of CVD in patients with DN [92].
Acute kidney injury to CKD transition
Acute kidney injury (AKI) is defined as the sudden declination of renal function within a few hours or days. The maladaptive repair in AKI can predispose individuals to progress toward CKD, particularly in the context of kidney aging. Previous studies suggest that gut dysbiosis in AKI accelerates the transition from AKI to CKD. In vitro experiment, Chen et al. demonstrated that IS is associated with hypoxia–reperfusion (H/R) induced G2/M cell cycle arrest, EMT and endoplasmic reticulum stress induction, which may lead to the maladaptive repair of AKI [93]. The cells that are arrested in the G2/M phase may secret pro-inflammatory factors, leading to microvascular rarefaction and collagen disposition in kidney [94]. Moreover, AKI is characterized by severe proximal tubule injury, including the downregulation of organic anion transporter (OAT)-1 and OAT-3, subsequently causing the decreased excretion of uremic toxins such as IS. In addition, the impaired gut barrier caused by both AKI and dysbiosis can indirectly expedite the transition from AKI to CKD. Butyrate is able to activate hypoxia-inducible factor (HIF) -1α in intestinal epithelial cells (IECs). HIF-1α promotes mucin expression and secretion, therefore maintaining the stability of gut barrier [95]. However, the SCFAs are reduced due to gut dysbiosis in CKD, compromising gut barrier and causing the leakage of gut-derived uremic toxins. Yang et al. demonstrated that AKI-induced dysbiosis can contribute to the progression of AKI. The results showed that dysbiosis is associated with increased colonocyte apoptosis, inflammation, and altered tight junction proteins, which may lead to the translocation of endotoxins and further potentiate the inflammation in AKI [96, 97].
Hemodialysis/Peritoneal dialysis
ESRD patients require renal replacement treatment (RRT), such as hemodialysis (HD) and peritoneal dialysis (PD). HD and PD retard the progression of ESRD through clearance of the retention water and removal of some uremic toxins, consequently maintaining the balance of the internal environment.
Infection is an important complication, both in HD and PD. Studies suggest that the gut microbiota may be a source of infectious pathogens. For example, Pseudomonas aeruginosa and Enterobacteriaceae increase in PD patients [98, 99]. P. aeruginosa has been found to be associated with around 40% of catheter removals attributed to infections, while Enterobacteriaceae is responsible for 12.0% of peritonitis cases in PD patients [100, 101]. Moreover, the DNA of P. aeruginosa was found in blood from different vascular access of HD patients [102].
During dialysis, the intestinal barrier is compromised. Increased intraperitoneal pressure in PD contributes to intestinal hypoperfusion [103]. Similarly, HD is always accompanied by reduced hepato-splanchnic perfusion, which leads to intestinal ischemia [104]. Therefore, the subsequent migration of bacteria and its metabolites exerts negative effects on HD and PD patients. For instance, circulating IS and pCS are elevated in HD and PD patients. The harmful effects of IS and pCS on the human body have been reviewed elsewhere, such as endothelial damage, EMT and inflammation [105]. Based on these mechanisms, researchers find evidence that pCS is associated with the morbidity of CVD and all-cause mortality in HD patients [106]. Moreover, pCS and IS can predict the progression of CKD [107].
In addition, constipation is common in dialysis patients. The decrease of commensal bacteria can contribute to the occurrence of constipation. Lactobacillus and Bifidobacterium are associated with faster colonic transit [108]. However, they decreased in PD patients, which may confer negative effects on gut healthy [109]. The mechanisms of intestinal microbiome progress the disease is enormously complex and only partly understood. We need more research to unravel the mysteries.
Microbiota in CKD
The gut microbiome is different in healthy individuals and patients with CKD. Research has revealed an elevated presence of both aerobic and anaerobic microorganisms in the duodenum and jejunum of CKD patients compared to healthy individuals [110, 111]. Moreover, many results demonstrate a notable reduction in the diversity of gut microbiota among CKD patients [5, 112]. Furthermore, a recent study conducted in China enrolled 489 fecal samples to find the alterations in the gut microbiota of patients with CKD compared to healthy individuals [113]. As CKD progressed, researchers observed significant increases in the abundance of genera Thalassospira, Akkermansia, and Blautia, while the genus RF9_norank exhibited a decrease. Particularly, Akkermansia is positively correlated with BUN and Scr, indicating its potential as a valuable diagnostic or therapeutic target in CKD. Moreover, the microbiota involved in the metabolism of ascorbate and aromatic amino acids is enriched in CKD, which aligns with the observed increase in uremic toxins in CKD patients [113].
In addition to the gut, the composition of microbiota in blood holds significant importance. Blood is traditionally supposed to be sterile, however, a recent study challenged this view, providing evidence supporting the existence of blood microbiome in healthy individuals [114]. Shah et al. compared the blood microbiome of patients with CKD and healthy controls. The results showed that the α diversity is decreased in the CKD group, similar to the change in the gut microbiota of CKD. Moreover, Proteobacteria is enriched in the CKD group. This bacteria phylum increased in blood and gut in many chronic diseases. Therefore, decreased diversity of blood microbiota and enriched Proteobacteria in the blood may be the factors contributing to CKD progression [115]. Merino-Ribas et al. explored the blood microbiome in PD patients with or without vascular calcification. They found an increase in Devosia in the blood of PD patients with vascular calcification. Vascular calcification is associated with high mortality risk in PD patients, suggesting that Devosia may be a biomarker or involved in the disease progression [116]. However, many questions remain to be addressed. The precise origin of blood microbiota has yet to be definitively determined. The leaky gut barrier and translocation of microbiota in oral and skin may be the sources [117]. Moreover, liver clearance and the immune system have important effects on the blood microbiome, emphasizing the necessity of incorporating these factors in future investigations [118].
The research on urine microbiome in patients with CKD is still not fully explored. A study enrolled 77 patients with non-dialysis-dependent CKD in order to understand the characteristic of urine microbiome in stage 3–5 CKD. The findings revealed a reduction in the diversity of the midstream urine microbiome concomitant with the decline in eGFR [119]. The possible mechanism may be the altered secretion of uromodulin, which favors the excretion of bacteria [119]. Since secondary urinary tract infection is common in patients with CKD, more attention should be paid to the urine microbiome.
Gut-kidney-heart axis
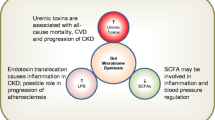
Among the complications of CKD, CVD stands as a prominent cause of mortality in CKD patients. The pathogenesis of CVD in CKD is multifactorial, involving several mechanisms, including (1) volume overload; (2) overactivation of RAAS; (3) inflammation and oxidative stress [120]. The detailed interaction between the heart and kidney in CKD has been well reviewed elsewhere [121]. Additionally, the gut microbiome also precipitates the development of CVD, particularly by producing uremic toxins. Thereafter, accumulated gut-derived uremic toxins are transferred to circulation through the impaired gut barrier and compromise the cardiovascular system.
The gut-derived uremic toxins exhibit the capacity to provoke vascular damage and promote the development of atherosclerosis. Gut bacteria metabolize choline, choline-containing compounds, betaine, and l-carnitine into trimethylamine (TMA), which is oxidized by flavin-dependent monooxygenase isoforms 1 and 3 (FMO1 and FMO3) to form TMAO in liver [122]. Seldin et al. have elucidated the capacity of TMAO to upregulate the expression of inflammatory genes in endothelial and smooth muscle cells by activating the NF-κB signaling pathway. Moreover, TMAO enhances the endothelial adhesion of leukocytes and contributes to atherosclerosis [123]. In addition, both TMAO and LPS promote atherosclerosis by increasing the expression of osteopontin (OPN) and activating macrophages of aortic [124]. OPN is an independent traditional risk factor of CVD [125]. In addition to TMAO, both IS and IAA possess the ability to increase the expression of TF protein via the AhR pathway, which is associated with several cardiovascular diseases such as atherosclerosis [126]. As reported by Campillo et al., IS and p-cresol can lead to monocytes across the extracellular matrix (ECM) by activating the integrin-linked kinase (ILK) /AKT signaling pathway. The activation of ILK/AKT signaling results in the formation of podosome, which is involved in ECM degradation. The increased degradation of the matrix favors the migration of monocytes and the interaction between migrated monocytes and vascular may cause injury [127].
