
Abstract
Breast cancer (BC) as one of the most common causes of human deaths among women, is always considered one of the global health challenges. Despite various advances in diagnostic and therapeutic methods, a significant percentage of BC patients have a poor prognosis due to the lack of therapeutic response. Therefore, investigating the molecular mechanisms involved in BC progression can improve the therapeutic and diagnostic strategies in these patients. Cytokine and growth factor-dependent signaling pathways play a key role during BC progression. In addition to cytokines and growth factors, long non-coding RNAs (lncRNAs) have also important roles in regulation of such signaling pathways. Therefore, in the present review we discussed the role of lncRNAs in regulation of PI3K/AKT, MAPK, and TGF-β signaling pathways in breast tumor cells. It has been shown that lncRNAs mainly have an oncogenic role through the promotion of these signaling pathways in BC. This review can be an effective step in introducing the lncRNAs inhibition as a probable therapeutic strategy to reduce tumor growth by suppression of PI3K/AKT, MAPK, and TGF-β signaling pathways in BC patients. In addition, considering the oncogenic role and increased levels of lncRNAs expressions in majority of the breast tumors, lncRNAs can be also considered as the reliable diagnostic markers in BC patients.
Similar content being viewed by others
Background
Breast cancer (BC) is the second-most common cause of cancer-related deaths that is considered as a health challenge among women [1]. Novel diagnostic and therapeutic strategies have decreased the mortality rate of breast cancer. However, approximately 279,100 new cases and 42,690 deaths due to breast cancer have occurred worldwide in 2020 [2]. Although, chemotherapy, radiotherapy, and surgery are major treatment options for the breast cancer, these modalities are not effective for the metastatic breast cancer patients [3, 4]. In fact, tumor metastasis is responsible for about 90% of cancer-related mortalities. Prevention of the tumor metastasis has been a major obstacle in the treatment of breast cancer. Therefore, investigating the underlying mechanisms of breast cancer metastasis is required to introduce novel and efficient therapeutic targets to reduce breast tumor invasion and metastasis. Early detection of breast cancer is critical for effective treatment. Diagnostic methods typically involve examination, ultrasonography, magnetic resonance imaging, mammography, and biopsy. Besides, constitutional treatments usually entail mastectomy, lumpectomy, hormone therapy, chemotherapy, and radiotherapy [5]. Non-coding RNAs (ncRNA) are considered as the key regulators of BC development and metastasis that can be suggested as the novel therapeutic approachs [6,7,8]. Long non-coding RNAs (lncRNAs) are a group of noncoding RNAs that are implicated in pivotal cellular processes in several tumors via interactions with protein, RNA, and DNA [9]. LncRNAs function as microRNA sponges in the cytoplasm and regulate gene expression post-transcriptionally through guiding RNA-binding proteins that are involved in active polysomes and mRNA decay [10,11,12,13]. In the nucleus, lncRNAs modulate RNA processing, transcription, and chromatin remodeling [14]. They are involved in various processes, including organogenesis, embryogenesis, and tumorigenesis [15, 16]. LncRNAs are implicated in the growth, metastasis, aggressiveness, migration, and programmed cell death of various cancers [17, 18]. Impared expression of lncRNAs has been detected in multiple human cancers, and they are pivotal regulators of tumor occurrence and progression via diverse signaling pathways [19, 20]. They are potent markers in early tumor diagnosis and targeted therapy [21,22,23]. Regarding the role of lncRNAs in regulation of signaling pathways in breast tumor cells, in the present review we discussed their role in regulation of PI3K/AKT, TGF-β, and MAPK signaling pathways during breast tumor progression and metastasis to suggest them as the reliable diagnostic and therapeutic options among BC patients (Table 1).
PI3K/AKT signaling pathway
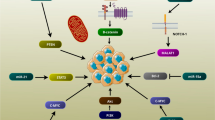
PI3K/AKT/mTOR pathway is a crucial axis in regulation of various pathophysiological cellular processes such as cell proliferation, metabolism, and tumor progression [24, 25]. Majority of the growth factors, cytokines, and mitogens affects the cellular growth via the PI3K/AKT pathway. This signaling pathway is activated via various receptors such as receptor tyrosine kinases (RTKs), cytokine receptors, and G-protein-coupled receptors (GPCRs) that promote PI3K to produce PIP3. Subsequently, PIP3 activates the AKT to regulate cellular metabolism and growth via the modulation of various effectors such as GSK3β and mTOR. Despite the extracellular stimuli, AKT can also be activated by the other signaling pathways including WNT and TGF-β [25]. PI3K/Akt pathway functions as an oncogenic signaling axis in the progression of various cancers [26, 27]. PI3K hyper activation is critical in the pathogenesis of breast cancer that modulates cell survival, motility, growth, and metabolism [28, 29]. PI3K/Akt/mTOR pathway has a key role in endocrine resistance of breast tumor cells. Therefore, inhibitors of this pathway can be used in combination with other therapeutic modalities in breast cancer. It has been reported that PIK3CA mutations increased sensitivity toward the PI3K inhibitors [30]. LncRNAs have key roles during breast tumor progression by regulation of PI3K/AKT signaling pathway (Fig. 1). hnRNPA2B1 is an essential modulator of several normal processes such as mRNA stability and translation, RNA trafficking, and mRNA splicing [31]. LINC01133 increased cell proliferation in TNBC through PI3K-independent activation of AKT. LINC01133 stimulated PROTOR1 as a part of the mTORC2 complex via hnRNPA2B1 sponging that induced AKT [32]. The stimulated mTOR signaling pathway has been correlated with poor prognosis and reduced survival in BC patients [33]. Interactions between Polycomb Repressive Complex 2 (PRC2) and HOTAIR change the chromatin structure to accelerate the tumor cells metastasis [34, 35]. HOTAIR inhibition significantly down regulated the mTOR, AKT, and PI3K. The precise mechanism by which HOTAIR triggers the expression of PI3K, Akt, and mTOR is not fully understood; however, it is hypothesized that HOTAIR regulates transcription factors [36]. Doxorubicin (DOX) is one of the well-known and most effective drugs in BC; however, tumor cells may develop drug resistance that results in treatment failure [37,38,39]. HOTAIR inhibition reduced DOX resistance in BC cells via suppression of the PI3K/AKT/mTOR pathway following the negative regulation of PI3K, AKT and mTOR. HOTAIR inhibition also induced apoptosis in DOXR-MCF-7 cells via regulating Bax, Bcl-2, and caspase-3 [40]. KB-1980E6.3 up regulation was significantly associated with breast tumor progression and poor prognosis. KB-1980E6.3 increased the growth, migration, and aggressiveness of BC cells. KB-1980E6.3 was also significantly correlated with MMP-2, MMP-9, and vimentin. Moreover, it stimulated the PI3K/AKT pathway through AKT and PI3K phosphorylations [41]. GHET1 inhibition mitigated cell growth and invasion while promoted programmed cell death. GHET1 downregulation inhibited the c-Myc, which resulted in PI3K/AKT inactivation. Therefore, its inhibition reduced BC progression through PI3K/AKT repression. GHET1 inhibition also hindered the MCF-7 cell migration via negative regulation of MMP-2 and MMP-9. Accordingly, inhibition of GHET1 attenuated PI3K/AKT, c-Myc, and their downstream effectors such as MMP-2/9 and CCND1 [42]. FOXD2-AS1 has been found to be upregulated in bladder, gastric, and ovarian cancers and was also involved in the tumor cells migration, growth, invasion, and prognosis [43,44,45]. There was significant FOXD2-AS1 upregulation in antiadriamycin-resistant breast tumor cells and tissues. FOXD2-AS1 also down regulated the p-AKT and pPI3K to inhibit the PI3K/AKT signaling pathway in BC cells. Furthermore, inhibition of FOXD2-AS1 negatively regulated the growth, aggressiveness, and migration of BC cells while induced apoptosis and chemosensitivity [46]. There was SOX21-AS1 up regulation in BC tissues that was correlated with tumor stage, grade, metastasis, and clinical outcomes. SOX21-AS1 suppression significantly reduced EMT, cell growth, and invasion in BC cells. Repression of SOX21-AS1 also inhibited the PI3K/AKT signaling pathway, which underscored their potential interaction. Hence, SOX21-AS1 increased cell proliferation, invasion, and EMT via targeting the PI3K/AKT pathway in BC [47]. CREBZF belongs to the ATF/CREB family of transcription factors that regulates p53 mediated apoptosis [48]. The MBNL1-AS1 functioned as a tumor suppressor in BC through miR-423-5p/CREBZF axis that regulated PI3K/AKT pathway [49].

