
Abstract
Multiple system atrophy (MSA) is a heterogenous, uniformly fatal neurodegenerative ɑ-synucleinopathy. Patients present with varying degrees of dysautonomia, parkinsonism, cerebellar dysfunction, and corticospinal degeneration. The underlying pathophysiology is postulated to arise from aberrant ɑ-synuclein deposition, mitochondrial dysfunction, oxidative stress and neuroinflammation. Although MSA is regarded as a primarily sporadic disease, there is a possible genetic component that is poorly understood. This review summarizes current literature on genetic risk factors and potential pathogenic genes and loci linked to both sporadic and familial MSA, and underlines the biological mechanisms that support the role of genetics in MSA. We discuss a broad range of genes that have been associated with MSA including genes related to Parkinson’s disease (PD), oxidative stress, inflammation, and tandem gene repeat expansions, among several others. Furthermore, we highlight various genetic polymorphisms that modulate MSA risk, including complex gene–gene and gene-environment interactions, which influence the disease phenotype and have clinical significance in both presentation and prognosis. Deciphering the exact mechanism of how MSA can result from genetic aberrations in both experimental and clinical models will facilitate the identification of novel pathophysiologic clues, and pave the way for translational research into the development of disease-modifying therapeutic targets.
Similar content being viewed by others

Introduction
Multiple system atrophy (MSA) comprises a group of clinically heterogenous, uniformly fatal, progressive neurodegenerative conditions associated with dysautonomia, parkinsonism, cerebellar dysfunction and corticospinal degeneration [1,2,3,4]. MSA is broadly categorized into the Cerebellar subtype (MSA-C) and Parkinsonism subtype (MSA-P), depending on the predominant neurological presentation [5]. Recently, the International Parkinson and Movement Disorder Society (MDS) revised the diagnostic criteria for MSA using an evidence-based and consensus-based approach [6]. These criteria classify MSA into four groups with varying diagnostic certainty: neuropathologically established MSA, clinically established MSA, clinically probable MSA, and possible prodromal MSA.
MSA is regarded as an ɑ-synucleinopathy, with its neuropathological hallmark being glial cytoplasmic inclusions (GCI) in oligodendrocytes [7,8,9]. The exact pathogenesis is poorly understood, but has been postulated to arise from ɑ-synuclein overexpression and accelerated uptake in neurons and oligodendrocytes, impaired ɑ-synuclein degradation from autophagic and proteasomal dysfunction, mitochondrial dysfunction, oxidative stress, and neuroinflammation [10].
MSA is widely regarded as a primarily sporadic disease, with a possible genetic component (Fig. 1). Familial forms are rare, with pooled estimates of heritability approximated to be 2.09–6.65% [11] alongside case reports of multiplex families with both autosomal dominant and autosomal recessive inheritance patterns [12,13,14,15,16].

Both genetic and environmental factors influence MSA risk. Genetic factors, whose expression is influenced by epigenetics and ethnicity, include genes associated with monogenic forms of PD, genes related to oxidative stress, genes with repeat expansions, genes related to inflammation, other genes related to PD and genes identified through MSA GWAS. The pathophysiology is further complicated by complex gene–gene and gene-environment interactions that have yet to be fully elucidated. GWAS genome-wide association study, MSA Multiple System Atrophy, PD Parkinson’s Disease
Current knowledge of the genetics of MSA is limited. To address this gap, we provide a concise review of published literature on genetic risk factors and potential pathogenic genes and loci linked to both sporadic and familial MSA, and outline the biological basis and evidence that support the genetic underpinnings in its pathophysiology. Furthermore, we highlight complex gene–gene and gene-environment interactions which influence the disease phenotype and have clinical significance in both presentation and prognosis.
SNCA
The SNCA gene (ɑ-synuclein, 4q22.1) encodes ɑ-synuclein, a protein that is found mainly in the presynaptic terminals of neurons and contributes to synaptic transmission [17, 18]. Similar to Parkinson’s disease (PD), SNCA has been of great interest since MSA is classified as an ɑ-synucleinopathy and GCI mainly contains filamentous, insoluble ɑ-synuclein. To date, no pathogenic SNCA mutation have been associated with monogenic forms of MSA. Although several case studies have reported rare mutations (including G51D, A53E), they have not been replicated in larger cohorts [19,20,21].
Studies looking at SNCA SNPs have been more promising. Scholz et al. [22] identified SNCA variants rs11931074 and rs3857059 to be significantly associated with MSA in a European population, with the former association also observed by Ross et al. [23]. A separate European study found another two SNCA variants that were linked to MSA, rs3822086 and rs3775444 [24]. However, these findings could not be replicated in Asian populations [25,26,27] and this could, in part, be related to the differences in the frequency of the risk alleles in different populations since the prevalence of the rs11931074 “T” allele is considerably higher in Asian populations (51–58%) than European populations (2–10%) [25]. All Asian studies also recruited clinically-diagnosed MSA patients only, compared to the European studies which included pathologically-diagnosed MSA patients. Other SNCA variants, including specific SNPs linked to PD (rs2736990 and rs356220) and a set of tagging SNPs estimated to represent 95% of haplotype diversity have not been shown to modify MSA risk [28,29,30].
SNCA copy number variations (CNV) have been associated with MSA, with copy number gains and resulting increase in SNCA expression leading to greater ɑ-synuclein inclusions in both the non-neuronal and neuronal cells of MSA subjects [31,32,33]. This was correlated with earlier onset of disease, reflecting the clinical implications of gene dosage on disease presentation. An earlier study of 58 MSA cases did not observe any SNCA gene multiplication, but there were limitations in the methodology as only whole gene multiplication was evaluated [34]. Larger cohorts are needed to draw relations between SNCA CNVs and MSA.
Of note, a genome-wide association study conducted in patients with MSA of European ancestry failed to detect any association between SNCA and MSA [35]. This may be attributed to interpopulation heterogeneity of SNCA, as observed by the authors.
LRRK2
LRRK2 (leucine-rich repeat kinase, 12q12), also referred to as dardarin or PARK8, is a large protein that has both kinase and GTPase activity [36]. Mutations are associated with autosomal dominant and sporadic late-onset PD, with incomplete and age-variable penetrance [37,38,39]. As pathological studies have revealed significant pleomorphism at the cellular level (including Lewy bodies and tau/ubiquitin inclusions) [40], various groups have investigated the association of LRRK2 mutations and other neurodegenerative conditions.
LRRK2 G2019S is the most commonly occurring pathogenic mutation, especially among the Ashkenazi Jewish, North African Arab and Spanish populations [41,42,43]. Studies thus far have failed to establish an association between LRRK2 G2019S and MSA [44,45,46], although interestingly a recent case report detected the mutation in a Caucasian subject who had pathologically-diagnosed MSA [47].
In a large combined US-UK series, LRRK2 M2397T polymorphism was protective for MSA, with a stronger association observed in the US cohort and for MSA-P/MSA-mixed patients [48]. A similar negative correlation was observed for G1624G, M1646T and N2081D within the US group, and N551K and R1398H within the UK group, but observed associations did not reach statistical significance.
Other LRRK2 variants (R1628P, G2385R) have also been investigated but no association has been found [49,50,51,52], with the exception of a case report of a rare variant Ile1371Val in an MSA patient [53].
GBA
GBA (glucocerebrosidase, 1q21) homozygous mutations are associated with Gaucher Disease, and more than two hundred pathogenic variants have been identified [54]. Pathogenic GBA variants have been demonstrated to increase the risk of developing PD [55,56,57,58,59] and dementia with Lewy bodies [60, 61]. A large-scale multicenter study identified twenty heterozygous GBA SNPs amongst MSA patients, of which nine are known to be pathogenic for Gaucher Disease (R120W, G202R, F213I, N370S, G377S, D409H, L444P, L444R, RecNciI) [62]. The pooled results across the North American, European, and Japanese series were statistically significant, but only the North American cohort reached significance when analyzed separately. One possible explanation could be the relatively large proportion of Ashkenazi Jews in North America compared to other parts of the world (with the exception of Israel), given that Gaucher Disease (especially type 1) has higher incidence in the Ashkenazi Jewish population compared to other ethnicities [63]. This was further supported by US studies which found significant associations between GBA SNPs and MSA, with one study noting that 3 out of the 6 Ashkenazi Jews in the study carried GBA mutations [64, 65]. Comparatively, there was no significant relationship between disease-causing GBA variants and MSA in European and Asian populations [66,67,68,69,70]. Functional studies suggest that lysosomal dysfunction as a result of GBA deficiency dysregulates ɑ-synuclein processing and induces its aggregation [71, 72]. This relationship is further complicated by other molecular regulatory mechanisms, such as the Thyroid Hormone Receptor Interacting Protein 12 (TRIP12), which ubiquinates glucocerebrosidase and influences GBA expression [73].