The vicious cycle among the gut, kidney and heart is intricate, and only a portion of mechanisms is currently elucidated. In addition to metabolites, how gut microbiota itself affects the gut-kidney-heart axis is needed to explore. Nevertheless, targeting this vicious cycle to develop novel interventions holds tremendous potential for yielding significant benefits for patients.
Advances in techniques and methodologies for gut microbiome in CKD research
In the 1970s, scientists detected gut microbiota by culture–based techniques [128]. However, most of the gut flora cannot be cultured in the experimental condition. In the past decades, sequencing technologies and data analysis pipelines develop rapidly and are applied widely, enabling us to put insight into the composition, and capability of the human intestinal microbiome [129]. The pros and cons of technologies have been reviewed elsewhere [130].
With the advance of tools, the understanding of gut microbiome in patients with CKD has been improved a lot. Numerous scholars have reported the composition of gut microbiota by analyzing 16S rRNA sequence data. Compared with healthy control, Klebsiella and Enterobacteriaceae increased, while Blautia and Roseburia decreased in patients with CKD [113]. Further, Wu et al. showed the differences in gut microbiota within different CKD stages. Escherichia_Shigella is significantly enriched in advanced CKD, while Dialiste, Lachnospiraceae_ND3007_group, Pseudobutyrivibrio, Roseburia, Ruminiclostridium spp. decrease with CKD progression [54]. In addition, the comparison between pre-dialysis and dialysis patients is also worthy of attention. A recent study showed that dialysis increases some commensal bacteria and elicits some pathogenic bacteria at the same time compared with non-dialysis ESRD patients [131]. The shotgun approach is sequencing all microbial genomes in the samples. It can provide data with higher resolution than 16S rRNA sequencing and help understand the metabolic activity of microbiota [132]. Recently, Wang et al. explored the taxonomic and function of the microbiome in patients with ESRD utilizing shotgun metagenome sequencing. They found that the amino acid biosynthesis is decreased and amino acid degradation is increased. Moreover, the enzyme involved in aromatic amino acid degradation and secondary bile acid biosynthesis is enriched. This evidence may suggest the source of accumulated uremic toxins in ESRD [133].
Besides, multi-omics analyses combined data from multiple biological sources such as genomics, metagenomics, transcriptomics, proteomics, and metabolomics promote a mechanistic understanding of gut-kidney axis on CKD progression. Opdebeeck et al. found that IS and pCS can induce calcification in aorta and peripheral arteries of rats. The proteomic and bioinformatic analysis identified the proteins in these vessels suggesting that inflammation, coagulation and glucometabolism pathways are correlated with the IS- and pCS-induced calcification [134]. In the metabolomics study, Wu et al. also found that caproic acid is highly positively correlated with eGFR, indicating its role in alleviating the severity of CKD [112]. Using a proteomics approach, Karaduta et al. found that CKD mice exhibited a similar pattern of gut proteins as shown in healthy mice after receiving resistant starch. They further verified the protective role of resistant starch in renal injury [135]. Lobel et al. also applied proteomics to study the relationship between diet, gut and kidney. They identified that TnaA is a highly S-sulfhydrated protein, which is able to catalyze tryptophan to indole. They demonstrated that high sulfated amino acid dietary can inhibit the activity of TnaA by posttranslational modification, reducing the production of the IS precursor and consequently alleviating the progression of CKD [136]. By integrating these different omics data, researchers can attain profound insights into the intricate molecular mechanisms involved in microbiome-host crosstalk.
In addition to exploring the relationship between gut microbiome and CKD, omics contribute to finding the causes and novel diagnosis markers of various kidney diseases. Sethi et al. analyzed the proteomic profile of 12 cases of C3 glomerulonephritis (C3GN). The results showed that alternative pathway (AP) dysregulation can lead to C3GN by causing glomerular accumulation of AP and TCC proteins [137]. Dasari et al. detected proteins in glomerular biopsy specimens of patients by liquid chromatography and tandem mass spectroscopy (LC–MS/MS). They found that DnaJ homolog subfamily B member 9 (DNAJB9) is a specific protein in fibrillary glomerulonephritis, which indicates the role of DNAJB9 in pathogenesis and its potential to be a biomarker [138]. Moreover, metabolomics is used to explore the underlying metabolic pathway in CKD progression. Ma et al. revealed that the overexpression of APOL1 gene, previously demonstrated to be associated with CKD progression, can lead to alterations in the tricarboxylic acid cycle, increased fatty acid oxidation, and compromised redox homeostasis [139].
Potential therapies
Given the close interplay between the gut microbiome and CKD, some therapies target bacteria and its metabolites, which may alter the gut milieu and ameliorate the progression of CKD and its complications.
Probiotics
Probiotics are living microorganisms. It has been accepted that probiotics confer health benefits on gut environment. The underlying mechanisms for the beneficial effects of probiotics on gut health are multifaceted and include the following: (1) Immunomodulation, where specific bacterial strains can exert anti-inflammatory effects by inhibiting NF-κB signaling [140]; (2) Enhancement of the intestinal barrier, as probiotic treatment has been shown to inhibit changes in tight junctions (TJs), which are vital components of the gut barrier [141]; and (3) Resistance to pathogens, as Bacillus subtilis LF11 has been demonstrated to protect epithelial cells by reducing the attachment and invasion of Salmonella [142]. Some probiotics secret bacteriocins to play an antimicrobial role [143].
There are studies showing the beneficial effects of probiotics on CKD. Simenhoff et al. treated 8 HD patients with oral Lactobacillus acidophilus and the results showed reduced serum dimethylamine (DMA) which damages the organ vascular [17, 110]. Moreover, oral administration of Bifidobacterium longum in capsule reduced serum IS levels [144]. Recently, Zhu et al. investigated the therapeutic effects of the probiotic Lactobacillus casei Zhang in both mouse models and patients with CKD. The findings revealed that the administration of L. casei Zhang improved the inflammatory response of local macrophages and tubular epithelia cells by increasing the level of serum SCFAs [145]. Further, the study observed a slower decrease in renal function in patients with CKD stages 3–5 after taking L. casei Zhang [145]. However, due to the extensive variation in diet, disease condition and age of individuals, the therapeutic precision of the probiotics needs to be fully considered.
Prebiotics
Prebiotics are non-digestible food ingredients that improve human health by stimulating the growth of commensal bacteria, such as Lactobacillus and Bifidobacterium [146]. Common prebiotics include resistant starch, inulin, and oligosaccharides such as fructo-oligosaccharides (FOS) and galacto-oligosaccharides (GOS) [147]. The prebiotics can be metabolized by gut microbiota to produce SCFAs, which in turn can exert beneficial effects. SCFAs play a crucial role in providing an energy source of gut microbiota, regulating the luminal pH and inhibiting the growth of pathogens [148]. Furthermore, GOS protects disrupted gut epithelial and accelerates the TJ reassembly [149]. Oligofructose inulin treatment for four weeks reduced the serum level of pCS in HD patients, with the effect lasting for at least four weeks [150]. Moreover, the association between the ratio of total protein to total fiber and the levels of both IS and pCS is found to be significant, and incorporating this index into the eGFR regression model can improve its predictive power for the levels of uremic toxins [151].