Role of lncRNAs during breast tumor progression by regulation of PI3K/AKT signaling pathway. (Created with BioRender.com)
PTEN as a well-known tumor suppressor inhibits the PI3K/AKT axis via dephosphorylation of PIP3 [50]. ZFAS1 enhanced apoptosis while mitigated cell growth and migration via miR-589 sponging that up regulated PTEN to inhibit the PI3K/AKT pathway [51]. There were PTENP1 and PTEN downregulations in BC tissues in comparison with normal margins that was correlated with a higher TNM stage and decreased overall survival in BC patients. PTENP1 inhibited tumor proliferation, colony formation, invasion, and tumor growth in BC. It regulated the chemoresistance, apoptosis, metastasis, and proliferation of BC cells by regulation of miR-20a/PTEN axis that inhibited PI3K/Akt pathway [52]. DUXAP8 exerts an oncogenic function in various cancers via targeting EZH2 and PTEN [53, 54]. PTENP1 suppressed BC cell invasion, colony formation, and survival while induced cell death via miR-19 sponging and subsequent regulation of PTEN/PI3K/Akt axis [55]. DUXAP8 induced the radioresistance of BC cells via stimulation of the PI3K/AKT/mTOR pathway and inhibition of EZH2 target genes including RHOB and E-cadherin [56]. Laminin subunit gamma 1 (LAMC1) is an extracellular matrix protein that has key roles in basement membranes, cell proliferation, movement, and development [57]. It also enhances the development of hepatocellular carcinoma via the PTEN/AKT axis [58]. SHNG6 induced the breast tumor cell growth and movement via miR-543 sponging and subsequent activation of LAMC1/PI3K/AKT axis [59].
Tamoxifen (TAM) is a widely used endocrine therapy that operates as an antagonist of estrogen in BC patients [5, 6]. Although, tamoxifen therapy is effective for most of the ER+ breast cancers, many patients eventually develop resistance to tamoxifen [7, 8]. Therefore, understanding the underlying mechanism of TAM resistance will decrease its adverse effects and facilitate overcoming resistance and sensitizing breast tumors. Enhancer of zeste homolog 2 (EZH2) is a member of polycomb proteins that is involved in the modulation of tumorigenesis [28, 29]. There was remarkable UCA1 upregulation in tamoxifen-resistant breast cancer relative to sensitive samples. Loss of UCA1 in LCC2 cells interrupted the cell cycle at G2/M phase and dysregulated p21 and CCND1. EZH2 negatively regulated p21 transcription via H3K27me3, which was induced by UCA1 in BC cells. Inhibition of UCA1 significantly reduced CREB and p-CREB expression levels. There was a positive association between the expression levels of AKT and UCA1. Therefore, UCA1 modulated the CREB via targeting the AKT and PI3K/AKT signaling pathways [60]. TTN-AS1 induced the breast tumor cell proliferation, aggressiveness, and tamoxifen resistance by targeting miR-107/ZNRF2 axis. ZNRF2 inhibition activated the PI3K/AKT pathway, which resulted in increased tamoxifen resistance in BC cells [61].
Triple Negative Breast Cancer (TNBC) as the most aggressive form of BC is characterized by a lack of estrogen receptor (ER), epidermal growth factor receptor 2 (HER-2), and progesterone receptor (PR) that results in the failure of targeted therapies in these patients [62]. About two-thirds of TNBC patients have a poor chemotherapy response [63]. IGF binds with IGF1R receptor to activate anti-apoptotic pathways and cell growth via the PI3K/AKT and RAS/MAPK signaling axes [64]. The downstream pathways of IGF1R induce the stemness and epithelial-to-mesenchymal transition (EMT) [65]. IGF1R is associated with the expression of EMT-related markers (Twist and Snail) and self-renewal factors (SOX2, OCT4, and NANOG) [66,67,68,69,70,71,72]. Additionally, IGF1R promotes cell growth and inhibits apoptosis via the Ras/Raf/MEK, PI3K/AKT/mTOR pathways [64, 73]. The HULC-IGF1R axis increased the growth and metastasis of breast tumor cells. HULC also enhanced IGF1R transcription through acetylation of H3K9 histone, intergenic chromosomal loop construction, and interaction with cis-acting elements. Accordingly, the HULC-IGF1R axis increased cisplatin resistance by up regulation of stem cell markers [74]. There was significant downregulation of IGF2-AS in BC plasma, tissues, and cell lines. IGF2-AS down regulated the IGF2 via DNMT1 that inhibited PI3K/AKT/mTOR and tumor progression in BC [75]. There was GAS5 downregulation in TNBC tissues that was associated with an invasive morphology in TNBC patients. GAS5 impeded cell growth while enhanced apoptosis in TNBC cells. GAS5 also attenuated TNBC development via miR-196a-5p sponging, which subsequently overexpressed FOXO1 and inhibited PI3K/AKT phosphorylation [76]. ZEB2-AS1 induced the growth, metastasis, and EMT in TNBC cells through regulating the PI3K/Akt/GSK3β/Zeb2 axis and polymerization of F‐actin [77].
Hormone receptor (HR)-positive BC patients may develop intrinsic and acquired resistance to hormone therapies, which is responsible for tumor relapses in these patients [78]. PI3K or mTOR inhibitors are used to restrain drug resistance, underscoring the critical role of PI3K/AKT/mTOR in endocrine therapy resistance [79, 80]. BDNF-AS stimulated the mTOR signaling pathway in endocrine-resistant BCs and TNBCs by RNH1 protein degradation. MEF2A is a transcription factor that up regulates the BDNF-AS. Interaction between RNH1 and RISC was involved in the destruction of mTOR mRNA. TRIM21 is an E3 ligase that degraded RNH1. BDNF-AS functioned as molecular glue for the TRIM21/RNH1 interaction that resulted in RNH1 degradation [81]. Trastuzumab as an anti-HER2 monoclonal antibody deactivates the downstream pathways and inhibits the production of HER2 dimers, which results in the suppression of tumor growth while apoptosis induction [82]. Trastuzumab resistance is one of the leading causes of treatment failure in HER2-positive BC patients. Therefore, developing new targeted therapies to overcome chemoresistance is an ideal strategy that improves the survival rate of HER2-positive BC patients [83]. HOTAIR mediated PTEN methylation activated PI3K/AKT to promote trastuzumab resistance. HOTAIR inhibition downregulated CCND1, p-MAPK, and p-AKT while upregulated P27 and PTEN. The upregulation of TGF-β, Snail, and Vimentin and impaired expression of CDH1 were reported in resistant BC cells. HOTAIR regulated acquired resistance through epigenetic changes, including demethylation of TGF-β and PTEN methylation, which increased the HER2-independent MEK/MAPK activity and subsequent proliferation and invasion of malignant cells [84].
Myc oncogene is a pivotal transcription factor that is implicated in tumor cell reprogramming and apoptosis [85, 86]. Lin28B facilitates the Myc nuclear translocation to increase AKT phosphorylation [87]. There was remarkable upregulation of Linc00839 in BC tissues that was corelated with an unfavorable prognosis. Linc00839 was regulated by Myc, which in turn modulated the expression of Lin28B and Myc proteins. Moreover, Linc00839 induced chemoresistance and growth via the PI3K/AKT signaling pathway and also phosphorylated the P38, STAT3, and Akt proteins. Therefore, Linc00839 ameliorated the breast tumor cell proliferation and chemoresistance through the Lin28B-induced Myc upregulation and PI3K/AKT activation [88]. RAD54B exhibits oncogenic function due to its pivotal role in the DNA repair and genomic instability [89]. AC012213.3 inhibition remarkebaly restrained the growth and invasion of BC cells. AC012213.3 regulated the RAD54B/PI3K/AKT axis to exert its oncogenic function in tumor cells. There was also AC012213.3 upregulation in BC tissues that was correlated with survival. Moreover, AC012213.3 targeted RAD54B, which resulted in tumor progression [90]. RNA-binding proteins (RBPs) are the key posttranscriptional regulators that have pivotal roles in tumor progression. Pumilio RNA binding family member 2 (PUM2) is a PUF family member of RBPs that modulates malignant tumors [91]. SCAMP1-TV2 suppression impeded the malignant characteristics of BC cells via decreasing their attachment to PUM2 and inducing the binding of PUM2 to INSM1 which finally downregulated the INSM1. Reduced expression of INSM1 inhibited SASH1 which suppressed the PI3K/AKT pathway in breast tumor cells [92].