COQ2 and other oxidative stress-related genes
COQ2 (coenzyme Q2, polyprenyltransferase, 4q21.23) encodes an important enzyme in the Coenzyme Q10 (CoQ) biosynthetic pathway, with loss-of-function mutations resulting in CoQ deficiency and consequent increase in mitochondrial oxidative stress with reduction in ATP synthesis [74, 75]. Reduction in COQ2 expression with corresponding decrease in CoQ and ATP levels have been shown in both the brain tissue and plasma of MSA patients, implicating CoQ biosynthesis in the pathogenesis of MSA [76,77,78].
The Multiple-System Atrophy Research Collaboration (MSARC) first published findings of a possible association between COQ2 and MSA after identifying a homozygous mutation (M128V-V393A/M128V-V393A) and compound heterozygous mutations (R387X/V393A) in COQ2 in two multiplex Japanese families [79]. The allele frequency of the V393A variant was found to be higher in MSA patients than controls within the Japanese series (4.8% vs 1.6%), but the variant was not found in any of the MSA patients or healthy controls in the European or North American series. In addition, this observation was made mainly within the MSA-C subgroup. Although the results were not always reproducible [80,81,82,83,84], other East Asia population case-control studies and meta-analyses showed a significant association between the V393A variant and MSA-C patients [85,86,87], suggesting that this genetic susceptibility is, at least in part, specific to certain populations and ethnicities [88,89,90,91,92]. The rarity of V393A polymorphism in the Caucasian population may also explain the lack of replicability.
Several other genetic polymorphisms have been reported (S107T, M128R, M128V, R387X, R197H, S146N, L402F, R173H, A32A, L25V, N386I, L162F), however larger sample sizes are needed to confirm their association [80,81,82,83, 85, 90].
Other genes involved in oxidative stress have been evaluated. Soma et al. [93] examined eight genes (CHOP, ATF3, CEBPB, SQSTM1, CARS, SLC1A4, ATF4, EIF4EBP1) involved in oxidative stress pathways and found significant associations between SLC1A4 rs759458 and MSA. Secondary analysis further uncovered several haplotypes of SLC1A4, SQSTM1 and EIF4EBP1 that altered MSA risk. Oxidative stress involves multiple complex pathways with various gene–gene interactions, thus requires more studies to further elucidate these mechanisms.
MAPT
The MAPT gene (microtubule associated protein tau, 17q21.31) encodes tau, a protein which confers and maintains neuronal microtubule stability, and whose aberrant deposition in neuronal or glial cells results in neurodegenerative disorders known as tauopathies [94,95,96,97,98,99]. There are 2 extended MAPT haplotypes H1 and H2, with H1 further divided into subhaplotypes (e.g., H1c, H1b, etc.) [100]. MAPT has been shown to affect susceptibility to PD [101], Alzheimer’s Disease [102,103,104], frontotemporal dementia [105], progressive supranuclear palsy [106], corticobasal degeneration [107] and dementia with Lewy bodies [108].
An association between H1 haplotype (rs1052553) “A” allele and MSA has been reported by some investigators, but not for the H1c subhaplotype (rs242557) [109]. The same group conducted a follow-up study [110] using a larger sample size and six tagging SNPs to capture > 95% of the haplotype diversity and define over twenty H1 haplotypes [111, 112]. When analyzing individual SNPs, three variants (rs242557, rs3785883 and rs8070723) modulated MSA risk amongst pathologically-diagnosed patients. In the haplotype analysis, two risk haplotypes (H1x and H1J) were identified amongst pathologically-diagnosed patients, although this differed from the one (H1U) identified amongst clinically-diagnosed patients. Separately, two protective haplotypes (H2 and H1E) were identified, with the H2 haplotype showing a significant association for MSA-C and MSA-mixed subtypes only.
Chen et al. studied MAPT rs242557 in a Chinese population but did not find any relation with MSA, which could be attributed to ethnic differences or diagnostic inaccuracies since this study included clinically-diagnosed patients only.
SCA-related genes, C9orf72, and other repeat expansions
Spinocerebellar Ataxia (SCA) refers to a broad group of genetic disorders where cerebellar ataxia is a common feature, with CAG repeat expansions as the most common underlying genetic anomaly [113, 114]. Trinucleotide repeat expansions in SCA-implicated genes have been shown to increase the risk of amyotrophic lateral sclerosis and depression [115,116,117,118,119], and affect disease severity in Alzheimer’s disease [120].
Studies have identified intermediate and pathologic expansions in SCA-related genes in MSA patients. This seems to vary between ethnicities as ATXN1 (Ataxin 1) and ATXN2 (Ataxin 2) (corresponding to SCA-1 and SCA-2 respectively) were implicated in an Italian population [121], whereas the majority of patients in a Korean population had repeat expansions in TBP (TATA-box binding protein) (corresponding to SCA-17) [122]. It is unclear if the larger number of CAG repeats in normal alleles of ATXN1 and ATXN2 amongst Caucasians is a contributory factor [123]. These expansions seem to be more associated with MSA-C than MSA-P, but more data are still needed as cases have been reported in both groups [124]. Interestingly, studies have also shown a higher mean CAG repeat length in MSA patients compared to controls [121, 125, 126].
Caution is needed in drawing definitive conclusions as all the included studies relied primarily on a clinical diagnosis of MSA based on consensus criteria, which may lead to diagnostic inaccuracies since both conditions can present with cerebellar dysfunction and parkinsonian features [127, 128]. This raises the possibility of misdiagnosis [129, 130] or dual pathologies [131, 132] rather than an underlying genetic association.
The hexanucleotide GGGGCC repeat expansion in C9orf72 (chromosome 9 open reading frame 72, 9p21.2) is most commonly associated with amyotrophic lateral sclerosis and frontotemporal dementia [133,134,135], but has also been detected in rare cases of PD, Alzheimer’s Disease, psychosis and atypical parkinsonism [136,137,138,139,140,141,142,143]. Goldman et al. were one of the first to report a link between C9orf72 repeat expansion and MSA in a pair of siblings carrying the mutation, and who were each diagnosed with clinical MSA and ALS respectively [144]. Subsequent publications, which comprised Caucasian and Asian cohorts and included pathologically-diagnosed MSA patients, could not replicated the findings [145,146,147,148,149]. However, a recently published Italian study found heterozygous mutations in the pathological range in two patients and intermediate/premutation range in four patients [150]. Given the small sample size (n = 100) and lack of neuropathological diagnosis of this study, more work needs to be done to further elucidate the role of C9orf72 repeat expansions in MSA.
RFC1 (Replication Factor C Subunit 1) biallelic intronic repeat expansions is associated with cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS) [151]. Given the similar clinical presentation with MSA-C, pentanucleotide repeat polymorphisms of RFC1 were investigated in recent studies. Wan et al. found biallelic and heterozygous AAGGG repeat expansions in three and thirteen clinically-diagnosed MSA patients respectively, but the association did not reach statistical significance [152]. Other studies, including one consisting exclusively of pathologically-diagnosed MSA patients only, did not identify any RFC1 repeat expansions [153, 154].
Trinucleotide repeats in NOTCH2NLC is responsible for neuronal intranuclear inclusion disease (NIID) [155]. Pathogenic GGC repeat expansions (at least 100 repeats) were identified in 2.6% of clinically-diagnosed MSA patients in one study [156], but were not present in any of the MSA patients in another study [157]. In the former, the patients with GGC repeat expansions had longer disease duration, slower progression and ɑ-synuclein-negative skin biopsies, suggesting a non-MSA condition or an underlying dual pathology.
Inflammation-related genes
Neuroinflammation has been a purported mechanism in the pathogenesis of MSA [158,159,160]. The consequent microglial activation, cytokine and chemokine release, and pro-inflammatory conditions are thought to accelerate ɑ-synuclein aggregation and oligodendroglial apoptosis [161, 162].
Genes encoding various interleukins (IL), TNF-ɑ and other inflammatory mediators have thus been an area of interest. The high producer allele “C” of gene polymorphism TNF-ɑ-1031C/T (rs1799964) was found to increase risk of MSA in both genotype distribution and minor allele frequency [163, 164]. IL-1ɑ-889 (rs1800587) allele “T”-carrying genotypes, associated with higher transcriptional activity, were also overrepresented in MSA with a positive gene dose effect in Caucasians [165]. However, this was not observed in two separate Asian series [164, 166].
The IL-8-251 (rs4073) “T” allele was found to increase risk of MSA in a dose-dependent manner despite having lower transcriptional activity than the “A” allele, a relationship that strengthened in individuals who also carry the intercellular adhesion molecule-1 (ICAM-1: E469K) “KK” genotype [167]. IL-1β-511 (rs16944) low producer allele “A” was also noted in greater frequency amongst MSA patients compared to allele “G” [166], and even contributed to earlier onset of disease [164]. This underscores the complexity of neuroinflammatory responses, especially since some cytokines play critical roles in neuronal regeneration and can confer early protection against neurodegeneration [168, 169].