In addition to non-digestible food ingredients, oral sorbents are used to reduce toxins levels. AST-120 is a sorbent with porous carbon particles. Several reports have demonstrated its beneficial effects on ameliorating CKD progression. A study showed that AST-120 reduces serum IS levels and ameliorates oxidative stress in cardiac tissue in CKD rats [152]. Recently, Huang et al. demonstrated that IS could inhibit mitophagy and subsequently cause damage to the intestinal barrier [153]. Mitophagy plays a protective role in healthy cells [154]. The study further revealed that AST-120 can provide protection against mitophagic impairment and intestinal barrier injury in CKD mice models [153]. Furthermore, the efficacy of AST-120 has also been demonstrated in human studies. A study by Nakamura et al. enrolled fifty patients with chronic renal failure, and the effects of AST-120 therapy were evaluated [155]. The results revealed that AST-120 therapy significantly reduced the levels of IL-6 and proteinuria, and inhibited the increase of serum creatinine levels [155]. However, it was observed that the therapy was not effective in preventing the decrease of GFR [155]. More clinical studies have been reviewed elsewhere [156]. Because of the challenges associated with accurately determining the sample size and duration of the study necessary, the evaluation of the effects of AST-120 on the renal hard endpoint is insufficient in many studies. Therefore, more well-designed studies need to be performed.
Synbiotics
Multiple combinations of probiotics and prebiotics have been applied in clinical practices, including OAT fiber/L. plantarum, and FOS/L. sporogens [157]. Synbiotics have the collaborative effects of the prebiotics and probiotics, which has higher efficacy. Probiotics are capable of releasing bacteriocins that inhibit bacteria involved in p-cresol production, while prebiotics can promote the growth of commensal bacteria [158]. A study has reported the efficacy of Probinul-neutro® in reducing serum p-cresol levels in patients with CKD when administered thrice daily for four weeks [159]. Another single-center, double-blind, placebo-controlled, randomized crossover trial study has found that synbiotics significantly reduce serum pCS but not IS in patients with CKD [160]. Moreover, synbiotics can increase Bifidobacterium and decrease Ruminococcaceae in their stool microbial community [160]. Recently, synbiotics have been shown to improve dysbiosis, decrease the amount of fecal indole and ameliorate CKD progression in CKD rat models [161].
Dietary manipulation
A current well-accepted hypothesis is that high-protein diets may be harmful to kidney [162]. The underlying mechanisms include causing hyperfiltration in kidney and upregulating the renal expression of TNF-α and IL-6 [163, 164]. In the Singapore Chinese Health Study, the researcher found that ESRD risk increases with an increased intake of red meat [165]. Moreover, most foods with high protein contain phosphate, which will disturb the serum phosphate level in the early stages of CKD [166]. On the contrary, the restriction of dietary protein can reduce sclerosis in kidney [167]. Therefore, many experts recommend a plant-dominant low-protein diet for patients with CKD [168]. In a meta-analysis, researchers found that CKD patients had lower mortality when adopting a healthy diet rich in fruit and vegetables, fish, legumes, cereals, whole grains, and fiber, and less in red meat and products containing sodium and refined sugars [152]. Furthermore, high-fiber diets contribute to the growth of beneficial bacteria such as Bacteroidetes that are capable of producing SCFAs [153]. These SCFAs can improve gut dysbiosis. Given the powerful role of diet–microbiota–host crosstalk, dietary manipulation could be an effective therapeutic tool [154].
Genetic engineering
In 2015, Sonnenburg proposed the concept of ‘‘smart’’ bacteria that can secrete anti-inflammatory molecules [169]. In recent years, genetic engineering technology has been also applied to reduce toxins and ameliorate disease. Devlin et al. discovered a family of tryptophanases widely distributed in the gut commensal Bacteroides [170]. Through the deletion of this gene, they were able to eliminate indole production in vivo [170]. Moreover, researchers have successfully designed microencapsulated genetically engineered live E.coli DH5 cells [171]. They demonstrated the effective reduction of urea levels in uremic rats upon oral administration of the products [171]. Besides, Steidler et al. designed Lactococcus lactis to secret IL-10 in the colon [172]. When this engineered Lactococcus lactis was orally administered, it was found to have a beneficial effect on reducing inflammation in colitis mouse models [172]. Recently, Ting et al. showed an innovative method to deplete a specific bacterium from gut flora. They designed E. cloacae strains expressing nanobodies, called programmed inhibitor cells (PICs) [173]. PICs recognize target bacteria via surface nanobodies and eradicate it from gut microbiota [173]. Even though microbiota engineering is far from clinical application, it may be a promising way to achieve precision medicine.
Fecal microbiota transplantation
Fecal microbiota transplantation (FMT) involves the transfer of gut microbiota from a healthy donor to a patient by infusing donor feces into the patient. It aims to restore the disturbed gut microbiota in patients. Therapeutic effects of FMT have been first demonstrated in the treatment of recurrent Clostridium difficile infection (CDI) [174]. The US Food and Drug Administration (FDA) permits the administration of FMT to CDI that does not respond to standard therapies [175]. Subsequently, there are increasing reports of the efficacy of FMT on other diseases [176]. A study found that FMT can decrease albuminuria and modulate renal phenotype in antibiotic-treated humanized IgAN mice [177]. In addition to effectiveness, a study has investigated the occurrence of adverse events (AEs) of FMT. A systematic analysis of cases published between 1983 and 2015 suggested an overall incidence of AEs was 28.5% (310/1089) [178]. Most of the AEs were mild, including abdominal discomfort, diarrhea, transient fever, nausea, vomiting and constipation [178]. The findings indicate that FMT is generally safe. However, rare but severe AEs have been reported, such as death, severe infections, and relapse of inflammatory bowel diseases (IBD) [178]. The risk of infections is associated with the inability of screening latent infection of donors [178]. Therefore, FDA has issued a warning on the risk of FMT [179]. More rigorous studies are required to assess the short- and long-term risks and efficacy of FMT.
Challenges and perspectives
As a result of technological advancements, the intricacies of the gut microbiome have been partly elucidated, revealing the involvement of gut microbiota and its metabolites in the progression of CKD. In particular, PBUTs have been identified as contributors to immune dysfunction with adverse effects on CKD and its complications. Thus, novel interventions that aim to mitigate or even eliminate these detrimental effects should be developed. As noted, genetic engineering is a promising technology. Targeting enzymes that involve in PBUTs production by utilizing genetic engineering may be a feasible avenue. Integrating this approach with the assessment of efficacy in ameliorating immune dysfunction and CKD progression may help with the design of therapeutic strategies.
Current treatments aim to reduce the levels of PBUTs and regulate dysbiosis through dietary restriction and synbiotics. However, these therapies are limited by inter-individual variabilities in diet, lifestyle, age, and medication. Therefore, personalized therapeutic strategies based on a patient’s status are needed. Long-term and continuous follow-up is also required to yield clinically meaningful results. Despite these challenges, the gut microbiome in patients with CKD represents an exciting area of research. The microbiome-based treatments show promise in improving clinical practice and outcomes of patients with CKD.
Availability of data and materials
Not applicable.
References
Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–33.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65.
Lynch SV, Pedersen O. The Human intestinal microbiome in health and disease. N Engl J Med. 2016;375:2369–79.
Chen Y-Y, Chen D-Q, Chen L, Liu J-R, Vaziri ND, Guo Y, Zhao YY. Microbiome–metabolome reveals the contribution of gut–kidney axis on kidney disease. J Transl Med. 2019;17:5.
Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, DeSantis TZ, Ni Z, Nguyen T-H, Andersen GL. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83:308–15.
Wong J, Piceno YM, DeSantis TZ, Pahl M, Andersen GL, Vaziri ND. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am J Nephrol. 2014;39:230–7.
Vanholder R, De Smet R, Waterloos MA, Van Landschoot N, Vogeleere P, Hoste E, Ringoir S. Mechanisms of uremic inhibition of phagocyte reactive species production: characterization of the role of p-cresol. Kidney Int. 1995;47:510–7.
Vaziri ND, Goshtasbi N, Yuan J, Jellbauer S, Moradi H, Raffatellu M, Kalantar-Zadeh K. Uremic plasma impairs barrier function and depletes the tight junction protein constituents of intestinal epithelium. Am J Nephrol. 2012;36:438–43.