TGF-β signaling pathway
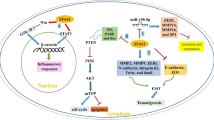
Transforming growth factor β (TGF-β) signaling is an important pathway during the development of different tumors. Deregulation of TGF-β pathway facilitates tumor cell proliferation, dissemination, metastasis, and immune scape [93]. TGF-β ligand binds to the TGF-βII/I receptors that phosphorylates and activates the SMAD2/3 (R-SMADs). Activated R-SMADs form a complex with SMAD4 and translocates to the nucleus to regulate the transcription of TGF-β target genes [94]. TGF-β has key role during breast cancer metastasis. Regarding the role of TGF-β in regulation of EMT process and stemness, it has a pivotal role in modulation of breast cancer stem cells [95]. LncRNAs have pivotal roles during breast tumor progression by regulation of TGF-β signaling pathway (Fig. 2). SMAD6 and SMAD7 inactivate TGF-β pathway via antagonistic signals and feedback loops [96]. SMAD7 is an important modulator of TGF-β signaling that inhibits the pathway through several proceses [97]. It functions within the cytoplasm by interfering with SMAD2/3 for the binding site of TGFβR1, thereby preventing the SMAD2/3 phosphorylation and suppressing signal transduction [98]. SMAD7 also facilitates the recruitment of SMURF1 and SMURF2 to TGF-βR1 and subsequent receptor degradation [99]. ARHGAP5-AS1 inhibited breast tumor cell migration via stabilizing SMAD7 [100]. CCAT2 suppression restrained tumor growth and invasion while induced apoptosis in BC cells via targeting the TGF-β signaling pathway. CCAT2 downregulation negatively regulated the TGF-β, α-SMA, and Smad2 proteins in BC cells [101]. There were linc-ROR upregulations in BC cell lines and tissues that was associated with an unfavorable prognosis. Linc-ROR also increased in-vivo tumor growth and invasion in BC via up regulation of the key components in TGF-β pathway [102]. HNF1A-AS1 induced TAM resistance in BC cells via the miR-363/SERTAD3 axis that promoted TGF-β/Smad [103]. TGFBR2 and TGF-β combination stimulates the TGF-β/Smad pathway that results in p21 and p15 up regulations, while c-Myc down regulation [104]. There was LINC00052 upregulation in BC that sponged miR-145-5p to induce TGF-βR2 expression [105]. CASC2 reduced BC progression by down regulation of TGF-β, Smad2, and a-SMA [106].

Role of lncRNAs during breast tumor progression by regulation of TGF-β signaling pathway. (Created with BioRender.com)
Epithelial-mesenchymal transition (EMT) process attenuates cell–cell adhesion via downregulation of the epithelial markers such as E-cadherin, whereas it induces cell mobility and the expression of mesenchymal markers including fibronectin, CDH2, and vimentin. EMT has also a pivotal role in breast tumor cell invasion and metastasis [107, 108]. TGF-β induces and preserves EMT process to promote tumor metastasis [109,110,111]. Runt-related transcription factor 2 (RUNX2) is a well-known regulator of osteoblast morphology and osteogenesis. The role of RUNX2 in tumorigenesis has been also investigated in recent years [112,113,114,115,116,117]. RUNX2 is involved in TGF-β-mediated EMT in which it is up regulated by TGF-β1 [115]. TGF-β1 inhibited the acetylation of the ANCR promoter and promoted HDAC3 enrichment at the ANCR promoter. ANCR modulated the TGF-β pathway via negative regulation of RUNX2. TGF-β also down regulated the ANCR that reduced BC cell invasion and migration. ANCR mitigated TGF-β1-induced EMT by RUNX2 down regulation [118]. LncRNA TPA provoked the aggressiveness and metastasis of BC through the initiation of EMT by targeting TGF-β signaling pathway [119]. RUNX2 provokes the EMT process by downregulation of CDH1 while up regulation of Vimentin and Snail2. TGF-β promotes BC metastasis by RUNX2 upregulation [120]. There was lncRNA-NORAD upregulation in BC, which was correlated with tumor growth, invasion, and poor prognosis. LncRNA-NORAD modulated TGF-β pathway to up regulate the RUNX2 [121]. Several pleiotropic transcription factors, such as Twist, ZEB1/2, Slug, and Snail, regulate the EMT process by promoting the mesenchymal while suppressing epithelial markers. Multiple intracellular and extracellular pathways also regulate the expression of these critical transcription factors [122]. TGF-β signaling pathway regulates a broad range of downstream genes, such as EMT transcription factors, via Smad and non-Smad pathways [123, 124]. TGF-β regulates the interaction between Smad and ERK signaling pathways, which enhance and preserve the expression of Slug [125, 126]. There were significant upregulations of UCA1.1 and AC026904 by TGF-β pathways. UCA1.1 and AC026904 enhanced the expression of Slug, thereby inducing EMT process and tumor metastasis. AC026904.1 played as a ceRNA to stimulate SLUG, whereas UCA1 up regulated the Slug via miR-203a and miR-1 spongings [127]. TGF-β promoted the DOX resistance and EMT process by UCA1 up regulation in breast tumor cells [128].
Cancer-associated fibroblasts (CAFs) as the pivotal components of the tumor microenvironment are involved in tumor growth, angiogenesis, invasion, and chemoresistance through cytokines and growth factors such as PDGF, b-FGF, VEGF, and TGF-β1 [129,130,131,132,133]. CDK5 has been reported to be a critical regulator of TGF-β1-mediated EMT during breast cancer progression [134]. CAFs increased the metastasis of BC cells via TGF-β1, which regulates the stroma-tumor cell interaction. CAFs also activated HOTAIR to promote EMT. HOTAIR was a direct transcriptional target of SMAD2/3/4. CAFs induced HOTAIR transcription to promote H3K27 trimethylation of EGR-1 and CDK5RAP1 promoters that up regulated the CDK5 and increased EMT. CAFs also activated HOTAIR, which in turn reinforced the EMT process. SMAD2/3/4 directly regulated the HOTAIR transcription. CAFs induced EMT and metastasis in BC cells via regulation of TGF-β1-mediated interactions between cancer cells and stromal cells [135]. Annexin A1 (ANXA1) belongs to the Ca2+ -dependent phospholipid-binding protein family that is involved in regulation of the leukocytes mediated immune responses [136]. ANXA1 is also engaged in regulation of signaling pathways to affect the tumor cell growth, invasion, angiogenesis, and apoptosis [137]. DCST1-AS1 attached to ANXA1 to promote TGF-β-mediated EMT in BC cells. DCST1-AS1 also increased the paclitaxel and doxorubicin resistances of BT-549 cells through targeting ANXA1. Additionally, DCST1-AS1 inhibition affected TGF-β-induced MMP2/9 releasing in MDA-MD-231 cells. DCST1-AS1 regulated the IGF2BP1; thereby DCST1-AS1 may perform its regulatory function on ANXA1 mRNA via targeting IGF2BP1. DCST1-AS1 enhanced TGF-β-mediated EMT and induced resistance to paclitaxel and doxorubicin via regulation of ANXA1 in TNBC cells [138].
RELA (p65, NF-κB3) is a NF-κB family member that modulates the proliferation and malignancy of several tumors via pro-survival and pro-inflammatory factors [139]. There was HOXA-AS2 upregulation in BC that was correlated with invasion, lumph node involvement, TNM staging, and survival. HOXA-AS2 inhibition significantly suppressed breast tumor cell growth via targeting miR-520c-3p/RELA and TGFBR2 axis [140]. RPs are a type of RNA-binding protein that are found in all cells [141]. RPL22 is a 60S ribosomal subunit and is associated with bacterial macrolide resistance by its mutation [142]. RPL22 promotes TGF-β pathway during tumor progression [143,144,145]. There was significant reduced expression of ADAMTS9-AS2 in TNBC samples compared to normal tissues that was correlated with tumor size, lymph node involvement, a higher TNM stage, patient age, and worse prognosis. ADAMTS9-AS2 attenuated the growth and invasion of TNBC cells via the TGF-β-mediated regulation of the ADAMTS9-AS2/RPL22 axis [146]. LncRNAs interact with PRC, SWI/SNF, and Pol II machinery to regulate gene expression inside the nucleus [34, 147,148,149,150]. Chromatin remodeling plays a crucial role in regulation of gene expression via proteins or protein complexes. SWI/SNF complex as a chromatin remodeller moves the nucleosomes and makes the DNA more accessible via recruitment of transcription factors to certain DNA regions [151]. There was TGFB2-AS1 downregulation in the metastatic TNBC patients. TGFB2-AS1/SMARCA4 interaction suppressed the SWI/SNF that was followed by TGFB2 and SOX2 down regulations. There was also up regulation of mesenchymal markers (slug, vimentin, and fibronection) while downregulation of epithelial markers (β-catenin) in breast tumor cells [152].