TREM2 (Triggering Receptor Expressed On Myeloid Cells 2) encodes a receptor that binds TYROBP (TYRO protein tyrosine kinase-binding protein) and forms a signaling complex that is involved in microglial activation, neuroinflammation and cytokine production [170]. TREM2 variants, specifically rs75932628 (p.R47H), have been implicated in neurodegenerative disorders such as PD [171], Alzheimer’s Disease [172, 173], amyotrophic lateral sclerosis [174] and frontotemporal lobe dementia [175]. This substitution was shown to be associated with increased risk of MSA in a Caucasian population, although this relationship weakened after adjusting for age and sex [176]. It is possible that this loss-of-function mutation affects myelin homeostasis and reduces clearance of myelin debris, causing microglial activation. Although a separate study in a Chinese population only found one patient carrying the T allele [177], this may be due to the relative rarity of this polymorphism amongst Asians [178].
Shadrin et al. recently employed a genome-wide genetic pleiotropy-informed approach to investigate the link between MSA and seven autoimmune diseases [179], and found substantial polygenic overlap between inflammatory bowel disease and MSA with three shared genetic loci (rs4957144 in the first intron of C7, rs12740041 and rs116843836 upstream of DENND1B and RSPO4 respectively). A transgenic mice model further showed that C7 expression in the midbrain was dysregulated. The effects for rs4957144 and rs12740041 on MSA and IBD were in opposite directionality, suggesting that these shared genes likely have complicated and differing pathogenic mechanisms on these diseases. It further lends credibility to the gut-brain axis theory and a connection between chronic bowel immune dysfunction and neuroinflammation [180,181,182].
The rs3135500 variant in the NOD2 gene, which activates nuclear factor κB (NK-κB) mediated inflammation, was shown to increase risk of MSA and correlate with increased peripheral mononuclear cell mRNA NO2 and plasma NOD2 protein levels [183, 184]. Acute phase reactant alpha 1-antichymotrypsin (ACT), encoded by the SERPINA3 gene, was also found in higher levels in the cerebrospinal fluid of MSA patients and correlated with a greater distribution of AA genotype compared to healthy controls [185]. This genotype also manifested phenotypically with earlier onset of symptoms and greater progression of disease compared to non-AA genotypes.
Genetic polymorphisms in IL-1R2, IL-1RA, IL-6, IL-10, TGF-β1, WNT3, HLA-DRB5 were not found to have significantly affected risk for developing MSA [163, 166, 186].
Other Parkinson’s disease (PD)-related genes
There are overlapping mechanisms and common pathways in the pathogenesis of PD and MSA, given the shared clinical and histopathological features, coexistence of both diseases within the same pedigree [15], and higher rates of parkinsonism among 1st-degree relatives of MSA [14, 187]. The susceptibility risk of genetic polymorphisms known to be related to PD have thus been studied in MSA cohorts.
One study investigated dopamine metabolism-related gene polymorphisms known to alter PD risk of phenotype (DDC rs921451, TH rs6356, COMT rs4680, MAOB rs1799836, DBH rs1611115) [188]. DDC rs921451 minor allele “C” was associated with an increased risk of MSA, especially in male subjects, while haplotype analysis showed the “T-T” haplotype in TH rs6356 and DDC rs921451 risk alleles reduced the risk for MSA. DDC rs921451 T > C was associated with reduced expression or activity of DDC, an enzyme involved in dopamine and norepinephrine synthesis [189, 190]. This may consequently result in features of parkinsonism and autonomic dysfunction, which are cardinal features in MSA [191, 192].
Another study found a possible increased risk of MSA amongst female patients carrying the NMD3 rs34016896 minor allele, which has been shown to correlate with nigral neuronal loss [193, 194], although it has not been shown to conclusively increase PD risk [195, 196].
Other genes and polymorphisms related to PD have been studied, but no associations have been found with MSA: PARK2 (parkin) [197], PINK1 [197], SREBF1 (rs11868035) [198], GPNMB (rs156429) [199], FBXO7 [200], SLC1A2 (rs3794087) [201], TMEM230 [202, 203], TMEM106B (rs1990622, rs3173615) [204], VMAT2 (rs363371, rs363324) [204], LINGO1 (rs11856808, rs9652490) [205], LINGO2 (rs10968280, rs13362909, rs7033345) [205], RAB7L1 (rs1572931) [206], CHCHD2 [207], DCTN1 [208] and ATP13A2 [209].
MSA genome-wide association studies
The only genome-wide association study conducted on MSA to date was published by Sailer et al. in 2016 [35]. It included subjects of European ancestry recruited from European and North American centers, with 291 out of 918 being pathologically-diagnosed cases. The study identified four loci of interest, FBXO47, ELOVL7, EDN1, and MAPT, but none surpassed the Bonferroni threshold for multiple testing. Notably, COQ2 and SNCA specifically were not found to significantly modify MSA risk in this cohort. Wenick et al. investigated ELOVL7 in a group of pathologically-diagnosed MSA patients, but could not identify any significant association [210].
Gene-environment interactions and epigenetics
Analyzing the genetic risk profile of the disease without observing its interplay with environmental factors would be overly simplistic. There have been various studies reporting the association between MSA and various environmental risk factors (including organic solvents, plastic monomers/additives, pesticides, agricultural activities, metals and smoking) [187, 211,212,213,214], but data on gene-environment interactions are limited. A recent case–control study found differential risks between individuals who shared the same SNPs and had varied exposures to different environmental factors including smoking, alcohol, drinking well water and pesticide exposure [215]. Such-gene-environment interactions will be clinically relevant since different genotypes may accentuate or attenuate the impact of certain environmental factors on MSA risk. Similarly, epigenetics is becoming increasingly implicated in the development of MSA through regulation of gene expression. Recent genome-wide studies have shown altered DNA methylation profiles between MSA patients and healthy controls (including hypomethylation of SNCA), some of which are modified by various environmental exposures [216, 217]. More work needs to be done to delineate such complex relationships.
Genetic model organisms
Animal models have been used to gain insight into the genetic basis of MSA [218]. For example, transgenic mouse models involving the overexpression of ɑ-synuclein in oligodendrocytes have been able to replicate MSA pathology and facilitate the understanding of GCI-linked neurodegeneration. However, this overexpression model may not fully recapitulate the processes seen in human model of MSA [219, 220]. Another transgenic mouse model showed that overexpression of ɑ1B-adrenergic receptors produced a MSA-like disorder with features of parkinsonism, autonomic dysfunction and ɑ-synuclein aggregation in oligodendrocytes [221]. However, the relevance and association of ɑ1B-adrenergic receptors to human MSA is unclear [222]. At present, there are no ideal MSA genetic models that have been developed and this should be a priority for investigators.
Discussion and limitations
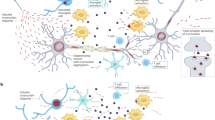
We present a comprehensive review of genes associated with MSA (Table 1). We highlight the possible biological mechanisms, outline complex gene–gene and gene-environment interactions, and show how genetic variations influence disease phenotype (Fig. 2).

Pathophysiological mechanisms and underlying genetic aberrations that modulate MSA risk. The pathological hallmark of MSA is the presence of glial cytoplasmic inclusions. Our understanding of disease biology is inadequate, but purported mechanisms include ɑ-synuclein overexpression and accelerated uptake, oxidative stress, and microglial activation. These mechanisms are modulated by a range of genes, which have been shown to influence disease phenotype. This is further complicated by gene–gene interactions (e.g. IL-8 and ICAM-1), gene-environmental interactions (e.g. between COQ2 and organic solvents/pesticides) and epigenetics (e.g. DNA hypomethylation of SNCA). MSA-C Multiple System Atrophy (Cerebellar subtype), MSA-P Multiple System Atrophy (Parkinsonian subtype), MSA-Mixed Multiple System Atrophy (Mixed subtype)
Despite the interesting observations from genetic studies in MSA, these must be interpreted with caution. To date, no large familial MSA pedigrees and monogenic forms have been identified. The genetic association studies reporting links with several genetic variants and loci do not determine an exact cause-effect relationship. Certain genes may contain innumerous disease-causing variants and haplotypes, thus preventing genome-wide association studies from detecting association signals from truly pathogenic genes. Most of these gene variants appear to confer a small or minimal effect size in the population, suggesting the possibility of other genetic determinants and contribution from environmental factors. Each gene may have an underlying set of gene regulators, or may in turn regulate other genes, hence adding further variables to an already convoluted genetic landscape. The sample sizes for most studies are small and do not have sufficient power to identify small differences. Furthermore, given that the phenotype of MSA is wide and varied, studies replying on solely clinical features assessed at a single time point may not be accurate. Most reported studies recruit clinically-diagnosed MSA patients based on the consensus statement proposed by Gilman et al. [5], as opposed to neuropathological criteria. One estimate places the clinical diagnostic accuracy of MSA at only 62% [223]. Thus, these studies may contain a sizeable minority of patients who do not actually have MSA, but rather a MSA-mimic such as other Parkinson-Plus syndromes with differing genetic susceptibility.