Satoh M, Hayashi H, Watanabe M, Ueda K, Yamato H, Yoshioka T, Motojima M. Uremic toxins overload accelerates renal damage in a rat model of chronic renal failure. Nephron Exp Nephrol. 2003;95:e111-118.
Sun C-Y, Chang S-C, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin–angiotensin–aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE. 2012;7: e34026.
Watanabe H, Miyamoto Y, Honda D, Tanaka H, Wu Q, Endo M, Noguchi T, Kadowaki D, Ishima Y, Kotani S, et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013;83:582–92.
Jager KJ, Kovesdy C, Langham R, Rosenberg M, Jha V, Zoccali C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int. 2019;96:1048–50.
KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease https://kdigo.org/guidelines/ckd-evaluation-and-management/
Chronic kidney disease (CKD) surveillance system. 2021 Accessed 30 Sep. https://nccd.cdc.gov/ckd/default.aspx
McCullough KP, Morgenstern H, Saran R, Herman WH, Robinson BM. Projecting ESRD incidence and prevalence in the United States through 2030. J Am Soc Nephrol. 2019;30:127.
Remuzzi G, Perico N, Macia M, Ruggenenti P. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int. 2005;68:S57–65.
Fujii H, Goto S, Fukagawa M. Role of uremic toxins for kidney, cardiovascular, and bone dysfunction. Toxins. 2018;10:E202.
Betjes MGH. Immune cell dysfunction and inflammation in end-stage renal disease. Nat Rev Nephrol. 2013;9:255–65.
Lorne E, Dupont H, Abraham E. Toll-like receptors 2 and 4: initiators of non-septic inflammation in critical care medicine? Intensive Care Med. 2010;36:1826–35.
Gollapudi P, Yoon J-W, Gollapudi S, Pahl MV, Vaziri ND. Leukocyte toll-like receptor expression in end-stage kidney disease. Am J Nephrol. 2010;31:247–54.
Pecoits-Filho R, Heimbürger O, Bárány P, Suliman M, Fehrman-Ekholm I, Lindholm B, Stenvinkel P. Associations between circulating inflammatory markers and residual renal function in CRF patients. Am J Kidney Dis. 2003;41:1212–8.
Sela S, Shurtz-Swirski R, Cohen-Mazor M, Mazor R, Chezar J, Shapiro G, Hassan K, Shkolnik G, Geron R, Kristal B. Primed peripheral polymorphonuclear leukocyte: a culprit underlying chronic low-grade inflammation and systemic oxidative stress in chronic kidney disease. J Am Soc Nephrol. 2005;16:2431–8.
Jeng Y, Lim PS, Wu MY, Tseng T-Y, Chen CH, Chen HP, Wu TK. Proportions of proinflammatory monocytes are important predictors of mortality risk in hemodialysis patients. Mediators Inflamm. 2017;2017: e1070959.
Yadav AK, Jha V. CD4+CD28null cells are expanded and exhibit a cytolytic profile in end-stage renal disease patients on peritoneal dialysis. Nephrol Dialysis Transpl Off Publ Eur Dialysis Transpl Assoc Eur Renal Assoc. 2011;26:1689–94.
Pahl MV, Vaziri ND, Yuan J. Adler SG upregulation of monocyte/macrophage HGFIN (Gpnmb/Osteoactivin) expression in end-stage renal disease. Clin J Am Soc Nephrol CJASN. 2010;5:56–61.
Betjes MGH, de Wit EEA, Weimar W, Litjens NHR. Circulating pro-inflammatory CD4posCD28null T cells are independently associated with cardiovascular disease in ESRD patients. Nephrol Dialysis Transpl Off Publ Eur Dialysis Transpl Assoc Eur Renal Assoc. 2010;25:3640–6.
Talal S, Mona K, Karem A, Yaniv L, Reut H-M, Ariel S, Moran A-K, Harel E, Campisi-Pinto S, Mahmoud A-A, et al. Neutrophil degranulation and severely impaired extracellular trap formation at the basis of susceptibility to infections of hemodialysis patients. BMC Med. 2022;20:364.
Verkade MA, van de Wetering J, Klepper M, Vaessen LMB, Weimar W, Betjes MGH. Peripheral blood dendritic cells and GM-CSF as an adjuvant for hepatitis B vaccination in hemodialysis patients. Kidney Int. 2004;66:614–21.
Syed-Ahmed, Narayanan M. Immune dysfunction and risk of infection in chronic kidney disease. Adv Chronic Kidney Dis. 2019;26:8–15.
Bowry SK, Kotanko P, Himmele R, Tao X, Anger M. The membrane perspective of uraemic toxins: which ones should, or can, be removed? Clin Kidney J. 2021;14:i17–31.
Ho-dac-Pannekeet MM, Koopmans JG, Struijk DG, Krediet RT. Restriction coefficients of low molecular weight solutes and macromolecules during peritoneal dialysis. Adv Peritoneal Dialysis Conf Peritoneal Dialysis. 1997;13:72–6.
Evenepoel P, Bammens B, Verbeke K, Vanrenterghem Y. Superior dialytic clearance of beta(2)-microglobulin and p-cresol by high-flux hemodialysis as compared to peritoneal dialysis. Kidney Int. 2006;70:794–9.
Krieter DH, Kerwagen S, Rüth M, Lemke H-D, Wanner C. Differences in dialysis efficacy have limited effects on protein-bound uremic toxins plasma levels over time. Toxins. 2019;11:47.
Snauwaert E, Van Biesen W, Raes A, Glorieux G, Walle JV, Roels S, Vanholder R, Askiti V, Azukaitis K, Bayazit A, Canpolat N, Fischbach M, Saoussen K, Litwin M, Obrycki L, Paglialonga F, Ranchin B, Samaille C, Schaefer F, Schmitt CP, Spasojevic B, Stefanidis CJ, Shroff R, Eloot S. Haemodiafiltration does not lower protein-bound uraemic toxin levels compared with haemodialysis in a paediatric population. Nephrol Dialysis Trans Off Publ Eur Dialysis Transpl Assoc Eur Renal Assoc. 2020;35(648):656.
DeMoss RD, Moser K. Tryptophanase in diverse bacterial species. J Bacteriol. 1969;98:167–71.
Stockinger B, Meglio PD, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol. 2014;32:403–32.
Lemos DR, McMurdo M, Karaca G, Wilflingseder J, Leaf IA, Gupta N, Miyoshi T, Susa K, Johnson BG, Soliman K, et al. Interleukin-1β activates a MYC-dependent metabolic switch in kidney stromal cells necessary for progressive tubulointerstitial fibrosis. J Am Soc Nephrol. 2018;29:1690–705.
Kim HY, Yoo T-H, Hwang Y, Lee GH, Kim B, Jang J, Yu HT, Kim MC, Cho J-Y, Lee CJ, et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci Rep. 2017;7:3057.
Kim HY, Yoo T-H, Cho J-Y, Kim HC, Lee WW. Indoxyl sulfate–induced TNF-α is regulated by crosstalk between the aryl hydrocarbon receptor, NF-κB, and SOCS2 in human macrophages. FASEB J. 2019;33:10844–58.
Lee C-T, Kuo C-C, Chen Y-M, Hsu C-Y, Lee W-C, Tsai Y-C, Ng H-Y, Kuo L-C, Chiou TT-Y, Yang Y-K, et al. Factors associated with blood concentrations of indoxyl sulfate and p—cresol in patients undergoing peritoneal dialysis peritoneal dialysis international. J Int Soc Peritoneal Dialysis. 2010;30:456–63.
Pletinck A, Glorieux G, Schepers E, Cohen G, Gondouin B, Landschoot MV, Eloot S, Rops Voorde A, Vriese AD, et al. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J Am Soc Nephrol. 2013;24(1981):1994.