MAPK signaling pathway
MAPK is a pivotal pathway for the regulation of tumor invasion in multiple malignant cancers [153,154,155,156]. The genes involved in the MAPK pathway play important roles in numerous biological processes, including apoptosis, proliferation, and differentiation [157, 158]. MAPK cascades transmit and amplify the extracellular stimuli such as growth factors and steroid hormones which are associated with cell proliferation and apoptosis. ERK is the most important effector of the MAPK during breast cancer progression. Steroid hormones can activate MAPK. It has been reported that MAPK activation was higher in about half of breast tumors compared with normal margins [159]. LncRNAs have key roles during breast tumor progression by regulation of MAPK signaling pathway (Fig. 3). MEK4 activates JNK via phosphorylation that promotes its nuclear accumulation to activate the ELK21, ATF-22, and c-Jun transcription factors [160]. CBR3-AS1 upregulation was correlated with poor prognosis in ADR-resistant BC cell lines and patients. CBR3-AS1 increased resistance to ADR in BC cells through targeting the miR-25-3p/MEK4/JNK1 axis and intensifying the MAPK pathway [161]. PTENP1 suppressed the migration and growth of BC cells via the AKT signaling pathway and cell cycle associated genes such as CDK2 and cyclin A2. PTENP1 also inhibited the growth and migration of BC cells via the MAPK signaling pathway by repressing the phosphorylation of key proteins in this pathway including Erk1/2 and p38 [162]. LncCAMTA1 promoted the BC progression via miR-20b/VEGF axis that induced JAK/STAT3 and MAPK/ERK pathways [163]. PRNCR1 inhibition impaired cell survival and increased apoptosis and the number of cells in the G0/G1 phase following increased Bax and decreased Bcl-2 expressions. PRNCR1 inhibition reduced CCND2 expression, while suppression of miR-377 restored CCND2 expression levels. CCND2 also increased p38 MAPK and MEK1 phosphorylation. Therefore, PRNCR1/miR-377/CCND2 axis repressed cell apoptosis while enhanced cell proliferation in breast cancer via accelerating the MEK/MAPK pathway [164]. Around 70–80% of breast cancer cases are ER+ that plays a vital role in tumor growth and patients survival [165]. Therefore, adjuvant targeted therapies with tamoxifen or aromatase inhibitors are the primary treatment options for ER+ BC patients [166]. However, resistance to hormonal therapy can occur due to the development of estrogen-independent growth. Linc-RoR induced tamoxifen resistance and estrogen-independent growth in BC cells. It activated the MAPK/ERK signaling axis via destabilizing DUSP7 as a suppressor of ERK, indicating the regulatory function of linc-RoR on the MAPK/ERK axis under estrogen deprivation [167]. MiRNA-host gene lncRNAs (lnc-miRHGs) are certain lncRNAs that contain miRNAs within their DNA sequences [168]. OTX1, as a homeobox gene is a pivotal regulator of early human fetal retina and mammary gland development [169]. ERK/MAPK signaling pathway plays a critical role in tumor cell growth, differentiation, angiogenesis, and metastasis [170, 171]. There was MIR100HG upregulation in TNBC tissues and cell lines that increased proliferation, invasion, and migration via targeting the miR-5590-3p/OTX1 axis. MIR100HG inhibition also repressed the ERK/MAPK pathway in TNBC cells [172].

Role of lncRNAs during breast tumor progression by regulation of MAPK signaling pathway. (Created with BioRender.com)
The premetastatic niche (PMN) of primary tumors is a pivotal factor for the metastasis and colonization of tumor cells in certain secondary tissues via organotropism, immunosuppression, angiogenesis, and vascular permeability [173]. CAFs as the principal stromal constituents of the tumor microenvironment regulate tumor progression [174]. CAFs induce tumor proliferation and metastasis via metabolic and extracellular remodeling, cytokines, and exosomes in the primary tumor microenvironment [175,176,177,178,179]. CAFs are pivotal modulators of angiogenesis by WNT5a, WNT2, SDF1, PDGFC, and VEGFA release [180,181,182,183]. There was significant SNHG5 up regulation in primary breast CAFs that modulated generation of PMN through angiogenesis. The interaction of SNHG5 and IGF2BP2 stabilized the ZNF281 mRNA in an m6A-dependent manner. ZNF281 modulated CCL2 and CCL5 expressions in CAFs, which resulted in p38 signaling activation in endothelial cells and subsequent PMN construction in the metastatic environment. Recruitment of IGF2BP2 by SNHG5 elevated the ZNF281 levels via m6A regulation, which induced CCL2/5 transcription and secretion to form PMN. CCL2 and CCL5 that were released by CAFs stimulated the p38 MAPK axis in endothelial cells, thereby regulating PMN generation [184].
Conclusions
Cytokine and growth factor-dependent signaling pathways play a key role during BC progression. In addition to growth factors and cytokines, lncRNAs have also pivotal roles in regulation of these signaling pathways. Therefore, here we discussed the role of lncRNAs in regulation of PI3K/AKT, MAPK, and TGF-β signaling pathways in breast tumor cells. It has been reported that lncRNAs mainly have an oncogenic role through the activation of these signaling pathways during BC progression. Therefore, the inhibition of lncRNAs can be introduced as a suitable therapeutic strategy to reduce the breast tumor growth by suppression of PI3K/AKT, MAPK, and TGF-β signaling pathways. Regarding, the tissue-specific characteristics of lncRNAs, they can also be suggested as the next generation tumor markers. Considering the up regulation of the majority of lncRNAs in tumor tissue and serum of BC patients, lncRNAs expression profiling can be introduced as an efficient non-invasive diagnostic method among these patients. However, regarding the role of lncRNAs in chronic disorders such as diabetes and metabolic disorders, it is required to assess the circulating levels of various lncRNAs in breast cancer patients to introduce a multi lncRNA panel marker instead of a single lncRNA as a non-invasive diagnostic method in these patients. While, the oncogenic lncRNAs can be inactivated using the antisense methods, tumor suppressive lncRNAs can be synthetically-engineered and employed to inhibit breast tumor growth. However, as the breast cancer is a heterogenic malignancy, the personalized medicine is required to use the lncRNAs as the therapeutic factors in breast cancer patients. Therefore, more clinical trials and in-vivo studies are required to bring the lncRNAs as the diagnostic and therapeutic options into the clinics for the breast cancer patients.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ANXA1:
-
Annexin A1
- BC:
-
Breast cancer
- CAFs:
-
Cancer-associated fibroblasts
- DOX:
-
Doxorubicin
- EZH2:
-
Enhancer of zeste homolog 2
- HER-2:
-
Epidermal growth factor receptor 2
- EMT:
-
Epithelial-to-mesenchymal transition
- ER:
-
Estrogen receptor
- GPCRs:
-
G-protein-coupled receptors
- HR:
-
Hormone receptor
- LAMC1:
-
Laminin subunit gamma 1
- lncRNAs:
-
Long non-coding RNAs
- ncRNA:
-
Non-coding RNAs
- PRC2:
-
Polycomb Repressive Complex 2
- PMN:
-
Premetastatic niche
- PR:
-
Progesterone receptor
- PUM2:
-
Pumilio RNA binding family member 2
- RTKs:
-
Receptor tyrosine kinases
- RBPs:
-
RNA-binding proteins
- RUNX2:
-
Runt-related transcription factor 2
- TAM:
-
Tamoxifen
- TGF-β:
-
Transforming growth factor β
- TNBC:
-
Triple negative breast cancer
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics 2021. CA Cancer J Clin. 2021;71(1):7–33.
Monnot GC, Romero P. Rationale for immunological approaches to breast cancer therapy. Breast. 2018;37:187–95.