Conclusion and future directions
Although MSA is largely sporadic, genetic studies have allowed us to understand potential genetic factors that underpin the disease. Current studies have suggested possible associations between MSA risk and a wide range of gene mutations and polymorphisms. These genes include those linked to other common neurodegenerative conditions and those which are known to play a major functional role on oxidative stress, neuroinflammation, and protein degradation. However, thus far no monogenic forms of MSA have been identified. Multinational and multicenter studies with longitudinal follow up data will be helpful in identifying rare gene variants with small effect sizes and delineating heterogeneity between various age, sex and ethnic subgroups. In addition, large scale epidemiologic cohorts will also facilitate the identification of gene–gene and gene-environmental interactions. Functional studies in both animal and human models of the various identified genetic variants/mutations can identify novel pathophysiologic clues which may lead to development of disease-modifying therapeutic targets [224,225,226].
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- CBD:
-
Corticobasal degeneration
- CNV:
-
Copy number variation
- DLB:
-
Dementia with lewy bodies
- FTD:
-
Frontotemporal dementia
- GCI:
-
Glial cytoplasmic inclusions
- MSA:
-
Multiple system atrophy
- NIID:
-
Neuronal intranuclear inclusion disease
- PD:
-
Parkinson’s disease
- PSP:
-
Progressive supranuclear palsy
- SCA:
-
Spinocerebellar ataxia
- SNP:
-
Single nucleotide polymorphism
References
Poewe W, Stankovic I, Halliday G, Meissner WG, Wenning GK, Pellecchia MT, et al. Multiple system atrophy. Nat Rev Dis Prim. 2022;8:56.
O’Sullivan SS, Massey LA, Williams DR, Silveira-Moriyama L, Kempster PA, Holton JL, et al. Clinical outcomes of progressive supranuclear palsy and multiple system atrophy. Brain. 2008;131:1362–72.
Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015;372:249–63.
Fanciulli A, Stankovic I, Krismer F, Seppi K, Levin J, Wenning GK. Multiple system atrophy. Int Rev Neurobiol. 2019;149:137–92.
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6.
Wenning GK, Stankovic I, Vignatelli L, Fanciulli A, Calandra-Buonaura G, Seppi K, et al. The movement disorder society criteria for the diagnosis of multiple system atrophy. Mov Disord. 2022;37:1131–48.
Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and shy-drager syndrome). J Neurol Sci. 1989;94:79–100.
Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. 1998;44:415–22.
Armstrong RA, Cairns NJ, Lantos PL. Multiple system atrophy (MSA): topographic distribution of the alpha-synuclein-associated pathological changes. Parkinsonism Relat Disord. 2006;12:356–62.
Monzio Compagnoni G, Di Fonzo A. Understanding the pathogenesis of multiple system atrophy: state of the art and future perspectives. Acta Neuropathol Commun. 2019;7:113.
Federoff M, Price TR, Sailer A, Scholz S, Hernandez D, Nicolas A, et al. Genome-wide estimate of the heritability of multiple system atrophy. Parkinsonism Relat Disord. 2016;22:35–41.
Wüllner U, Abele M, Schmitz-Huebsch T, Wilhelm K, Benecke R, Deuschl G, et al. Probable multiple system atrophy in a German family. J Neurol Neurosurg Psychiatry. 2004;75:924–5.
Hara K, Momose Y, Tokiguchi S, Shimohata M, Terajima K, Onodera O, et al. Multiplex families with multiple system atrophy. Arch Neurol. 2007;64:545–51.
Vidal J-S, Vidailhet M, Derkinderen P, Tzourio C, Alpérovitch A. Familial aggregation in atypical Parkinson’s disease: a case control study in multiple system atrophy and progressive supranuclear palsy. J Neurol. 2010;257:1388–93.
Fujioka S, Ogaki K, Tacik PM, Uitti RJ, Ross OA, Wszolek ZK. Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord. 2014;20(Suppl 1):S29-34.
Itoh K, Kasai T, Tsuji Y, Saito K, Mizuta I, Harada Y, et al. Definite familial multiple system atrophy with unknown genetics. Neuropathology. 2014;34:309–13.
Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernández MA, Balbuena-Olvera AJ, Morales-Moreno ID, Argüero-Sánchez R, et al. Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front Neurosci. 2019;13:1399.
Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature. 2020;585:464–9.
Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, et al. α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013;125:753–69.
Kiely AP, Ling H, Asi YT, Kara E, Proukakis C, Schapira AH, et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener. 2015;10:41.
Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging. 2014;35:2180.
Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65:610–4.
Ross OA, Vilariño-Güell C, Wszolek ZK, Farrer MJ, Dickson DW. Reply to: SNCA variants are associated with increased risk of multiple system atrophy. Ann Neurol. 2010. https://doi.org/10.1002/ana.21786.
Al-Chalabi A, Dürr A, Wood NW, Parkinson MH, Camuzat A, Hulot J-S, et al. Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS ONE. 2009;4:e7114.
Yun JY, Lee W-W, Lee J-Y, Kim HJ, Park SS, Jeon BS. SNCA variants and multiple system atrophy. Ann Neurol. 2010. https://doi.org/10.1002/ana.21889.
Sun Z, Xiang X, Tang B, Chen Z, Peng H, Xia K, et al. SNP rs11931074 of the SNCA gene may not be associated with multiple system atrophy in Chinese population. Int J Neurosci. 2015;125:612–5.
Chen Y, Wei Q-Q, Ou R, Cao B, Chen X, Zhao B, et al. Genetic variants of snca are associated with susceptibility to Parkinson’s disease but not amyotrophic lateral sclerosis or multiple system atrophy in a Chinese population. PLoS ONE. 2015;10:e0133776.
Morris HR, Vaughan JR, Datta SR, Bandopadhyay R, Rohan De Silva HA, Schrag A, et al. Multiple system atrophy/progressive supranuclear palsy: alpha-synuclein, synphilin, tau, and APOE. Neurology. 2000;55:1918–20.
Ozawa T, Healy DG, Abou-Sleiman PM, Ahmadi KR, Quinn N, Lees AJ, et al. The alpha-synuclein gene in multiple system atrophy. J Neurol Neurosurg Psychiatry. 2006;77:464–7.
Guo XY, Chen YP, Song W, Zhao B, Cao B, Wei QQ, et al. SNCA variants rs2736990 and rs356220 as risk factors for Parkinson’s disease but not for amyotrophic lateral sclerosis and multiple system atrophy in a Chinese population. Neurobiol Aging. 2014;35:2882.e1-2882.e6.
Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson E-M, Schüle B, et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68:916–22.
Mokretar K, Pease D, Taanman J-W, Soenmez A, Ejaz A, Lashley T, et al. Somatic copy number gains of α-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain. 2018;141:2419–31.
Perez-Rodriguez D, Kalyva M, Leija-Salazar M, Lashley T, Tarabichi M, Chelban V, et al. Investigation of somatic CNVs in brains of synucleinopathy cases using targeted SNCA analysis and single cell sequencing. Acta Neuropathol Commun. 2019;7:219.
Lincoln SJ, Ross OA, Milkovic NM, Dickson DW, Rajput A, Robinson CA, et al. Quantitative PCR-based screening of alpha-synuclein multiplication in multiple system atrophy. Parkinsonism Relat Disord. 2007;13:340–2.
Sailer A, Scholz SW, Nalls MA, Schulte C, Federoff M, Price TR, et al. A genome-wide association study in multiple system atrophy. Neurology. 2016;87:1591–8.
Berwick DC, Heaton GR, Azeggagh S, Harvey K. LRRK2 biology from structure to dysfunction: research progresses, but the themes remain the same. Mol Neurodegener. 2019;14:49.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7.
Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76:672–80.
Correia Guedes L, Ferreira JJ, Rosa MM, Coelho M, Bonifati V, Sampaio C. Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: a systematic review. Parkinsonism Relat Disord. 2010;16:237–42.
Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004;62:1619–22.
Lesage S, Dürr A, Tazir M, Lohmann E, Leutenegger A-L, Janin S, et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006. https://doi.org/10.1056/NEJMc055540.
Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006. https://doi.org/10.1056/NEJMc055509.
Infante J, Rodríguez E, Combarros O, Mateo I, Fontalba A, Pascual J, et al. LRRK2 G2019S is a common mutation in Spanish patients with late-onset Parkinson’s disease. Neurosci Lett. 2006;395:224–6.
Hernandez D, Paisan Ruiz C, Crawley A, Malkani R, Werner J, Gwinn-Hardy K, et al. The dardarin G 2019 S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci Lett. 2005;389:137–9.
Ozelius LJ, Foroud T, May S, Senthil G, Sandroni P, Low PA, et al. G2019S mutation in the leucine-rich repeat kinase 2 gene is not associated with multiple system atrophy. Mov Disord. 2007;22:546–9.