Hubbard TD, Murray IA, Perdew GH. Indole and tryptophan metabolism: endogenous and dietary routes to ah receptor activation. Drug Metab Dispos. 2015;43:1522–35.
de Melo M, Curi T, Miyasaka C, Palanch A, Curi R. Effect of indole acetic acid on oxygen metabolism in cultured rat neutrophil. General Pharmacol Vascul Syst. 1998;31:573–8.
Salopek-Sondi B, Piljac-Žegarac J, Magnus V, Kopjar N. Free radical–scavenging activity and DNA damaging potential of auxins IAA and 2-methyl-IAA evaluated in human neutrophils by the alkaline comet assay. J Biochem Mol Toxicol. 2010;24:165–73.
Dou L, Sallée M, Cerini C, Poitevin S, Gondouin B, Jourde-Chiche N, Fallague K, Brunet P, Calaf R, Dussol B, et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J Am Soc Nephrol. 2015;26:876–87.
Schepers E, Glorieux G, Vanholder R. The gut the forgotten organ in uremia? Blood Purif. 2010;29:130–6.
Schepers E, Meert N, Glorieux G, Goeman J, Van der Eycken J, Vanholder R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol Dial Transplant. 2007;22:592–6.
Azevedo MLV, Bonan NB, Dias G, Brehm F, Steiner TM, Souza WM, Stinghen AEM, Barreto FC, Elifio-Esposito S, Pecoits-Filho R, Moreno-Amaral AN. p-Cresyl sulfate affects the oxidative burst, phagocytosis process, and antigen presentation of monocyte-derived macrophages. Toxicol Lett. 2016;263:1–5.
Shiba T, Kawakami K, Sasaki T, Makino I, Kato I, Kobayashi T, Uchida K, Kaneko K. Effects of intestinal bacteria-derived p-cresyl sulfate on Th1-type immune response in vivo and in vitro. Toxicol Appl Pharmacol. 2014;274:191–9.
Kawakami K, Makino I, Kato I, Uchida K, Onoue M. p-Cresol inhibits IL-12 production by murine macrophages stimulated with bacterial immunostimulant. Immunopharmacol Immunotoxicol. 2009;31:304–9.
Shiba T, Makino I, Sasaki T, Fukuhara Y, Kawakami K, Kato I, Kobayashi T. p-Cresyl sulfate decreases peripheral B cells in mice with adenine-induced renal dysfunction. Toxicol Appl Pharmacol. 2018;342:50–9.
Beaman M, Michael J, MacLennan IC, Adu D. T-cell-independent and T-cell-dependent antibody responses in patients with chronic renal failure. Nephrol Dialysis Transpl Off Publ Eur Dialysis Trans Assoc Eur Renal Assoc. 1989;4:216–21.
Evenepoel P, Meijers BKI, Bammens BRM, Verbeke K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. 2009;76:S12–9.
Wu IW, Lin CY, Chang LC, Lee CC, Chiu CY, Hsu HJ, Sun CY, Chen YC, Kuo YL, Yang CW, et al. Gut microbiota as diagnostic tools for mirroring disease progression and circulating nephrotoxin levels in chronic kidney disease: discovery and validation study. Int J Biol Sci. 2020;16:420–34.
Bolati D, Shimizu H, Higashiyama Y, Nishijima F, Niwa T. Indoxyl sulfate induces epithelial-to-mesenchymal transition in rat kidneys and human proximal tubular cells. Am J Nephrol. 2011;34:318–23.
Poveda J, Sanchez-Niño MD, Glorieux G, Sanz AB, Egido J, Vanholder R, Ortiz A. p-cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol Dialysis Transpl Off Publ Eur Dialysis Trans Assoc Eur Renal Assoc. 2014;29:56–64.
Sun C-Y, Chang S-C, Wu M-S. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012;81:640–50.
Sun C-Y, Hsu H-H, Wu M-S. p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol Dial Transplant. 2013;28:70–8.
Wiley SR, Winkles JA. A member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 2003;14:241–9.
Li H-B, Xu M-L, Xu X-D, Tang Y-Y, Jiang H-L, Li L, Xia W-J, Cui N, Bai J, Dai Z-M, et al. Faecalibacterium prausnitzii attenuates CKD via butyrate-renal GPR43 axis. Circ Res. 2022;131:e120–34.
Gonzalez A, Krieg R, Massey HD, Carl D, Ghosh S, Gehr TWB, Ghosh SS. Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol Dial Transpl. 2019;34:783–94.
Al-Harbi NO, Nadeem A, Ahmad SF, Alotaibi MR, AlAsmari AF, Alanazi WA, Al-Harbi MM, El-Sherbeeny AM, Ibrahim KE. Short chain fatty acid, acetate ameliorates sepsis-induced acute kidney injury by inhibition of NADPH oxidase signaling in T cells. Int Immunopharmacol. 2018;58:24–31.
Mikami D, Kobayashi M, Uwada J, Yazawa T, Kamiyama K, Nishimori K, Nishikawa Y, Nishikawa S, Yokoi S, Kimura H, et al. 2020. Short-chain fatty acid mitigates adenine-induced chronic kidney disease via FFA2 and FFA3 pathways. Biochimica et Biophysica Acta (BBA) Molecular and Cell Biology of Lipids. 65: 158666.
Marzocco S, Fazeli G, Di Micco L, Autore G, Adesso S, Dal Piaz F, Heidland A. Supplementation of short-chain fatty acid, sodium propionate, in patients on maintenance hemodialysis: beneficial effects on inflammatory parameters and gut-derived uremic toxins, a pilot study (PLAN Study). J Clin Med. 2018;7:315.
Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368:2402–14.
Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG, Julian BA. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22:1795–803.
Hu X, Du J, Xie Y, Huang Q, Xiao Y, Chen J, Yan S, Gong Z, Ouyang S. Fecal microbiota characteristics of Chinese patients with primary IgA nephropathy: a cross-sectional study. BMC Nephrol. 2020;21:97.
De Angelis M, Montemurno E, Piccolo M, Vannini L, Lauriero G, Maranzano V, Gozzi G, Serrazanetti D, Dalfino G, Gobbetti M, Gesualdo L. Microbiota and metabolome associated with immunoglobulin a nephropathy (IgAN). PLoS ONE. 2014;9: e99006.
Qin W, Zhong X, Fan JM, Zhang YJ, Liu XR, Ma XY. External suppression causes the low expression of the Cosmc gene in IgA nephropathy. Nephrol Dialysis Transpl Off Publ Eur Dialysis Transpl Assoc Eur Renal Assoc. 2008;23:1608–14.
Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci USA. 2002;99:16613–8.
Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Investig. 1999;104:73–81.
McCarthy DD, Kujawa J, Wilson C, Papandile A, Poreci U, Porfilio EA, Ward L, Lawson MAE, Macpherson AJ, McCoy KD, et al. Mice overexpressing BAFF develop a commensal flora-dependent. IgA-Assoc Nephropathy J Clin Investig. 2011;121:3991–4002.
Han L, Fang X, He Y, Ruan XZ. Forefronts symposium iga nephropathy, the gut microbiota, and gut−kidney crosstalk. Kidney Int Rep. 2015;1(189):196.
Zhong Z, Tan J, Tan L, Tang Y, Qiu Z, Pei G, Qin W. Modifications of gut microbiota are associated with the severity of IgA nephropathy in the Chinese population. Int Immunopharmacol. 2020;89: 107085.
Chemouny JM, Gleeson PJ, Abbad L, Lauriero G, Le Boedec E, Roux K, Monot C, Bredel M, Bex-Coudrat J, Sannier A, et al. Modulation of the microbiota by oral antibiotics treats immunoglobulin a nephropathy in humanized mice. Nephrol Dial Transpl. 2019;34(1135):1144.
Xie Y, Bowe B, Mokdad AH, Xian H, Yan Y, Li T, Maddukuri G, Tsai C-Y, Floyd T, Al-Aly Z. Analysis of the global burden of disease study highlights the global, regional, and national trends of chronic kidney disease. Kidney Int. 2018;94(567):581.