Moghbeli M. Genetic and molecular biology of breast cancer among Iranian patients. J Transl Med. 2019;17(1):218.
Ataollahi MR, Sharifi J, Paknahad MR, Paknahad A. Breast cancer and associated factors: a review. J Med Life. 2015;8(Spec Iss 4):6–11.
Lin FM, Kumar S, Ren J, Karami S, Bahnassy S, Li Y, et al. SUMOylation of HP1α supports association with ncRNA to define responsiveness of breast cancer cells to chemotherapy. Oncotarget. 2016;7(21):30336–49.
Arieti F, Huet T, Gabus-Darlix C, Thore S. Structural perspective on the steroid receptor RNA activator, a ncRNA with potential implication in breast cancer. Int J Mol Med. 2013;32:S61.
Moghbeli M, Zangouei AS, Nasrpour Navaii Z, Taghehchian N. Molecular mechanisms of the microRNA-132 during tumor progressions. Cancer Cell Int. 2021;21(1):439.
Yang G, Lu X, Yuan L. LncRNA A link between RNA and cancer. Biochim Biophys Acta (BBA)—Gene Reguly Mech. 2014;1839(11):1097–109.
Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–52.
Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, et al. LincRNA-p21 suppresses target mRNA translation. Mol Cell. 2012;47(4):648–55.
Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491(7424):454–7.
Yao RW, Wang Y, Chen LL. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21(5):542–51.
He P, Zhang C, Ji Y, Ge MK, Yu Y, Zhang N, et al. Epithelial cells-enriched lncRNA SNHG8 regulates chromatin condensation by binding to Histone H1s. Cell Death Differ. 2022;29(8):1569–81.
Jariwala N, Sarkar D. Emerging role of lncRNA in cancer: a potential avenue in molecular medicine. Ann Transl Med. 2016;4(15):286.
Rahmani Z, Mojarrad M, Moghbeli M. Long non-coding RNAs as the critical factors during tumor progressions among Iranian population: an overview. Cell Biosci. 2020;10:6.
Xu CG, Yang MF, Ren YQ, Wu CH, Wang LQ. Exosomes mediated transfer of lncRNA UCA1 results in increased tamoxifen resistance in breast cancer cells. Eur Rev Med Pharmacol Sci. 2016;20(20):4362–8.
Khalili-Tanha G, Moghbeli M. Long non-coding RNAs as the critical regulators of doxorubicin resistance in tumor cells. Cell Mol Biol Lett. 2021;26(1):39.
Lin C, Yang L. Long noncoding RNA in cancer: wiring signaling circuitry. Trends Cell Biol. 2018;28(4):287–301.
Hamidi AA, Khalili-Tanha G, Nasrpour Navaei Z, Moghbeli M. Long non-coding RNAs as the critical regulators of epithelial mesenchymal transition in colorectal tumor cells: an overview. Cancer Cell Int. 2022;22(1):71.
Qiu MT, Hu JW, Yin R, Xu L. Long noncoding RNA: an emerging paradigm of cancer research. Tumour Biol. 2013;34(2):613–20.
Eades G, Zhang YS, Li QL, Xia JX, Yao Y, Zhou Q. Long non-coding RNAs in stem cells and cancer. World J Clin Oncol. 2014;5(2):134–41.
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–69.
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–44.
Navaei ZN, Khalili-Tanha G, Zangouei AS, Abbaszadegan MR, Moghbeli M. PI3K/AKT signaling pathway as a critical regulator of Cisplatin response in tumor cells. Oncol Res. 2021;29(4):235–50.
Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, Kundu GC. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol Cancer. 2018;17(1):34.
Moghbeli M, Makhdoumi Y, Soltani Delgosha M, Aarabi A, Dadkhah E, Memar B, et al. ErbB1 and ErbB3 co-over expression as a prognostic factor in gastric cancer. Biol Res. 2019;52(1):2.
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. 2017;45:62–71.
Paplomata E, O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014;6(4):154–66.
Liu Y, Shi SL. The roles of hnRNP A2/B1 in RNA biology and disease. Wiley Interdiscip Rev RNA. 2021;12(2):e1612.
Tu Z, Hu Y, Raizada D, Bassal MA, Tenen DG, Karnoub AE. Long noncoding RNA-mediated activation of PROTOR1/PRR5-AKT signaling shunt downstream of PI3K in triple-negative breast cancer. Proc Natl Acad Sci USA. 2022;119(43):e2203180119.
Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13(10):433–42.
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–6.
Gupta S, Iljin K, Sara H, Mpindi JP, Mirtti T, Vainio P, et al. FZD4 as a mediator of ERG oncogene-induced WNT signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Can Res. 2010;70(17):6735–45.
Sadeghalvad M, Mansouri K, Mohammadi-Motlagh HR, Noorbakhsh F, Mostafaie A, Alipour S, et al. Long non-coding RNA HOTAIR induces the PI3K/AKT/mTOR signaling pathway in breast cancer cells. Rev Assoc Med Bras 1992. 2022;68(4):456–62.
Wang H, Yu Y, Jiang Z, Cao WM, Wang Z, Dou J, et al. Next-generation proteasome inhibitor MLN9708 sensitizes breast cancer cells to doxorubicin-induced apoptosis. Sci Rep. 2016;6:26456.
Palmieri C, Krell J, James CR, Harper-Wynne C, Misra V, Cleator S, et al. Rechallenging with anthracyclines and taxanes in metastatic breast cancer. Nat Rev Clin Oncol. 2010;7(10):561–74.
Wu X, Fu Y, Wang Y, Wan S, Zhang J. Gaining insight into crizotinib resistance mechanisms caused by L2026M and G2032R mutations in ROS1 via molecular dynamics simulations and free-energy calculations. J Mol Model. 2017;23(4):141.
Li Z, Qian J, Li J, Zhu C. Knockdown of lncRNA-HOTAIR downregulates the drug-resistance of breast cancer cells to doxorubicin via the PI3K/AKT/mTOR signaling pathway. Exp Ther Med. 2019;18(1):435–42.
He L, Tang L, Wang R, Liu L, Zhu P, Jiang K, et al. Long noncoding RNA KB-1980E6.3 promotes breast cancer progression through the PI3K/AKT signalling pathway. Pathol Res Pract. 2022;234:153891.
Han M, Wang Y, Gu Y, Ge X, Seng J, Guo G, et al. lncRNA GHET1 knockdown suppresses breast cancer activity in vitro and in vivo. Am J Transl Res. 2019;11(1):31–44.
Gao J, Liu F, Zhao X, Zhang P. Long non-coding RNA FOXD2-AS1 promotes proliferation, migration and invasion of ovarian cancer cells via regulating the expression of miR-4492. Exp Ther Med. 2021;21(4):307.
Xu TP, Wang WY, Ma P, Shuai Y, Zhao K, Wang YF, et al. Upregulation of the long noncoding RNA FOXD2-AS1 promotes carcinogenesis by epigenetically silencing EphB3 through EZH2 and LSD1, and predicts poor prognosis in gastric cancer. Oncogene. 2018;37(36):5020–36.
Su F, He W, Chen C, Liu M, Liu H, Xue F, et al. The long non-coding RNA FOXD2-AS1 promotes bladder cancer progression and recurrence through a positive feedback loop with Akt and E2F1. Cell Death Dis. 2018;9(2):233.
Nong Q, Yu S, Hu H, Hu X. Knockdown of lncRNA FOXD2-AS1 inhibits proliferation, migration, and drug resistance of breast cancer cells. Comput Math Methods Med. 2021;2021:9674761.
Sheng XY, Wang CH, Wang CF, Xu HY. Long-chain non-coding SOX21-AS1 promotes proliferation and migration of breast cancer cells through the PI3K/AKT signaling pathway. Cancer Manag Res. 2020;12:11005–14.
López-Mateo I, Villaronga M, Llanos S, Belandia B. The transcription factor CREBZF is a novel positive regulator of p53. Cell Cycle. 2012;11(20):3887–95.
Fang J, Jiang G, Mao W, Huang L, Huang C, Wang S, et al. Up-regulation of long noncoding RNA MBNL1-AS1 suppresses breast cancer progression by modulating miR-423-5p/CREBZF axis. Bioengineered. 2022;13(2):3707–23.