Cho J-W, Kim S-Y, Park S-S, Jeon BS. The G2019S LRRK2 Mutation is rare in Korean patients with Parkinson’s disease and multiple system atrophy. J Clin Neurol. 2009;5:29–32.
Riboldi GM, Palma J-A, Cortes E, Iida MA, Sikder T, Henderson B, et al. Early-onset pathologically proven multiple system atrophy with LRRK2 G2019S mutation. Mov Disord. 2019. https://doi.org/10.1002/mds.27710.
Heckman MG, Schottlaender L, Soto-Ortolaza AI, Diehl NN, Rayaprolu S, Ogaki K, et al. LRRK2 exonic variants and risk of multiple system atrophy. Neurology. 2014;83:2256–61.
Tan EK, Skipper L, Chua E, Wong M-C, Pavanni R, Bonnard C, et al. Analysis of 14 LRRK2 mutations in Parkinson’s plus syndromes and late-onset Parkinson’s disease. Mov Disord. 2006;21:997–1001.
Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, et al. Lrrk2 and lewy body disease. Ann Neurol. 2006;59:388–93.
Lu C-S, Chang H-C, Weng Y-H, Chen R-S, Bonifati V, Wu-Chou Y-H. Analysis of the LRRK2 Gly2385Arg variant in primary dystonia and multiple system atrophy in Taiwan. Parkinsonism Relat Disord. 2008;14:393–6.
Yuan X, Chen Y, Cao B, Zhao B, Wei Q, Guo X, et al. An association analysis of the R1628P and G2385R polymorphisms of the LRRK2 gene in multiple system atrophy in a Chinese population. Parkinsonism Relat Disord. 2015;21:147–9.
Lee K, Nguyen K-D, Sun C, Liu M, Zafar F, Saetern J, et al. LRRK2 p.Ile1371Val mutation in a case with neuropathologically confirmed multi-system atrophy. J Parkinsons Dis. 2018;8:93–100.
Ginns EI, Choudary PV, Tsuji S, Martin B, Stubblefield B, Sawyer J, et al. Gene mapping and leader polypeptide sequence of human glucocerebrosidase: implications for gaucher disease. Proc Natl Acad Sci USA. 1985;82:7101–5.
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among gaucher disease carriers. J Med Genet. 2004;41:937–40.
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2004;351:1972–7.
Bras J, Paisan-Ruiz C, Guerreiro R, Ribeiro MH, Morgadinho A, Januario C, et al. Complete screening for glucocerebrosidase mutations in parkinson disease patients from portugal. Neurobiol Aging. 2009;30:1515–7.
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–61.
Zhao F, Bi L, Wang W, Wu X, Li Y, Gong F, et al. Mutations of glucocerebrosidase gene and susceptibility to Parkinson’s disease: an updated meta-analysis in a European population. Neuroscience. 2016;320:239–46.
Mata IF, Samii A, Schneer SH, Roberts JW, Griffith A, Leis BC, et al. Glucocerebrosidase gene mutations: a risk factor for lewy body disorders. Arch Neurol. 2008;65:379–82.
Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, et al. A multicenter study of glucocerebrosidase mutations in dementia with lewy bodies. JAMA Neurol. 2013;70:727–35.
Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Dürr A, et al. Variants associated with gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. 2015;2:417–26.
Grabowski GA. Gaucher disease: gene frequencies and genotype/phenotype correlations. Genet Test. 1997;1:5–12.
Sklerov M, Kang UJ, Liong C, Clark L, Marder K, Pauciulo M, et al. Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov Disord Clin Pract. 2017;4:574–81.
Wernick AI, Walton RL, Koga S, Soto-Beasley AI, Heckman MG, Gan-Or Z, et al. GBA variation and susceptibility to multiple system atrophy. Parkinsonism Relat Disord. 2020;77:64–9.
Segarane B, Li A, Paudel R, Scholz S, Neumann J, Lees A, et al. Glucocerebrosidase mutations in 108 neuropathologically confirmed cases of multiple system atrophy. Neurology. 2009;72:1185–6.
Jamrozik Z, Lugowska A, Slawek J, Kwiecinski H. Glucocerebrosidase mutations p.L444P and p.N370S are not associated with multisystem atrophy, progressive supranuclear palsy and corticobasal degeneration in polish patients. J Neurol. 2010. https://doi.org/10.1007/s00415-009-5363-4.
Sun Q, Guo J, Han W, Zuo X, Wang L, Yao L, et al. Genetic association study of glucocerebrosidase gene L444P mutation in essential tremor and multiple system atrophy in mainland China. J Clin Neurosci Off J Neurosurg Soc Australas. 2013;20:217–9.
Srulijes K, Hauser A-K, Guella I, Asselta R, Brockmann K, Schulte C, et al. No association of GBA mutations and multiple system atrophy. Eur J Neurol. 2013. https://doi.org/10.1111/ene.12086.
Pihlstrøm L, Schottlaender L, Chelban V, Meissner WG, Federoff M, Singleton A, et al. Lysosomal storage disorder gene variants in multiple system atrophy. Brain. 2018. https://doi.org/10.1093/brain/awy124.
Bras J, Singleton A, Cookson MR, Hardy J. Emerging pathways in genetic Parkinson’s disease: potential role of ceramide metabolism in Lewy body disease. FEBS J. 2008;275:5767–73.
Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010;120:641–9.
Seo BA, Kim D, Hwang H, Kim MS, Ma S-X, Kwon S-H, et al. TRIP12 ubiquitination of glucocerebrosidase contributes to neurodegeneration in Parkinson’s disease. Neuron. 2021;109:3758-3774.e11.
Duberley KEC, Abramov AY, Chalasani A, Heales SJ, Rahman S, Hargreaves IP. Human neuronal coenzyme Q10 deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: implications for pathogenesis and treatment. J Inherit Metab Dis. 2013;36:63–73.
Desbats MA, Morbidoni V, Silic-Benussi M, Doimo M, Ciminale V, Cassina M, et al. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum Mol Genet. 2016;25:4256–65.
Schottlaender LV, Bettencourt C, Kiely AP, Chalasani A, Neergheen V, Holton JL, et al. Coenzyme Q10 levels are decreased in the cerebellum of multiple-system atrophy patients. PLoS ONE. 2016;11:e0149557.
Mitsui J, Matsukawa T, Yasuda T, Ishiura H, Tsuji S. Plasma coenzyme Q10 levels in patients with multiple system atrophy. JAMA Neurol. 2016;73:977–80.
Hsiao J-HT, Purushothuman S, Jensen PH, Halliday GM, Kim WS. Reductions in COQ2 expression relate to reduced ATP levels in multiple system atrophy brain. Front Neurosci. 2019;13:1187.
MSARC M-SARC. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med. 2013;369:233–44.
Jeon BS, Farrer MJ, Bortnick SF. Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014. https://doi.org/10.1056/NEJMc1311763.
Wen X-D, Li H-F, Wang H-X, Ni W, Dong Y, Wu Z-Y. Mutation analysis of COQ2 in Chinese patients with cerebellar subtype of multiple system atrophy. CNS Neurosci Ther. 2015;21:626–30.
Chen YP, Zhao B, Cao B, Song W, Guo X, Wei Q-Q, et al. Mutation scanning of the COQ2 gene in ethnic Chinese patients with multiple-system atrophy. Neurobiol Aging. 2015;36:1222.
Sun Z, Ohta Y, Yamashita T, Sato K, Takemoto M, Hishikawa N, et al. New susceptible variant of COQ2 gene in Japanese patients with sporadic multiple system atrophy. Neurol Genet. 2016;2:e54.
Mikasa M, Kanai K, Li Y, Yoshino H, Mogushi K, Hayashida A, et al. COQ2 variants in Parkinson’s disease and multiple system atrophy. J Neural Transm. 2018;125:937–44.
Lin C-H, Tan E-K, Yang C-C, Yi Z, Wu R-M. COQ2 gene variants associate with cerebellar subtype of multiple system atrophy in Chinese. Mov Disord. 2015. https://doi.org/10.1002/mds.26138.
Zhao Q, Yang X, Tian S, An R, Zheng J, Xu Y. Association of the COQ2 V393A variant with risk of multiple system atrophy in East Asians: a case-control study and meta-analysis of the literature. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol. 2016;37:423–30.
Porto KJ, Hirano M, Mitsui J, Chikada A, Matsukawa T, Ishiura H, et al. COQ2 V393A confers high risk susceptibility for multiple system atrophy in East Asian population. J Neurol Sci. 2021;429:117623.
Sharma M, Wenning G, Krüger R. Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014. https://doi.org/10.1056/NEJMc1311763.
Schottlaender LV, Houlden H. Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014. https://doi.org/10.1056/NEJMc1311763.
Ogaki K, Fujioka S, Heckman MG, Rayaprolu S, Soto-Ortolaza AI, Labbé C, et al. Analysis of COQ2 gene in multiple system atrophy. Mol Neurodegener. 2014;9:44.