Gross JL, de Azevedo MJ, Silveiro SP, Henrique Canani L, Luiza Caramori M, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28(164):176.
Urushihara M, Kagami S. Role of the intrarenal renin–angiotensin system in the progression of renal disease. Pediatr Nephrol. 2017;32(1471):1479.
Arora MK, Singh UK. Molecular mechanisms in the pathogenesis of diabetic nephropathy: an update. Vascul Pharmacol. 2013;58:259–71.
Chen-chen L, Ze-bo H, Wang R, Hong Ze, Jian L, Chen P, Zhang J, Li X, Yuan B, Huang S, Ruan X, Liu B, Ma K. Gut microbiota dysbiosis-induced activation of the intrarenal renin–angiotensin system is involved in kidney injuries in rat diabetic nephropathy. Acta Pharmacol Sin. 2020;41(1111):1118.
Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan L-X, Rey F, Wang T, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci. 2013;110:4410–5.
Hu ZB, Lu J, Chen PP, Lu CC, Zhang JX, Li XQ, Yuan BY, Huang SJ, Ruan XZ, Liu BC. Dysbiosis of intestinal microbiota mediates tubulointerstitial injury in diabetic nephropathy via the disruption of cholesterol homeostasis. Theranostics. 2020;10:2803–16.
Lu J, Chen PP, Zhang JX, Li XQ, Wang GH, Yuan BY, Huang SJ, Liu XQ, Jiang TT, Wang MY, et al. GPR43 deficiency protects against podocyte insulin resistance in diabetic nephropathy through the restoration of AMPKα activity. Theranostics. 2021;11:4728–42.
Dai H, Liu Q, Liu B. Research progress on mechanism of podocyte depletion in diabetic nephropathy. J Diabetes Res. 2017;2017:2615286.
The diabetic kidney. Totowa. NJ: Humana Press; 2006.
Iatcu CO, Steen A, Covasa M. Gut microbiota and complications of type-2 diabetes. Nutrients. 2022;14:166.
Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, Braverman S, Tengeler AC, Barak O, Elazar M, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 2018;359:1376–83.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72.
Linh HT, Iwata Y, Senda Y, Sakai-Takemori Y, Nakade Y, Oshima M, Nakagawa-Yoneda S, Ogura H, Sato K, Minami T, et al. Intestinal bacterial translocation contributes to diabetic kidney disease. J Am Soc Nephrol. 2022;33:1105–19.
Fischer JC, Bscheider M, Eisenkolb G, Lin C-C, Wintges A, Otten V, Lindemans CA, Heidegger S, Rudelius M, Monette S, et al. RIG-I/MAVS and STING signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci Trans Med. 2017;9:2513.
Kikuchi K, Saigusa D, Kanemitsu Y, Matsumoto Y, Thanai P, Suzuki N, Mise K, Yamaguchi H, Nakamura T, Asaji K, et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat Commun. 2019;10:1–17.
Yamamoto H, Tsuruoka S, Ioka T, Ando H, Ito C, Akimoto T, Fujimura A, Asano Y, Kusano E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006;69(1780):1785.
Chen J-H, Chao C-T, Huang J-W, Hung K-Y, Liu S-H, Tarng D-C, Chiang C-K. Early elimination of uremic toxin ameliorates AKI-to-CKD transition. Clin Sci. 2021;135:2643–58.
Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11:264–76.
Pral LP, Fachi JL, Corrêa RO, Colonna M, Vinolo MAR. Hypoxia and HIF-1 as key regulators of gut microbiota and host interactions. Trends Immunol. 2021;42:604–21.
Yang J, Kim CJ, Go YS, Lee HY, Kim M-G, Oh SW, Cho WY, Im S-H, Jo SK. Intestinal microbiota control acute kidney injury severity by immune modulation. Kidney Int. 2020;98:932–46.
Vallon V. Tubular transport in acute kidney injury: relevance for diagnosis. Prognosis Int Nephron. 2016;134:160–6.
Wang I-K, Lai H-C, Yu C-J, Liang C-C, Chang C-T, Kuo H-L, Yang Y-F, Lin C-C, Lin H-H, Liu Y-L, et al. Real-time PCR analysis of the intestinal microbiotas in peritoneal dialysis patients. Appl Environ Microbiol. 2012;78:1107–12.
Simões-Silva L, Araujo R, Pestana M, Soares-Silva I, Sampaio-Maia B. The microbiome in chronic kidney disease patients undergoing hemodialysis and peritoneal dialysis. Pharmacol Res. 2018;130:143–51.
Juergensen PH, Finkelstein FO, Brennan R, Santacroce S, Ahern MJ. Pseudomonas peritonitis associated with continuous ambulatory peritoneal dialysis: a six-year study. Am J Kidney Dis. 1988;11:413–7.
Szeto C, Chow VCY, Chow KM, Lai RWM, Chung KY, Leung CB, Kwan BCH, Li PKT. PK-T enterobacteriaceae peritonitis complicating peritoneal dialysis: a review of 210 consecutive cases. Kidney Int. 2006;69(1245):1252.
Bossola M, Sanguinetti M, Scribano D, Zuppi C, Giungi S, Luciani G, Torelli R, Posteraro B, Fadda G, Tazza L. Circulating bacterial-derived DNA fragments and markers of inflammation in chronic hemodialysis patients. CJASN. 2009;4:379–85.
Imholz ALT, Koomen GCM, Struijk DG, Arisz L, Krediet RT. Effect of an increased intraperitoneal pressure on fluid and solute transport during CAPD. Kidney Int. 1993;44:1078–85.
Ribitsch W, Schneditz D, Franssen CFM, Schilcher G, Stadlbauer V, Horina JH, Rosenkranz AR. Increased hepato-splanchnic vasoconstriction in diabetics during regular hemodialysis. PLoS ONE. 2015;10: e0145411.
Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J Am Soc Nephrol. 1897;2014:25.
Wu I-W, Hsu K-H, Hsu H-J, Lee C-C, Sun C-Y, Tsai C-J, Wu M-S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients–a prospective cohort study. Nephrology Dialysis Transpl Off Publ Eur Dialysis Transpl Assoc Eur Renal Assoc. 2012;27:1169–75.
Wu I-W, Hsu K-H, Lee C-C, Sun C-Y, Hsu H-J, Tsai C-J, Tzen C-Y, Wang Y-C, Lin C-Y, Wu M-S. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrology Dialysis Transpl Off Publ Eur Dialysis and Transpl Assoc Eur Renal Assoc. 2011;26:938–47.
Parthasarathy G, Chen J, Chen X, Chia N, O’Connor HM, Wolf PG, Gaskins HR. Relationship between microbiota of the colonic mucosa vs feces and symptoms, colonic transit, and methane production in female patients with chronic constipation. Gastroenterology. 2016;150:367-379.e361.
Quigley EMM. The enteric microbiota in the pathogenesis and management of constipation. Best Pract Res Clin Gastroenterol. 2011;25:119–26.
Simenhoff ML, Dunn SR, Zollner GP, Fitzpatrick ME, Emery SM, Sandine WE, Ayres JW. Biomodulation of the toxic and nutritional effects of small bowel bacterial overgrowth in end-stage kidney disease using freeze-dried Lactobacillus acidophilus. Miner Electrolyte Metab. 1996;22:92–6.
Hida M, Aiba Y, Sawamura S, Suzuki N, Satoh T, Koga Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron. 1996;74:349–55.
Wu I-W, Gao S-S, Chou H-C, Yang H-Y, Chang L-C, Kuo Y-L, Dinh MCV, Chung W-H, Yang C-W, Lai H-C, et al. Integrative metagenomic and metabolomic analyses reveal severity-specific signatures of gut microbiota in chronic kidney disease. Theranostics. 2020;10:5398–411.
Ren Z, Fan Y, Li A, Shen Q, Wu J, Ren L, Lu H, Ding S, Ren H, Liu C, et al. Alterations of the human gut microbiome in chronic kidney disease. Adv Sci. 2020;7:2001936.