Colakoglu T, Yildirim S, Kayaselcuk F, Nursal TZ, Ezer A, Noyan T, et al. Clinicopathological significance of PTEN loss and the phosphoinositide 3-kinase/Akt pathway in sporadic colorectal neoplasms: is PTEN loss predictor of local recurrence? Am J Surg. 2008;195(6):719–25.
Zhang S, Wang J, Yao T, Tao M. LncRNA ZFAS1/miR-589 regulates the PTEN/PI3K/AKT signal pathway in the proliferation, invasion and migration of breast cancer cells. Cytotechnology. 2020;72(3):415–25.
Gao X, Qin T, Mao J, Zhang J, Fan S, Lu Y, et al. PTENP1/miR-20a/PTEN axis contributes to breast cancer progression by regulating PTEN via PI3K/AKT pathway. J Exp Clin Cancer Res CR. 2019;38(1):256.
Sun M, Nie FQ, Zang C, Wang Y, Hou J, Wei C, et al. The pseudogene DUXAP8 promotes non-small-cell lung cancer cell proliferation and invasion by epigenetically silencing EGR1 and RHOB. Mol Ther. 2017;25(3):739–51.
Lin MG, Hong YK, Zhang Y, Lin BB, He XJ. Mechanism of lncRNA DUXAP8 in promoting proliferation of bladder cancer cells by regulating PTEN. Eur Rev Med Pharmacol Sci. 2018;22(11):3370–7.
Shi X, Tang X, Su L. Overexpression of long noncoding RNA PTENP1 Inhibits cell proliferation and migration via suppression of miR-19b in breast cancer cells. Oncol Res. 2018;26(6):869–78.
Lei C, Li S, Fan Y, Hua L, Pan Q, Li Y, et al. LncRNA DUXAP8 induces breast cancer radioresistance by modulating the PI3K/AKT/mTOR pathway and the EZH2-E-cadherin/RHOB pathway. Cancer Biol Ther. 2022;23(1):1–13.
Aumailley M. The laminin family. Cell Adh Migr. 2013;7(1):48–55.
Ye G, Qin Y, Wang S, Pan D, Xu S, Wu C, et al. Lamc1 promotes the Warburg effect in hepatocellular carcinoma cells by regulating PKM2 expression through AKT pathway. Cancer Biol Ther. 2019;20(5):711–9.
Wang YQ, Huang G, Chen J, Cao H, Xu WT. LncRNA SNHG6 promotes breast cancer progression and epithelial-mesenchymal transition via miR-543/LAMC1 axis. Breast Cancer Res Treat. 2021;188(1):1–14.
Li Z, Yu D, Li H, Lv Y, Li S. Long non-coding RNA UCA1 confers tamoxifen resistance in breast cancer endocrinotherapy through regulation of the EZH2/p21 axis and the PI3K/AKT signaling pathway. Int J Oncol. 2019;54(3):1033–42.
Fang J, Li K, Huang C, Xue H, Ni Q. LncRNA TTN-AS1 confers tamoxifen resistance in breast cancer via sponging miR-107 to modulate PI3K/AKT signaling pathway. Am J Transl Res. 2022;14(4):2267–79.
Chen JQ, Russo J. ERalpha-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochem Biophys Acta. 2009;1796(2):162–75.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig. 2011;121(7):2750–67.
Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–28.
Farabaugh SM, Boone DN, Lee AV. Role of IGF1R in breast cancer subtypes, stemness, and lineage differentiation. Front Endocrinol. 2015;6:59.
Taliaferro-Smith L, Oberlick E, Liu T, McGlothen T, Alcaide T, Tobin R, et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget. 2015;6(7):4757.
Dupont J, Fernandez AM, Glackin CA, Helman L, LeRoith D. Insulin-like growth factor 1 (IGF-1)-induced twist expression is involved in the anti-apoptotic effects of the IGF-1 receptor. J Biol Chem. 2001;276(28):26699–707.
Wada A, Yokoo H, Yanagita T, Kobayashi H. New twist on neuronal insulin receptor signaling in health, disease, and therapeutics. J Pharmacol Sci. 2005;99(2):128–43.
Kim H-J, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-κB and snail. Mol Cell Biol. 2007;27(8):3165–75.
Chiu Y-F, Wu C-C, Kuo M-H, Miao C-C, Zheng M-Y, Chen P-Y, et al. Critical role of SOX2–IGF2 signaling in aggressiveness of bladder cancer. Sci Rep. 2020;10(1):1–13.
Kuo Y-C, Au H-K, Hsu J-L, Wang H-F, Lee C-J, Peng S-W, et al. IGF-1R promotes symmetric self-renewal and migration of alkaline phosphatase+ germ stem cells through HIF-2α-OCT4/CXCR4 loop under hypoxia. Stem Cell Rep. 2018;10(2):524–37.
Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology. 2012;56(3):1004–14.
Hakuno F, Takahashi S-I. IGF1 receptor signaling pathways. J Mol Endocrinol. 2018;61(1):T69–86.
Zhou L, Li H, Sun T, Wen X, Niu C, Li M, et al. HULC targets the IGF1R–PI3K-AKT axis in trans to promote breast cancer metastasis and cisplatin resistance. Cancer Lett. 2022;548:215861.
Zhang Y, Yan H, Jiang Y, Chen T, Ma Z, Li F, et al. Long non-coding RNA IGF2-AS represses breast cancer tumorigenesis by epigenetically regulating IGF2. Exp Biol Med. 2021;246(4):371–9.
Li S, Zhou J, Wang Z, Wang P, Gao X, Wang Y. Long noncoding RNA GAS5 suppresses triple negative breast cancer progression through inhibition of proliferation and invasion by competitively binding miR-196a-5p. Biomed Pharmacother. 2018;104:451–7.
Zhang G, Li H, Sun R, Li P, Yang Z, Liu Y, et al. Long non-coding RNA ZEB2-AS1 promotes the proliferation, metastasis and epithelial mesenchymal transition in triple-negative breast cancer by epigenetically activating ZEB2. J Cell Mol Med. 2019;23(5):3271–9.
Early Breast Cancer Trialists’ Collaborative Group. A romatase inhibitors versus tamoxifen in early breast cancer patient-level meta-analysis of the randomised trials. Lancet. 2015;386(10001):1341–52.
AlFakeeh A, Brezden-Masley C. Overcoming endocrine resistance in hormone receptor-positive breast cancer. Curr Oncol. 2018;25(Suppl 1):S18-s27.
Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925–36.
Lin X, Dinglin X, Cao S, Zheng S, Wu C, Chen W, et al. Enhancer-driven lncRNA BDNF-AS induces endocrine resistance and malignant progression of breast cancer through the RNH1/TRIM21/mTOR cascade. Cell Rep. 2020;31(10):107753.
Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416(6878):279–80.
de Melo GD, Jardim DL, Marchesi MS, Hortobagyi GN. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget. 2016;7(39):64431–46.
Chen T, Liu Z, Zeng W, Huang T. Down-regulation of long non-coding RNA HOTAIR sensitizes breast cancer to trastuzumab. Sci Rep. 2019;9(1):19881.
Kim T, Jeon YJ, Cui R, Lee JH, Peng Y, Kim SH, et al. Role of MYC-regulated long noncoding RNAs in cell cycle regulation and tumorigenesis. J Natl Cancer Inst. 2015;107(4):diu505.
Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016;354(6315):4445.
Meder L, König K, Dietlein F, Macheleidt I, Florin A, Ercanoglu MS, et al. LIN28B enhanced tumorigenesis in an autochthonous KRAS(G12V)-driven lung carcinoma mouse model. Oncogene. 2018;37(20):2746–56.
Chen Q, Shen H, Zhu X, Liu Y, Yang H, Chen H, et al. A nuclear lncRNA Linc00839 as a Myc target to promote breast cancer chemoresistance via PI3K/AKT signaling pathway. Cancer Sci. 2020;111(9):3279–91.
McAndrew EN, McManus KJ. The enigmatic oncogene and tumor suppressor-like properties of RAD54B: Insights into genome instability and cancer. Genes Chromosom Cancer. 2017;56(7):513–23.
Zhong H, Zeng G, He L. Overexpression of the lncRNA AC012213.3 promotes proliferation, migration and invasion of breast cancer via RAD54B/PI3K/AKT Axis and is associated with worse patient prognosis. Cancer Manag Res. 2021;13:7213–23.