Ronchi D, Di Biase E, Franco G, Melzi V, Del Sorbo F, Elia A, et al. Mutational analysis of COQ2 in patients with MSA in Italy. Neurobiol Aging. 2016;45:213.e1-213.e2.
Procopio R, Gagliardi M, Brighina L, Nicoletti G, Morelli M, Ferrarese C, et al. Genetic mutation analysis of the COQ2 gene in Italian patients with multiple system atrophy. Gene. 2019. https://doi.org/10.1016/j.gene.2019.144037.
Soma H, Yabe I, Takei A, Fujiki N, Yanagihara T, Sasaki H. Associations between multiple system atrophy and polymorphisms of SLC1A4, SQSTM1, and EIF4EBP1 genes. Mov Disord. 2008;23:1161–7.
Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004;84:361–84.
Yoshida H, Goedert M. Phosphorylation of microtubule-associated protein tau by AMPK-related kinases. J Neurochem. 2012;120:165–76.
Kovacs GG. Tauopathies. Handb Clin Neurol. 2017;145:355–68.
Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59.
Zilka N, Korenova M, Novak M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol Germany. 2009;118:71–86.
Ferrer I, López-González I, Carmona M, Arregui L, Dalfó E, Torrejón-Escribano B, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol England. 2014;73:81–97.
Sánchez-Juan P, Moreno S, de Rojas I, Hernández I, Valero S, Alegret M, et al. The MAPT H1 haplotype is a risk factor for alzheimer’s disease in APOE ε4 non-carriers. Front Aging Neurosci. 2019;11:327.
Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12.
Chang C-W, Hsu W-C, Pittman A, Wu Y-R, Hardy J, Fung H-C. Structural study of the microtubule-associated protein tau locus of alzheimer’s disease in Taiwan. Biomed J. 2014;37:127–32.
Myers AJ, Kaleem M, Marlowe L, Pittman AM, Lees AJ, Fung HC, et al. The H1c haplotype at the MAPT locus is associated with alzheimer’s disease. Hum Mol Genet. 2005;14:2399–404.
Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. 2007;25:561–70.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5.
Höglinger GU, Melhem NM, Dickson DW, Sleiman PMA, Wang L-S, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705.
Yokoyama JS, Karch CM, Fan CC, Bonham LW, Kouri N, Ross OA, et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017;133:825–37.
Labbé C, Heckman MG, Lorenzo-Betancor O, Soto-Ortolaza AI, Walton RL, Murray ME, et al. MAPT haplotype H1G is associated with increased risk of dementia with Lewy bodies. Alzheimers Dement. 2016;12:1297–304.
Vilariño-Güell C, Soto-Ortolaza AI, Rajput A, Mash DC, Papapetropoulos S, Pahwa R, et al. MAPT H1 haplotype is a risk factor for essential tremor and multiple system atrophy. Neurology. 2011;76:670–2.
Labbé C, Heckman MG, Lorenzo-Betancor O, Murray ME, Ogaki K, Soto-Ortolaza AI, et al. MAPT haplotype diversity in multiple system atrophy. Parkinsonism Relat Disord. 2016;30:40–5.
Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. 2005;42:837–46.
Allen M, Kachadoorian M, Quicksall Z, Zou F, Chai HS, Younkin C, et al. Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimers Res Ther. 2014;6:39.
Manto M-U. The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum. 2005;4:2–6.
Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Prim. 2019;5:24.
Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–75.
Sproviero W, Shatunov A, Stahl D, Shoai M, van Rheenen W, Jones AR, et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging. 2017;51:178.e1-178.e9.
Ross OA, Rutherford NJ, Baker M, Soto-Ortolaza AI, Carrasquillo MM, DeJesus-Hernandez M, et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet. 2011;20:3207–12.
Conforti FL, Spataro R, Sproviero W, Mazzei R, Cavalcanti F, Condino F, et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology. 2012;79:2315–20.
Gardiner SL, van Belzen MJ, Boogaard MW, van Roon-Mom WMC, Rozing MP, van Hemert AM, et al. Large normal-range TBP and ATXN7 CAG repeat lengths are associated with increased lifetime risk of depression. Transl Psychiatry. 2017;7:e1143.
Gardiner SL, Harder AVE, Campman YJM, Trompet S, Gussekloo J, van Belzen MJ, et al. Repeat length variations in ATXN1 and AR modify disease expression in Alzheimer’s disease. Neurobiol Aging. 2019;73:230.e9-230.e17.
Mongelli A, Sarro L, Rizzo E, Nanetti L, Meucci N, Pezzoli G, et al. Multiple system atrophy and CAG repeat length: a genetic screening of polyglutamine disease genes in Italian patients. Neurosci Lett. 2018;678:37–42.
Kim H-J, Jeon BS, Shin J, Lee W-W, Park H, Jung YJ, et al. Should genetic testing for SCAs be included in the diagnostic workup for MSA? Neurology. 2014;83:1733–8.
Takano H, Cancel G, Ikeuchi T, Lorenzetti D, Mawad R, Stevanin G, et al. Close associations between prevalences of dominantly inherited spinocerebellar ataxias with CAG-repeat expansions and frequencies of large normal CAG alleles in Japanese and caucasian populations. Am J Hum Genet. 1998;63:1060–6.
Kim J-M, Hong S, Kim GP, Choi YJ, Kim YK, Park SS, et al. Importance of low-range CAG expansion and CAA interruption in SCA2 Parkinsonism. Arch Neurol. 2007;64:1510–8.
Zhou X, Wang C, Ding D, Chen Z, Peng Y, Peng H, et al. Analysis of (CAG)(n) expansion in ATXN1, ATXN2 and ATXN3 in Chinese patients with multiple system atrophy. Sci Rep. 2018;8:3889.
Wernick AI, Walton RL, Soto-Beasley AI, Koga S, Heckman MG, Valentino RR, et al. Frequency of spinocerebellar ataxia mutations in patients with multiple system atrophy. Clin Auton Res Off J Clin Auton Res Soc. 2021;31:117–25.
Lu C-S, Wu Chou Y-H, Kuo P-C, Chang H-C, Weng Y-H. The parkinsonian phenotype of spinocerebellar ataxia type 2. Arch Neurol. 2004;61:35–8.
Modoni A, Contarino MF, Bentivoglio AR, Tabolacci E, Santoro M, Calcagni ML, et al. Prevalence of spinocerebellar ataxia type 2 mutation among Italian Parkinsonian patients. Mov Disord. 2007;22:324–7.
Stamelou M, Quinn NP, Bhatia KP. “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy-a diagnostic guide. Mov Disord. 2013;28:1184–99.
Kim H-J, Stamelou M, Jeon B. Multiple system atrophy-mimicking conditions: diagnostic challenges. Parkinsonism Relat Disord. 2016;22(Suppl 1):S12-5.
Factor SA, Qian J, Lava NS, Hubbard JD, Payami H. False-positive SCA8 gene test in a patient with pathologically proven multiple system atrophy. Ann Neurol. 2005. https://doi.org/10.1002/ana.20389.
Nirenberg MJ, Libien J, Vonsattel J-P, Fahn S. Multiple system atrophy in a patient with the spinocerebellar ataxia 3 gene mutation. Mov Disord. 2007;22:251–4.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68.
Majounie E, Renton AE, Mok K, Dopper EGP, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–30.
Kertesz A, Ang LC, Jesso S, MacKinley J, Baker M, Brown P, et al. Psychosis and hallucinations in frontotemporal dementia with the C9ORF72 mutation: a detailed clinical cohort. Cogn Behav Neurol Off J Soc Behav Cogn Neurol. 2013;26:146–54.
Lindquist SG, Duno M, Batbayli M, Puschmann A, Braendgaard H, Mardosiene S, et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet. 2013;83:279–83.
Majounie E, Abramzon Y, Renton AE, Perry R, Bassett SS, Pletnikova O, et al. Repeat expansion in C9ORF72 in Alzheimer’s disease. N Engl J Med. 2012. https://doi.org/10.1056/NEJMc1113592.
Xi Z, Zinman L, Grinberg Y, Moreno D, Sato C, Bilbao JM, et al. Investigation of c9orf72 in 4 neurodegenerative disorders. Arch Neurol. 2012;69:1583–90.
Lesage S, Le Ber I, Condroyer C, Broussolle E, Gabelle A, Thobois S, et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain. 2013;136:385–91.
Cooper-Knock J, Frolov A, Highley JR, Charlesworth G, Kirby J, Milano A, et al. C9ORF72 expansions, parkinsonism, and Parkinson disease: a clinicopathologic study. Neurology. 2013;81:808–11.
Jiao B, Guo J-F, Wang Y-Q, Yan X-X, Zhou L, Liu X-Y, et al. C9orf72 mutation is rare in Alzheimer’s disease, Parkinson’s disease, and essential tremor in China. Front Cell Neurosci. 2013;7:164.
Nuytemans K, Bademci G, Kohli MM, Beecham GW, Wang L, Young JI, et al. C9ORF72 intermediate repeat copies are a significant risk factor for Parkinson disease. Ann Hum Genet. 2013;77:351–63.