Païssé S, Valle C, Servant F, Courtney M, Burcelin R, Amar J, Lelouvier B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion. 2016;56:1138–47.
Shah NB, Allegretti AS, Nigwekar SU, Kalim S, Zhao S, Lelouvier B, Servant F, Serena G, Thadhani RI, Raj DS, Fasano A. Blood microbiome profile in CKD : a pilot study. CJASN. 2019;14:692–701.
Merino-Ribas A, Araujo R, Pereira L, Campos J, Barreiros L, Segundo MA, Silva N, Costa CFFA, Quelhas-Santos J, Trindade F, et al. Vascular calcification and the gut and blood microbiome in chronic kidney disease patients on peritoneal dialysis: a pilot study. Biomolecules. 2022;12:867.
Sciarra F, Franceschini E, Campolo F, Venneri MA. The diagnostic potential of the human blood microbiome: are we dreaming or awake? Int J Mol Sci. 2023;24:10422.
Lelouvier B, Servant F, Païssé S, Brunet A-C, Benyahya S, Serino M, Valle C, Ortiz MR, Puig J, Courtney M, et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology. 2016;64:2015–27.
Kramer H, Kuffel G, Thomas-White K, Wolfe AJ, Vellanki K, Leehey DJ, Bansal VK, Brubaker L, Flanigan R, Koval J, et al. Diversity of the midstream urine microbiome in adults with chronic kidney disease. Int Urol Nephrol. 2018;50:1123–30.
Matsushita K, Ballew SH, Wang AYM, Kalyesubula R, Schaeffner E, Agarwal R. Epidemiology and risk of cardiovascular disease in populations with chronic kidney disease. Nat Rev Nephrol. 2022;18(696):707.
Braam B, Joles JA, DanishwarGaillard AHCA. Cardiorenal syndrome—current understanding and future perspectives. Nat Rev Nephrol. 2014;10:48–55.
Bennett BJ, de Aguiar TQ, Vallim ZW, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL, Lusis AJ. Trimethylamine-N-Oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17(49):60.
Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, Lusis AJ, Shih DM. Trimethylamine N-Oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc. 2016;5:e002767.
Fatkhullina AR, Peshkova IO, Dzutsev A, Aghayev T, McCulloch JA, Thovarai V, Badger JH, Vats R, Sundd P, Tang H-Y, et al. an interleukin-23-interleukin-22 axis regulates intestinal microbial homeostasis to protect from diet-induced atherosclerosis. Immunity. 2018;49:943-957.e949.
Lok ZSY, Lyle AN. Osteopontin in vascular disease. Arterioscler Thromb Vasc Biol. 2019;39:613–22.
Gondouin B, Cerini C, Dou L, Sallée M, Duval-Sabatier A, Pletinck A, Calaf R, Lacroix R, Jourde-Chiche N, Poitevin S, et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013;84:733–44.
Campillo S, Bohorquez L, Gutiérrez-Calabrés E, García-Ayuso D, Miguel V, Griera M, Calle Y, de Frutos S, Rodríguez-Puyol M, Rodríguez-Puyol D, Calleros L. Indoxyl sulfate- and P-cresol-induced monocyte adhesion and migration is mediated by integrin-linked kinase-dependent podosome formation. Exp Mol Med. 2022;54:226–38.
Moore WE, Holdeman LV. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl Microbiol. 1974;27:961–79.
Dave M, Higgins PD, Middha S. The human gut microbiome: current knowledge, challenges, and future directions. Transl Res J Lab Clin Med. 2012;160:246–57.
Gao B, Chi L, Zhu Y, Shi X, Tu P, Li B, Yin J, Gao N, Shen W, Schnabl B. An Introduction to next generation sequencing bioinformatic analysis in gut microbiome studies. Biomolecules. 2021;11:530.
Luo D, Zhao W, Lin Z, Wu J, Lin H, Li Y, Song J, Zhang J. The effects of hemodialysis and peritoneal dialysis on the gut microbiota of end-stage renal disease patients, and the relationship between gut microbiota and patient prognoses. Front Cell Infect Microbiol. 2021;11: 579386.
Sato N, Kakuta M, Hasegawa T, Yamaguchi R, Uchino E, Murashita K, Nakaji S, Imoto S, Yanagita M. Metagenomic profiling of gut microbiome in early chronic kidney disease. Nephrology Dialysis Trans Off Publ Eur Dialysis Trans Assoc Eur Renal Assoc. 2021;36:1675–84.
Wang X, Yang S, Li S, Zhao L, Hao Y, Qin J, Zhang L, Zhang C, Bian W, Zuo L, et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut. 2020;69:2131–42.
Opdebeeck B, Maudsley S, Azmi A, Maré A, Leger DW, Meijers DB, Verhulst A, Evenepoel P, D’Haese PC, Neven E. Indoxyl sulfate and p-cresyl sulfate promote vascular calcification and associate with glucose intolerance. J Am Soc Nephrol. 2019;30:751.
Karaduta O, Glazko G, Dvanajscak Z, Arthur J, Mackintosh S, Orr L, Rahmatallah Y, Yeruva L, Tackett A, Zybailov B. Resistant starch slows the progression of CKD in the 5/6 nephrectomy mouse model. Physiol Rep. 2020;8:e14610.
Lobel L, Cao YG, Fenn K, Glickman JN, Garrett WS. Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science. 2020;369(1518):1524.
Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, Theis JD, Dogan A, Smith RJH. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82(465):473.
Dasari S, Alexander MP, Vrana JA, Theis JD, Mills JR, Negron V, Sethi S, Dispenzieri A, Highsmith WEJ, Nasr Kurtin SH, DnaJ PJ. Heat shock protein family B member 9 is a novel biomarker for fibrillary. GN J Am Soc Nephrol. 2018;29:1–51.
Ma L, Palmer ND, Choi YA, Murea M, Snipes JA, Parks JS, Langefeld CD, Freedman I. BI APOL1 risk variants impair multiple mitochondrial pathways in a metabolomics analysis. Kidney. 2020;360(1):1353.
Fitzpatrick LR, Small J, Hoerr RA, Bostwick EF, Maines L, Koltun WA. In vitro and in vivo effects of the probiotic Escherichia coli strain M-17: immunomodulation and attenuation of murine colitis. Br J Nutr. 2008;100(530):541.
Resta-Lenert S, Barrett KE. Probiotics and commensals reverse TNF-alpha- and IFN-gamma-induced dysfunction in human intestinal epithelial cells. Gastroenterology. 2006;130:731–46.
Zhang R, Li Z, Gu X, Zhao J, Guo T, Kong J. Probiotic Bacillus subtilis LF11 protects intestinal epithelium against salmonella infection. Front Cell Infect Microbiol. 2022;12:837886.
Cotter PD, Hill C. Ross RP Bacteriocins: developing innate immunity for food. Nat Rev Microbiol. 2005;3:777–88.
Takayama F, Taki K, Niwa T. Bifidobacterium in gastro-resistant seamless capsule reduces serum levels of indoxyl sulfate in patients on hemodialysis. Am J Kidney Dis. 2003;41:S142–5.
Zhu H, Cao C, Wu Z, Zhang H, Sun Z, Wang M, Xu H, Zhao Z, Wang Y, Pei G, et al. The probiotic L. casei Zhang slows the progression of acute and chronic kidney disease. Cell Metab. 2021;33:1926-1942.e1928.
Quigley EMM Prebiotics and Probiotics in Digestive Health. Clinical gastroenterology and hepatology: the official clinical practice. J Am Gastroenterol Assoc. 2019;17:333–44.
Sarao LK, Arora M. Probiotics, prebiotics, and microencapsulation: a review. Crit Rev Food Sci Nutr. 2017;57:344–71.
Corrêa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MAR. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunol. 2016;5: e73.
Akbari P, Fink-Gremmels J, Willems RHAM, Difilippo E, Schols HA, Schoterman MHC, Garssen J, Braber S. Characterizing microbiota-independent effects of oligosaccharides on intestinal epithelial cells: insight into the role of structure and size. Eur J Nutr. 2017;56:1919–30.