Naudin C, Hattabi A, Michelet F, Miri-Nezhad A, Benyoucef A, Pflumio F, et al. PUMILIO/FOXP1 signaling drives expansion of hematopoietic stem/progenitor and leukemia cells. Blood. 2017;129(18):2493–506.
Tao W, Ma J, Zheng J, Liu X, Liu Y, Ruan X, et al. Silencing SCAMP1-TV2 inhibited the malignant biological behaviors of breast cancer cells by interaction with PUM2 to facilitate INSM1 mRNA degradation. Front Oncol. 2020;10:613.
Massagué J. TGFbeta in Cancer. Cell. 2008;134(2):215–30.
Tzavlaki K, Moustakas A. TGF-beta Signaling. Biomolecules. 2020;10(3):487.
Imamura T, Hikita A, Inoue Y. The roles of TGF-beta signaling in carcinogenesis and breast cancer metastasis. Breast Cancer. 2012;19(2):118–24.
Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19(23):2783–810.
Stolfi C, Marafini I, De Simone V, Pallone F, Monteleone G. The dual role of Smad7 in the control of cancer growth and metastasis. Int J Mol Sci. 2013;14(12):23774–90.
Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389(6651):631–5.
Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, et al. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276(16):12477–80.
Wang CL, Li JC, Zhou CX, Ma CN, Wang DF, Wo LL, et al. Long non-coding RNA ARHGAP5-AS1 inhibits migration of breast cancer cell via stabilizing SMAD7 protein. Breast Cancer Res Treat. 2021;189(3):607–19.
Wu ZJ, Li Y, Wu YZ, Wang Y, Nian WQ, Wang LL, et al. Long non-coding RNA CCAT2 promotes the breast cancer growth and metastasis by regulating TGF-β signaling pathway. Eur Rev Med Pharmacol Sci. 2017;21(4):706–14.
Hou L, Tu J, Cheng F, Yang H, Yu F, Wang M, et al. Long noncoding RNA ROR promotes breast cancer by regulating the TGF-β pathway. Cancer Cell Int. 2018;18:142.
Li Y, Liu L, Lv Y, Zhang Y, Zhang L, Yu H, et al. Silencing long non-coding RNA HNF1A-AS1 inhibits growth and resistance to TAM of breast cancer cells via the microRNA-363/SERTAD3 axis. J Drug Target. 2021;29(7):742–53.
Wang Y, Tan X, Tang Y, Zhang C, Xu J, Zhou J, et al. Dysregulated Tgfbr2/ERK-Smad4/SOX2 signaling promotes lung squamous cell carcinoma formation. Can Res. 2019;79(17):4466–79.
Dong M, Xu T, Li H, Li X. LINC00052 promotes breast cancer cell progression and metastasis by sponging miR-145-5p to modulate TGFBR2 expression. Oncol Lett. 2021;21(5):368.
Zhang Y, Zhu M, Sun Y, Li W, Wang Y, Yu W. Upregulation of lncRNA CASC2 suppresses cell proliferation and metastasis of breast cancer via inactivation of the TGF-β signaling pathway. Oncol Res. 2019;27(3):379–87.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119(6):1429–37.
Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24(37):5764–74.
Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P. TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci. 1999;112(Pt 24):4557–68.
Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16(4):1987–2002.
Boregowda RK, Medina DJ, Markert E, Bryan MA, Chen W, Chen S, et al. The transcription factor RUNX2 regulates receptor tyrosine kinase expression in melanoma. Oncotarget. 2016;7(20):29689–707.
Farina NH, Zingiryan A, Akech JA, Callahan CJ, Lu H, Stein JL, et al. A microRNA/Runx1/Runx2 network regulates prostate tumor progression from onset to adenocarcinoma in TRAMP mice. Oncotarget. 2016;7(43):70462–74.
Guo ZJ, Yang L, Qian F, Wang YX, Yu X, Ji CD, et al. Transcription factor RUNX2 up-regulates chemokine receptor CXCR4 to promote invasive and metastatic potentials of human gastric cancer. Oncotarget. 2016;7(15):20999–1012.
Niu DF, Kondo T, Nakazawa T, Oishi N, Kawasaki T, Mochizuki K, et al. Transcription factor Runx2 is a regulator of epithelial-mesenchymal transition and invasion in thyroid carcinomas. Lab Invest J Tech Methods Pathol. 2012;92(8):1181–90.
Pratap J, Lian JB, Javed A, Barnes GL, van Wijnen AJ, Stein JL, et al. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 2006;25(4):589–600.
van der Deen M, Akech J, Lapointe D, Gupta S, Young DW, Montecino MA, et al. Genomic promoter occupancy of runt-related transcription factor RUNX2 in osteosarcoma cells identifies genes involved in cell adhesion and motility. J Biol Chem. 2012;287(7):4503–17.
Li Z, Dong M, Fan D, Hou P, Li H, Liu L, et al. LncRNA ANCR down-regulation promotes TGF-β-induced EMT and metastasis in breast cancer. Oncotarget. 2017;8(40):67329–43.
Li Q, Mo W, Ding Y, Ding X. Study of lncRNA TPA in promoting invasion and metastasis of breast cancer mediated by TGF-β signaling pathway. Front Cell Dev Biol. 2021;9:688751.
Chimge NO, Baniwal SK, Little GH, Chen YB, Kahn M, Tripathy D, et al. Regulation of breast cancer metastasis by Runx2 and estrogen signaling: the role of SNAI2. Breast Cancer Res BCR. 2011;13(6):R127.
Zhou K, Ou Q, Wang G, Zhang W, Hao Y, Li W. High long non-coding RNA NORAD expression predicts poor prognosis and promotes breast cancer progression by regulating TGF-β pathway. Cancer Cell Int. 2019;19:63.
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84.
Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–72.
Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan ZK, et al. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Can Res. 2009;69(24):9228–35.
Aomatsu K, Arao T, Sugioka K, Matsumoto K, Tamura D, Kudo K, et al. TGF-β induces sustained upregulation of SNAI1 and SNAI2 through Smad and non-Smad pathways in a human corneal epithelial cell line. Invest Ophthalmol Vis Sci. 2011;52(5):2437–43.
Li GY, Wang W, Sun JY, Xin B, Zhang X, Wang T, et al. Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics. 2018;8(10):2846–61.
Wo L, Zhang B, You X, Hu Y, Gu Z, Zhang M, et al. Up-regulation of LncRNA UCA1 by TGF-β promotes doxorubicin resistance in breast cancer cells. Immunopharmacol Immunotoxicol. 2022;44(4):492–9.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17(2):135–47.
Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–72.
Servais C, Erez N. From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation. J Pathol. 2013;229(2):198–207.
Mueller MM, Fusenig NE. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4(11):839–49.
Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30(9):1002–19.
Liang Q, Li L, Zhang J, Lei Y, Wang L, Liu DX, et al. CDK5 is essential for TGF-β1-induced epithelial-mesenchymal transition and breast cancer progression. Sci Rep. 2013;3:2932.
Ren Y, Jia HH, Xu YQ, Zhou X, Zhao XH, Wang YF, et al. Paracrine and epigenetic control of CAF-induced metastasis: the role of HOTAIR stimulated by TGF-ß1 secretion. Mol Cancer. 2018;17(1):5.
Senchenkova EY, Ansari J, Becker F, Vital SA, Al-Yafeai Z, Sparkenbaugh EM, et al. Novel role for the AnxA1-Fpr2/ALX signaling axis as a key regulator of platelet function to promote resolution of inflammation. Circulation. 2019;140(4):319–35.
Sheikh MH, Solito E. Annexin A1: uncovering the many talents of an old protein. Int J Mol Sci. 2018;19(4):1045.
Tang L, Chen Y, Chen H, Jiang P, Yan L, Mo D, et al. DCST1-AS1 Promotes TGF-β-Induced epithelial-mesenchymal transition and enhances chemoresistance in triple-negative breast cancer cells via ANXA1. Front Oncol. 2020;10:280.
Lesina M, Wörmann SM, Morton J, Diakopoulos KN, Korneeva O, Wimmer M, et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J Clin Invest. 2016;126(8):2919–32.
Fang Y, Wang J, Wu F, Song Y, Zhao S, Zhang Q. Long non-coding RNA HOXA-AS2 promotes proliferation and invasion of breast cancer by acting as a miR-520c-3p sponge. Oncotarget. 2017;8(28):46090–103.
Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell. 2009;34(1):3–11.