Goldman JS, Quinzii C, Dunning-Broadbent J, Waters C, Mitsumoto H, Brannagan TH 3rd, et al. Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72. JAMA Neurol. 2014;71:771–4.
Schottlaender LV, Holton JL, Houlden H. Multiple system atrophy and repeat expansions in C9orf72. JAMA Neurol. 2014. https://doi.org/10.1001/jamaneurol.2014.1808.
Schottlaender LV, Polke JM, Ling H, MacDoanld ND, Tucci A, Nanji T, et al. Analysis of C9orf72 repeat expansions in a large series of clinically and pathologically diagnosed cases with atypical parkinsonism. Neurobiol Aging. 2015;36(1221):e1-6.
Scholz SW, Majounie E, Revesz T, Holton JL, Okun MS, Houlden H, et al. Multiple system atrophy is not caused by C9orf72 hexanucleotide repeat expansions. Neurobiol Aging. 2015;36(1223):e1-2.
Sun Z, Jiang H, Jiao B, Hou X, Shen L, Xia K, et al. C9orf72 hexanucleotide expansion analysis in Chinese patients with multiple system atrophy. Parkinsonism Relat Disord. 2015. https://doi.org/10.1016/j.parkreldis.2015.04.008.
Chen X, Chen Y, Wei Q, Ou R, Cao B, Zhao B, et al. C9ORF72 repeat expansions in Chinese patients with Parkinson’s disease and multiple system atrophy. J Neural Transm. 2016;123:1341–5.
Bonapace G, Gagliardi M, Procopio R, Morelli M, Quattrone A, Brighina L, et al. Multiple system atrophy and C9orf72 hexanucleotide repeat expansions in a cohort of Italian patients. Neurobiol Aging. 2022;112:12–5.
Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019;51:649–58.
Wan L, Chen Z, Wan N, Liu M, Xue J, Chen H, et al. Biallelic intronic AAGGG expansion of RFC1 is related to multiple system atrophy. Ann Neurol. 2020;88:1132–43.
Fan Y, Zhang S, Yang J, Mao C-Y, Yang Z-H, Hu Z-W, et al. No biallelic intronic AAGGG repeat expansion in RFC1 was found in patients with late-onset ataxia and MSA. Parkinsonism Relat Disord. 2020. https://doi.org/10.1016/j.parkreldis.2020.02.017.
Sullivan R, Yau WY, Chelban V, Rossi S, O’Connor E, Wood NW, et al. RFC1 intronic repeat expansions absent in pathologically confirmed multiple systems atrophy. Mov Disord. 2020. https://doi.org/10.1002/mds.28074.
Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet. 2019;51:1215–21.
Fang P, Yu Y, Yao S, Chen S, Zhu M, Chen Y, et al. Repeat expansion scanning of the NOTCH2NLC gene in patients with multiple system atrophy. Ann Clin Transl Neurol. 2020;7:517–26.
Xu K, Wan L, Chen Z, Wang C, Peng H, Hou X, et al. No genetic evidence for the involvement of GGC repeat expansions of the NOTCH2NLC gene in Chinese patients with multiple system atrophy. Neurobiol Aging. 2021;97:144.e5-144.e7.
Jellinger KA. Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov Disord. 2014;29:1720–41.
Vieira BDM, Radford RA, Chung RS, Guillemin GJ, Pountney DL. Neuroinflammation in multiple system atrophy: response to and cause of α-synuclein aggregation. Front Cell Neurosci. 2015;9:437.
Hoffmann A, Ettle B, Battis K, Reiprich S, Schlachetzki JCM, Masliah E, et al. Oligodendroglial α-synucleinopathy-driven neuroinflammation in multiple system atrophy. Brain Pathol. 2019;29:380–96.
Lim S, Chun Y, Lee JS, Lee S-J. Neuroinflammation in synucleinopathies. Brain Pathol. 2016;26:404–9.
Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1–13.
Nishimura M, Kuno S, Kaji R, Kawakami H. Influence of a tumor necrosis factor gene polymorphism in Japanese patients with multiple system atrophy. Neurosci Lett. 2005;374:218–21.
Zhou X, Wang C, Chen Z, Peng Y, Peng H, Hou X, et al. Association of TNF-α rs1799964 and IL-1β rs16944 polymorphisms with multiple system atrophy in Chinese Han population. Int J Neurosci. 2018;128:761–4.
Combarros O, Infante J, Llorca J, Berciano J. Interleukin-1A (-889) genetic polymorphism increases the risk of multiple system atrophy. Mov Disord. 2003;18:1385–6.
Nishimura M, Kawakami H, Komure O, Maruyama H, Morino H, Izumi Y, et al. Contribution of the interleukin-1beta gene polymorphism in multiple system atrophy. Mov Disord. 2002;17:808–11.
Infante J, Llorca J, Berciano J, Combarros O. Interleukin-8, intercellular adhesion molecule-1 and tumour necrosis factor-alpha gene polymorphisms and the risk for multiple system atrophy. J Neurol Sci. 2005;228:11–3.
Wang J, Bankiewicz KS, Plunkett RJ, Oldfield EH. Intrastriatal implantation of interleukin-1. Reduction of parkinsonism in rats by enhancing neuronal sprouting from residual dopaminergic neurons in the ventral tegmental area of the midbrain. J Neurosurg. 1994;80:484–90.
Hamelin L, Lagarde J, Dorothée G, Leroy C, Labit M, Comley RA, et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain England. 2016;139:1252–64.
Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener. 2017;12:56.
Benitez BA, Cruchaga C. TREM2 and neurodegenerative disease. N Engl J Med. 2013. https://doi.org/10.1056/NEJMc1306509.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–27.
Ulland TK, Colonna M. TREM2—a key player in microglial biology and Alzheimer disease. Nat Rev Neurol. 2018;14:667–75.
Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 variant p. R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014;71:449–53.
Guerreiro RJ, Lohmann E, Brás JM, Gibbs JR, Rohrer JD, Gurunlian N, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70:78–84.
Ogaki K, Heckman MG, Koga S, Martens YA, Labbé C, Lorenzo-Betancor O, et al. Association study between multiple system atrophy and TREM2 p.R47H. Neurol Genet. 2018;4:e257.
Chen Y, Chen X, Guo X, Song W, Cao B, Wei Q, et al. Assessment of TREM2 rs75932628 association with Parkinson’s disease and multiple system atrophy in a Chinese population. Neurol Sci Italy. 2015;36:1903–6.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Shadrin AA, Mucha S, Ellinghaus D, Makarious MB, Blauwendraat C, Sreelatha AAK, et al. Shared genetics of multiple system atrophy and inflammatory bowel disease. Mov Disord. 2021;36:449–59.
Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28:203–9.
Lema Tomé CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P. Inflammation and α-synuclein’s prion-like behavior in Parkinson’s disease–is there a link? Mol Neurobiol. 2013;47:561–74.
Pellegrini C, Antonioli L, Colucci R, Blandizzi C, Fornai M. Interplay among gut microbiota, intestinal mucosal barrier and enteric neuro-immune system: a common path to neurodegenerative diseases? Acta Neuropathol. 2018;136:345–61.
Cao B, Chen Y, Zhou Q, Zhang L, Ou R, Wei Q, et al. Functional variant rs3135500 in NOD2 increases the risk of multiple system atrophy in a Chinese population. Front Aging Neurosci. 2018;10:150.
Schwarz SC, Seufferlein T, Liptay S, Schmid RM, Kasischke K, Foster OJ, et al. Microglial activation in multiple system atrophy: a potential role for NF-kappaB/rel proteins. Neuroreport. 1998;9:3029–32.
Furiya Y, Hirano M, Kurumatani N, Nakamuro T, Matsumura R, Futamura N, et al. Alpha-1-antichymotrypsin gene polymorphism and susceptibility to multiple system atrophy (MSA). Brain Res Mol Brain Res. 2005;138:178–81.
Su W-M, Gu X-J, Hou Y-B, Zhang L-Y, Cao B, Ou R-W, et al. Association analysis of WNT3, HLA-DRB5 and IL1R2 polymorphisms in Chinese patients with Parkinson’s disease and multiple system atrophy. Front Genet. 2021;12:765833.
Nee LE, Gomez MR, Dambrosia J, Bale S, Eldridge R, Polinsky RJ. Environmental-occupational risk factors and familial associations in multiple system atrophy: a preliminary investigation. Clin Auton Res. 1991;1:9–13.
Chen Y, Ou R, Zhang L, Gu X, Yuan X, Wei Q-Q, et al. Contribution of five functional loci of dopamine metabolism-related genes to Parkinson’s disease and multiple system atrophy in a Chinese population. Front Neurosci. 2020;14:889.
Bell C, Mann R. Identification of dopaminergic nerves in humans. Am J Hypertens. 1990;3:4S-6S.