Meijers BKI, De Preter V, Verbeke K, Vanrenterghem Y, Evenepoel P. p -Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol Dial Transpl. 2010;25:219–24.
Rossi M, Johnson DW, Xu H, Carrero JJ, Pascoe E, French C, Campbell KL. Dietary protein-fiber ratio associates with circulating levels of indoxyl sulfate and p-cresyl sulfate in chronic kidney disease patients. Nutr Metab Cardiovasc Dis. 2015;25(860):865.
Kelly JT, Palmer SC, Wai SN, Ruospo M, Carrero J-J, Campbell KL, Strippoli GFM. Healthy dietary patterns and risk of mortality and ESRD in CKD: a meta-analysis of cohort studies. Clin J Am Soc Nephrol. 2017;12:272–9.
Marques FZ, Nelson E, Chu P-Y, Horlock D, Fiedler A, Ziemann M, Tan JK, Kuruppu S, Rajapakse NW, El-Osta A, et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135:964–77.
Zmora N, Suez J, Elinav E. You are what you eat diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol. 2019;16:35–56.
Fujii H, Nishijima F, Goto S, Sugano M, Yamato H, Kitazawa R, Kitazawa S, Fukagawa M. Oral charcoal adsorbent (AST-120) prevents progression of cardiac damage in chronic kidney disease through suppression of oxidative stress. Nephrol Dialysis Transpl Off Publ Eur Dialysis Transpl Assoc Eur Renal Assoc. 2009;24(2089):2095.
Huang Y, Zhou J, Wang S, Xiong J, Chen Y, Liu Y, Xiao T, Li Y, He T, Li Y, et al. Indoxyl sulfate induces intestinal barrier injury through IRF1-DRP1 axis-mediated mitophagy impairment. Theranostics. 2020;10:7384–400.
Markowiak P, Śliżewska K. Effects of probiotics, prebiotics, and synbiotics on human health. Nutrients. 2017;9:1021.
Saulnier DMA, Spinler JK, Gibson GR, Versalovic J. Mechanisms of probiosis and prebiosis considerations for enhanced functional foods. Curr Opin Biotechnol. 2009;20(135):141.
Guida B, Germanò R, Trio R, Russo D, Memoli B, Grumetto L, Barbato F, Cataldi M. Effect of short-term synbiotic treatment on plasma p-cresol levels in patients with chronic renal failure: a randomized clinical trial. Nutr Metab Cardiovasc Dis. 2014;24:1043–9.
Rossi M, Johnson DW, Morrison M, Pascoe EM, Coombes JS, Forbes JM, Szeto C-C, McWhinney BC, Ungerer JPJ, Campbell KL. Synbiotics easing renal failure by improving gut microbiology (SYNERGY): a randomized trial. Clin J Am Soc Nephrol. 2016;11:223–31.
Yang C-Y, Chen T-W, Lu W-L, Liang S-S, Huang H-D, Tseng C-P, Tarng D-C. Synbiotics alleviate the gut indole load and dysbiosis in chronic kidney disease. Cells. 2021;10:114.
Knight EL, Stampfer MJ, Hankinson SE, Spiegelman D. Curhan GC The impact of protein intake on renal function decline in women with normal renal function or mild renal insufficiency. Ann Intern Med. 2003;138:460–7.
Ko G-J, Rhee CM, Kalantar-Zadeh K, Joshi S. The effects of high-protein diets on kidney health and longevity. J Am Soc Nephrol. 2020;31:1667–79.
Tovar-Palacio C, Tovar AR, Torres N, Cruz C, Hernández-Pando R, Salas-Garrido G, Pedraza-Chaverri J, Correa-Rotter R. Proinflammatory gene expression and renal lipogenesis are modulated by dietary protein content in obese Zucker fa/fa rats. Am J Physiol-Renal Physiol. 2011;300:F263–71.
Lew Q-LJ, Jafar TH, Koh HWL, Jin A, Chow KY, Yuan J-M, Red KWP. Meat intake and risk of ESRD. J Am Soc Nephrol. 2017;28:304–12.
Kamper A-L, Strandgaard S. Long-term effects of high-protein diets on renal function. Annu Rev Nutr. 2017;37:347–69.
Hostetter TH, Meyer TW, Rennke HG, Brenner Noddin BM, Wttao JA, Sandstrom DJ. Chronic effects of dietary protein in the rat with intact and reduced renal mass. Kidney Int. 1986;30:509–17.
Sakaguchi Y, Kaimori J-Y. Isaka Y plant-dominant low protein diet: a potential alternative dietary practice for patients with chronic kidney disease. Nutrients. 2023;15:1002.
Microbiome SJL, Engineering. Nature. 2015;518:S10–S10.
Devlin AS, Marcobal A, Dodd D, Nayfach S, Plummer N, Meyer T, Pollard KS, Sonnenburg JL. Fischbach MA modulation of a circulating uremic solute via rational genetic manipulation of the gut microbiota. Cell Host Microbe. 2016;20:709–15.
Prakash S, Chang TMS. Microencapsulated genetically engineered live E. coli DH5 cells administered orally to maintain normal plasma urea level in uremic rats. Nature Med. 1996;2:883–7.
Steidler L, Hans W, Schotte L, Neirynck S, Obermeier F, Falk W, Fiers W, Remaut E. Treatment of murine colitis by lactococcus lactis secreting interleukin-10. Science. 2000;289:1352–5.
Ting S-Y, Martínez-García E, Huang S, Bertolli SK, Kelly KA, Cutler KJ, Su ED, Zhi H, Tang Q, Radey MC, et al. Targeted depletion of bacteria from mixed populations by programmable adhesion with antagonistic competitor cells. Cell Host Microbe. 2020;28:313-321.e316.
van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EdJ, Bartelsman JFWM, Tijssen JGP, Speelman P, Dijkgraaf MGW, Keller JJ. Duodenal infusion of donor feces for recurrent clostridium difficile. N Engl J Med. 2013;368(407):415.
FUAD Others. Guidance for industry biologics evaluation and research, us food and drug administration. Berlin: Silver Spring; 2013.
Gulati AS, Nicholson MR, Khoruts A, Kahn SA. Fecal microbiota transplantation across the lifespan balancing efficacy safety and innovation official journal of the American College of gastroenterology ACG. Am J Gastroenterol. 2023. https://doi.org/10.14309/ajg.0000000000002167.
Lauriero G, Abbad L, Vacca M, Celano G, Chemouny JM, Calasso M, Berthelot L, Gesualdo L, De Angelis M. Monteiro RC fecal microbiota transplantation modulates renal phenotype in the humanized mouse model of IgA nephropathy. Frontiers Immunol. 2021. https://doi.org/10.3389/fimmu.2021.694787.
Wang S, Xu M, Wang W, Cao X, Piao M, Khan S, Yan F, Cao H. Systematic review: adverse events of fecal microbiota transplantation. PLoS ONE. 2016;11: e0161174.
Important safety alert regarding use of fecal microbiota for transplantation and risk of serious adverse reactions due to transmission of multidrug resistant organisms https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/important-safety-alert-regarding-use-fecal-microbiota-transplantation-and-risk-serious-adverse
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science Foundation of China (No. 81870490 to P. Y.) Shanghai Pujiang Program (22PJ1409300 to P. Y.), S. Y. is supported by the Innovative Research Team of High-Level Local Universities in Shanghai (SHSMU-ZLCX20211700) and the Hospital Fund (XK2019007).
Author information
Authors and Affiliations
Contributions
Y. P., S. Y., and Z. T. conceived the ideas, wrote and revised the paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tang, Z., Yu, S. & Pan, Y. The gut microbiome tango in the progression of chronic kidney disease and potential therapeutic strategies. J Transl Med 21, 689 (2023). https://doi.org/10.1186/s12967-023-04455-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04455-2