
Abstract
Background
Acral melanoma (AM) is the most common subtype in Chinese melanoma patients with a very poor prognosis. However, our understanding of the disease pathogenesis and molecular landscape is limited by the few studies that have been conducted. Here, we profiled the clinical characteristics, mutational landscapes and tumor immune microenvironment of AM patients to gain insights into disease characteristics and potential treatment strategies.
Methods
A total of 90 AM patients were enrolled and their tissue samples were subjected to next-generation sequencing and multiplexed immunohistochemistry tests. Kaplan–Meier curves and log-rank tests were used to analyze the prognostic potential of various genetic aberrations and immune cell compositions in AM.
Results
The median disease-free survival was 21.3 months and estimated median overall survival (OS) was 60 months. More advanced stages, older ages and thickness of greater than 4 mm were associated with worse prognosis in AM patients (HR = 2.57, 95% CI 1.25–5.29, p = 0.01; HR = 2.77, 95% CI 1.22–6.28, p = 0.02; HR = 3.43, 95% CI 1.51–7.82, p < 0.01, respectively), while patients who received post-surgical treatments had better survival (HR = 0.36, 95% CI 0.17–0.76, p = 0.01). The most frequently altered genes included BRAF (14.5%), KIT (16.9%), NRAS (12%), NF1 (10.8%), APC (7.2%), and ARID2 (6%). Copy number variations (CNV) were commonly found in CCND1 (19.3%), CDK4 (19.3%), MDM2 (14.5%) and FGF19 (12%). CDK4 amplifications was independently associated with shorter OS in AM patients (HR = 3.61, 95% CI 1.38–9.46, p = 0.01). CD8 + T cells (p < 0.001) and M1 macrophages (p = 0.05) were more highly enriched in the invasive margin than in the tumor center. Patients with higher levels of M1 macrophage infiltration in the invasive margin derived markedly longer OS (HR = 0.43, 95% CI 0.20–0.95, p = 0.03). Interestingly, in CDK4-amplified patients, there tended to be a low level of M1 macrophage infiltration in the invasive margin (p = 0.06), which likely explains the poor prognosis in such patients.
Conclusions
Our study provided a comprehensive portrait of the clinicopathological features, genetic aberrations and tumor microenvironment profiles in AM patients and identified candidate prognostic factors, which may facilitate development of additional therapeutic options and better inform clinical management of AM patients. Based on these prognostic factors, further studies should focus on enhancing the infiltration of M1 macrophages, especially in CDK4-amplified AM patients.
Similar content being viewed by others

Introduction
The incidence of melanoma has been on the rise, with newly diagnosed cases increasing by 170% worldwide from 1990 to 2019 [1]. Acral melanoma (AM) is a rare and aggressive subtype that arises on the hands or feet. AM more commonly affects the Asian populations, with approximately 40% of Chinese melanoma patients having the acral subtype [2]. Complete surgical resection is considered the best option for cure in melanoma at early stages. For patients with advanced melanoma, the efficacy of surgical interventions or chemotherapy is very limited. The five-year survival rate of melanoma patients with stage I, II, III, and IV diseases were 94.1%, 44.0%, 38.4% and 4.6%, respectively [3]. Despite recent development of targeted therapy and immunotherapy, which have led to improved survival outcomes in melanoma patients [4], identification of prognostic factors might be helpful for gaining insight into the disease pathogenesis and further optimizing treatment strategies. A retrospective study of 522 Chinese melanoma patients showed that tumor stage and presence of ulceration were important prognostic factors [3]. Other clinical factors, including disease duration before diagnosis, Breslow thickness, mitotic rate, vascular invasion, regional lymph node metastasis and tumor stage, have been reported in association with AM patient survival [5]. On the other hand, limited studies have evaluated the prognostic values of molecular markers in melanoma. While some studies have indicated negative associations of prognosis with key driver alterations in NRAS, BRAF, and KIT in melanoma patients [2, 6,7,8], contradictory claim about the role of BRAF mutation in predicting outcomes in Korean patients with primary acral lentiginous melanoma (ALM) has also been made [9]. Even fewer studies of prognosis have been conducted in patients with AM. In such patients, the prognostic value of clinical features and genetic aberrations remains largely unknown.