Devos D, Lejeune S, Cormier-Dequaire F, Tahiri K, Charbonnier-Beaupel F, Rouaix N, et al. Dopa-decarboxylase gene polymorphisms affect the motor response to L-dopa in Parkinson’s disease. Parkinsonism Relat Disord. 2014;20:170–5.
Lau ACW, Diggle JL, Bring PP. Improvement in severe orthostatic hypotension following carbidopa dose reduction. Can J Neurol Sci Le J Can Des Sci Neurol. 2018. https://doi.org/10.1017/cjn.2017.284.
Redenšek S, Flisar D, Kojović M, Gregorič Kramberger M, Georgiev D, Pirtošek Z, et al. Dopaminergic pathway genes influence adverse events related to dopaminergic treatment in Parkinson’s disease. Front Pharmacol. 2019;10:8.
IPDGC IPDGC, WTCCC2 WTCCC 2. A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 2011;7:e1002142.
Shulman JM, Yu L, Buchman AS, Evans DA, Schneider JA, Bennett DA, et al. Association of Parkinson disease risk loci with mild parkinsonian signs in older persons. JAMA Neurol. 2014;71:429–35.
Hernandez DG, Nalls MA, Ylikotila P, Keller M, Hardy JA, Majamaa K, et al. Genome wide assessment of young onset Parkinson’s disease from Finland. PLoS ONE. 2012;7:e41859.
Liu Z-H, Guo J-F, Li K, Wang Y-Q, Kang J-F, Wei Y, et al. Analysis of several loci from genome-wide association studies in Parkinson’s disease in mainland China. Neurosci Lett. 2015;587:68–71.
Brooks JA, Houlden H, Melchers A, Islam AJ, Ding J, Li A, et al. Mutational analysis of parkin and PINK1 in multiple system atrophy. Neurobiol Aging. 2011;32(548):e5-7.
Yuan X, Cao B, Wu Y, Chen Y, Wei Q, Ou R, et al. Association analysis of SNP rs11868035 in SREBF1 with sporadic Parkinson’s disease, sporadic amyotrophic lateral sclerosis and multiple system atrophy in a Chinese population. Neurosci Lett. 2018;664:128–32.
Xu Y, Chen Y, Ou R, Wei Q-Q, Cao B, Chen K, et al. No association of GPNMB rs156429 polymorphism with Parkinson’s disease, amyotrophic lateral sclerosis and multiple system atrophy in Chinese population. Neurosci Lett. 2016;622:113–7.
Conedera S, Apaydin H, Li Y, Yoshino H, Ikeda A, Matsushima T, et al. FBXO7 mutations in Parkinson’s disease and multiple system atrophy. Neurobiol Aging. 2016;40:192.e1-192.e5.
Xu Y, Cao B, Chen Y, Ou R, Wei Q, Yang J, et al. SLC1A2 rs3794087 are associated with susceptibility to Parkinson’s disease, but not essential tremor, amyotrophic lateral sclerosis or multiple system atrophy in a Chinese population. J Neurol Sci. 2016;365:96–100.
Yang X, An R, Xi J, Zhen J, Chen Y, Huang H, et al. Sequence TMEM230 gene in patients with multiple system atrophy in a southwest Chinese population: a pilot study. J Neurol Sci. 2017. https://doi.org/10.1016/j.jns.2017.02.006.
Procopio R, Gagliardi M, Brighina L, Nicoletti G, Morelli M, Piatti M, et al. Analysis of the TMEM230 gene in patients with multiple system atrophy. J Neurol Sci. 2018. https://doi.org/10.1016/j.jns.2018.07.019.
Hu T, Chen Y, Ou R, Wei Q, Cao B, Zhao B, et al. Association analysis of polymorphisms in VMAT2 and TMEM106B genes for Parkinson’s disease, amyotrophic lateral sclerosis and multiple system atrophy. J Neurol Sci. 2017;377:65–71.
Chen Y, Cao B, Yang J, Wei Q, Ou RW, Zhao B, et al. Analysis and meta-analysis of five polymorphisms of the LINGO1 and LINGO2 genes in Parkinson’s disease and multiple system atrophy in a Chinese population. J Neurol. 2015;262:2478–83.
Guo X-Y, Chen Y-P, Song W, Zhao B, Cao B, Wei Q-Q, et al. An association analysis of the rs1572931 polymorphism of the RAB7L1 gene in Parkinson’s disease, amyotrophic lateral sclerosis and multiple system atrophy in China. Eur J Neurol. 2014;21:1337–43.
Yang X, An R, Zhao Q, Zheng J, Tian S, Chen Y, et al. Mutational analysis of CHCHD2 in Chinese patients with multiple system atrophy and amyotrophic lateral sclerosis. J Neurol Sci. 2016;368:389–91.
Procopio R, Gagliardi M, D’Amelio M, Brighina L, Nicoletti G, Morelli M, et al. DCTN1 mutation analysis in Italian patients with PSP, MSA, and DLB. Neurobiol Aging. 2020;93:143.e5-143.e7.
Gagliardi M, Procopio R, Nicoletti G, Morelli M, Brighina L, Quattrone A, et al. Mutation analysis of the ATP13A2 gene in patients with PD and MSA from Italy. J Neurol Sci. 2021. https://doi.org/10.1016/j.jns.2021.120031.
Wernick AI, Walton RL, Soto-Beasley AI, Koga S, Ren Y, Heckman MG, et al. Investigating ELOVL7 coding variants in multiple system atrophy. Neurosci Lett. 2021;749: 135723.
Chrysostome V, Tison F, Yekhlef F, Sourgen C, Baldi I, Dartigues JF. Epidemiology of multiple system atrophy: a prevalence and pilot risk factor study in Aquitaine. France Neuroepidemiol. 2004;23:201–8.
Vidal J-S, Vidailhet M, Elbaz A, Derkinderen P, Tzourio C, Alperovitch A. Risk factors of multiple system atrophy: a case-control study in French patients. Mov Disord. 2008;23:797–803.
Vanacore N, Bonifati V, Fabbrini G, Colosimo C, De Michele G, Marconi R, et al. Case-control study of multiple system atrophy. Mov Disord. 2005;20:158–63.
Tseng F-S, Deng X, Ong Y-L, Li H-H, Tan E-K. Multiple System Atrophy (MSA) and smoking: a meta-analysis and mechanistic insights. Aging. 2020;12:21959–70.
Kuo M-C, Lu Y-C, Tai C-H, Soong B-W, Hu F-C, Chen M-L, et al. COQ2 and SNCA polymorphisms interact with environmental factors to modulate the risk of multiple system atrophy and subtype disposition. Eur J Neurol. 2022;29:2956–66.
Bettencourt C, Foti SC, Miki Y, Botia J, Chatterjee A, Warner TT, et al. White matter DNA methylation profiling reveals deregulation of HIP1, LMAN2, MOBP, and other loci in multiple system atrophy. Acta Neuropathol. 2020;139:135–56.
Rydbirk R, Folke J, Busato F, Roché E, Chauhan AS, Løkkegaard A, et al. Epigenetic modulation of AREL1 and increased HLA expression in brains of multiple system atrophy patients. Acta Neuropathol Commun. 2020;8:29.
Stefanova N, Wenning GK. Animal models of multiple system atrophy. Clin Auton Res Off J Clin Auton Res Soc. 2015;25:9–17.
Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, et al. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–59.
Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, et al. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci Off J Soc Neurosci. 2005;25:10689–99.
Zuscik MJ, Sands S, Ross SA, Waugh DJ, Gaivin RJ, Morilak D, et al. Overexpression of the alpha1B-adrenergic receptor causes apoptotic neurodegeneration: multiple system atrophy. Nat Med. 2000;6:1388–94.
Papay R, Zuscik MJ, Ross SA, Yun J, McCune DF, Gonzalez-Cabrera P, et al. Mice expressing the alpha(1B)-adrenergic receptor induces a synucleinopathy with excessive tyrosine nitration but decreased phosphorylation. J Neurochem. 2002;83:623–34.
Koga S, Aoki N, Uitti RJ, van Gerpen JA, Cheshire WP, Josephs KA, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology. 2015;85:404–12.
Lemos M, Wenning GK, Stefanova N. Current experimental disease-modifying therapeutics for multiple system atrophy. J Neural Transm. 2021;128:1529–43.
Ndayisaba A, Jellinger K, Berger T, Wenning GK. TNFα inhibitors as targets for protective therapies in MSA: a viewpoint. J Neuroinflamm. 2019;16:80.
Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. 2020;16:97–107.
Funding
EKT is supported by grants from the National Medical Research Council (STaR and PD‑LCG 000207).
Author information
Authors and Affiliations
Contributions
EKT conceptualized the study. FST, JQXF and ASM conducted the literature review and wrote the manuscript, and EKT did revisions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no conflict of interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tseng, F.S., Foo, J.Q.X., Mai, A.S. et al. The genetic basis of multiple system atrophy. J Transl Med 21, 104 (2023). https://doi.org/10.1186/s12967-023-03905-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-03905-1