Lehtopolku M, Kotilainen P, Haanperä-Heikkinen M, Nakari UM, Hänninen ML, Huovinen P, et al. Ribosomal mutations as the main cause of macrolide resistance in Campylobacter jejuni and Campylobacter coli. Antimicrob Agents Chemother. 2011;55(12):5939–41.
Tian X, Sun D, Zhao S, Xiong H, Fang J. Screening of potential diagnostic markers and therapeutic targets against colorectal cancer. Onco Targets Ther. 2015;8:1691–9.
Guo B, Wu S, Zhu X, Zhang L, Deng J, Li F, et al. Micropeptide CIP2A-BP encoded by LINC00665 inhibits triple-negative breast cancer progression. EMBO J. 2020;39(1):e102190.
Suriyamurthy S, Baker D, Ten Dijke P, Iyengar PV. Epigenetic reprogramming of TGF-β signaling in breast cancer. Cancers. 2019;11(5):726.
Ni K, Huang Z, Zhu Y, Xue D, Jin Q, Zhang C, et al. The lncRNA ADAMTS9-AS2 regulates RPL22 to modulate TNBC progression via controlling the TGF-β signaling pathway. Front Oncol. 2021;11:654472.
Li XL, Subramanian M, Jones MF, Chaudhary R, Singh DK, Zong X, et al. Long noncoding RNA PURPL suppresses basal p53 levels and promotes tumorigenicity in colorectal cancer. Cell Rep. 2017;20(10):2408–23.
Espinoza CA, Allen TA, Hieb AR, Kugel JF, Goodrich JA. B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat Struct Mol Biol. 2004;11(9):822–9.
Creamer KM, Lawrence JB. XIST RNA: a window into the broader role of RNA in nuclear chromosome architecture. Philoso Trans Royal Soc London Ser B, Biol Sci. 2017;372(1733):20160360.
Jégu T, Blum R, Cochrane JC, Yang L, Wang CY, Gilles ME, et al. Xist RNA antagonizes the SWI/SNF chromatin remodeler BRG1 on the inactive X chromosome. Nat Struct Mol Biol. 2019;26(2):96–109.
Savas S, Skardasi G. The SWI/SNF complex subunit genes: their functions, variations, and links to risk and survival outcomes in human cancers. Crit Rev Oncol Hematol. 2018;123:114–31.
Zhou C, Wang D, Li J, Wang Q, Wo L, Zhang X, et al. TGFB2-AS1 inhibits triple-negative breast cancer progression via interaction with SMARCA4 and regulating its targets TGFB2 and SOX2. Proc Natl Acad Sci USA. 2022;119(39):e2117988119.
Al-Khayal K, Vaali-Mohammed MA, Elwatidy M, Bin Traiki T, Al-Obeed O, Azam M, et al. A novel coordination complex of platinum (PT) induces cell death in colorectal cancer by altering redox balance and modulating MAPK pathway. BMC Cancer. 2020;20(1):685.
Alimirah F, Pulido T, Valdovinos A, Alptekin S, Chang E, Jones E, et al. Cellular senescence promotes skin carcinogenesis through p38MAPK and p44/42MAPK signaling. Can Res. 2020;80(17):3606–19.
Franklin DA, Sharick JT, Ericsson-Gonzalez PI, Sanchez V, Dean PT, Opalenik SR, et al. MEK activation modulates glycolysis and supports suppressive myeloid cells in TNBC. JCI Insight. 2020. https://doi.org/10.1172/jci.insight.134290.
Chakraborty R, Abdel-Wahab O, Durham BH. MAP-Kinase-driven hematopoietic neoplasms: a decade of progress in the molecular age. Cold Spring Harbor Perspect Med. 2021;11(5):a034892.
Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int J Biochem Cell Biol. 2001;33(11):1047–63.
Bogoyevitch MA, Boehm I, Oakley A, Ketterman AJ, Barr RK. Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential. Biochem Biophys Acta. 2004;1697(1–2):89–101.
Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng MH, et al. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002;80(2):239–56.
Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995;20(3):117–22.
Zhang M, Wang Y, Jiang L, Song X, Zheng A, Gao H, et al. LncRNA CBR3-AS1 regulates of breast cancer drug sensitivity as a competing endogenous RNA through the JNK1/MEK4-mediated MAPK signal pathway. J Exp Clin Cancer Res CR. 2021;40(1):41.
Chen S, Wang Y, Zhang JH, Xia QJ, Sun Q, Li ZK, et al. Long non-coding RNA PTENP1 inhibits proliferation and migration of breast cancer cells via AKT and MAPK signaling pathways. Oncol Lett. 2017;14(4):4659–62.
Lu P, Gu Y, Li L, Wang F, Yang X, Yang Y. Long noncoding RNA CAMTA1 promotes proliferation and mobility of the human breast cancer cell line MDA-MB-231 via targeting miR-20b. Oncol Res. 2018;26(4):625–35.
Ouyang J, Liu Z, Yuan X, Long C, Chen X, Wang Y, et al. LncRNA PRNCR1 promotes breast cancer proliferation and inhibits apoptosis by modulating microRNA-377/CCND2/MEK/MAPK Axis. Arch Med Res. 2021;52(5):471–82.
Kümler I, Knoop AS, Jessing CA, Ejlertsen B, Nielsen DL. Review of hormone-based treatments in postmenopausal patients with advanced breast cancer focusing on aromatase inhibitors and fulvestrant. ESMO open. 2016;1(4):e000062.
Gu G, Dustin D, Fuqua SA. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr Opin Pharmacol. 2016;31:97–103.
Peng WX, Huang JG, Yang L, Gong AH, Mo YY. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol Cancer. 2017;16(1):161.
Sun Y, Jia X, Wang M, Deng Y. Long noncoding RNA MIR31HG abrogates the availability of tumor suppressor microRNA-361 for the growth of osteosarcoma. Cancer Manag Res. 2019;11:8055–64.
Larsen KB, Lutterodt MC, Møllgård K, Møller M. Expression of the homeobox genes OTX2 and OTX1 in the early developing human brain. J Histochem Cytochem. 2010;58(7):669–78.
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007.
Sheng W, Shi X, Lin Y, Tang J, Jia C, Cao R, et al. Musashi2 promotes EGF-induced EMT in pancreatic cancer via ZEB1-ERK/MAPK signaling. J Exp Clin Cancer Res CR. 2020;39(1):16.
Chen FY, Zhou ZY, Zhang KJ, Pang J, Wang SM. Long non-coding RNA MIR100HG promotes the migration, invasion and proliferation of triple-negative breast cancer cells by targeting the miR-5590-3p/OTX1 axis. Cancer Cell Int. 2020;20:508.
Liu Y, Cao X. Characteristics and significance of the pre-metastatic Niche. Cancer Cell. 2016;30(5):668–81.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18(2):99–115.
Wen S, Hou Y, Fu L, Xi L, Yang D, Zhao M, et al. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3-p38 MAPK signalling. Cancer Lett. 2019;442:320–32.
Xi L, Peng M, Liu S, Liu Y, Wan X, Hou Y, et al. Hypoxia-stimulated ATM activation regulates autophagy-associated exosome release from cancer-associated fibroblasts to promote cancer cell invasion. J Extracell Vesicles. 2021;10(11):e12146.
Tang X, Hou Y, Yang G, Wang X, Tang S, Du YE, et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. 2016;23(1):132–45.
Sun K, Tang S, Hou Y, Xi L, Chen Y, Yin J, et al. Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine. 2019;41:370–83.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–86.
Zhou X, Yan T, Huang C, Xu Z, Wang L, Jiang E, et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J Exp Clin Cancer Res CR. 2018;37(1):242.
Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. 2009;15(1):21–34.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48.
Unterleuthner D, Neuhold P, Schwarz K, Janker L, Neuditschko B, Nivarthi H, et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis. 2020;23(2):159–77.
Zeng H, Hou Y, Zhou X, Lang L, Luo H, Sun Y, et al. Cancer-associated fibroblasts facilitate premetastatic niche formation through lncRNA SNHG5-mediated angiogenesis and vascular permeability in breast cancer. Theranostics. 2022;12(17):7351–70.
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
AM were involved in search strategy and drafting. MM designed, revised, structured, and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Maharati, A., Moghbeli, M. Long non-coding RNAs as the critical regulators of PI3K/AKT, TGF-β, and MAPK signaling pathways during breast tumor progression. J Transl Med 21, 556 (2023). https://doi.org/10.1186/s12967-023-04434-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04434-7