In addition to clinical and genetic factors, the tumor immune microenvironment (TIME), which consists of a complex array of immune cells that carry out both anti-tumoral and immune-suppressive functions [10], has emerged as an area of intense research. The composition of immune cells in the TIME plays a critical role in tumor progression and can be exploited to forecast patient prognosis and improve treatment outcomes. It has been shown that CD8+ cell infiltration is associated with better survival in melanoma [11, 12]. Enrichment of CD4+ and CD8+ cells has been reported in responders to anti-PD-1 therapy in the metastatic setting [13]. Tumor-associated macrophage (TAM) is another important component of the TIME and can be mainly classified into proinflammatory M1 and anti-inflammatory M2 phenotypes. While several studies have attempted to analyze the modulating effect of TAM on melanoma development, its prognostic value has been inconclusive [14,15,16,17]. In addition, most of these studies have utilized CD68 as a marker for TAM, which is relatively nonspecific and generally used as a pan-macrophage marker. Thus, further characterization of the macrophage phenotype is also required.
Here, we performed next-generation sequencing (NGS) and multiplexed immunohistochemistry (mIHC) on baseline surgical samples from patients with primary AM to thoroughly characterize the mutational profile and immune landscape in association with patient prognosis. Through multi-dimensional analysis of clinicopathology, genetic aberrations and TIME, we investigated prognostic factors of AM, aiming to gain more insight into disease characteristics and potential treatment strategies.
Patients and methods
Patient population and samples collection
Ninety AM patients who admitted to Drum Tower Hospital from July 2010 and January 2021 were retrospectively included in this study. The clinicopathological information was collected, including age, sex, stage (TNM staging system, AJCC 8th Edition), Breslow thickness, local lymph node metastasis, ulceration, post-surgical treatment status and survival until the last follow-up or death. Archived formalin-fixed paraffin-embedded (FFPE) surgical-resected primary AM samples were available for all patients. And FFPE samples were confirmed by pathologists from the centralized clinical testing center before genetic testing.
Targeted NGS and genetic analysis
Qualified samples were subjected to targeted NGS by a Clinical Laboratory Improvement Amendments-certified and College of American Pathologists-accredited clinical testing laboratory (Nanjing Geneseeq Technology Inc., Nanjing, China) using a pan-cancer gene panel (GeneseeqPrime™, Geneseeq Technology Inc.). DNA extraction, library construction, and targeted capture enrichment were carried out following standard protocols as previously described with modifications [18, 19]. In brief, FFPE samples were de-paraffinized first with xylene before genomic DNA extraction using QIAamp DNA FFPE Tissue Kit (Qiagen Cat. No. 56404) according to the manufacturer’s instructions. Genomic DNA extracted from tumor samples was qualified using Nanodrop2000 (Thermo Fisher Scientific, Waltham, MA), then quantified using the dsDNA HS assay kit on a Qubit 3.0 fluorometer (Life Technology, US) according to the manufacturer’s recommendations. Targeted NGS libraries were prepared using the KAPA Hyper Prep kit (KAPA Biosystems) with an optimized manufacturer’s protocol for different sample types. Targeted hybridization enrichment was performed as previously described [20]. According to the manufacturer's instructions, the target-enriched libraries were then sequenced on a HiSeq4000 NGS platform (Illumina).
Sequencing data was first demultiplexed and subjected to FASTQ file quality control using Trimmomatic [21]. Only data without extra nucleotide bases and passed quality control (QC above 15) were subjected to the following analyses. Raw reads were mapped to the reference Human Genome (hg19) using Burrows-Wheeler Aligner (BWA-mem, v0.7.12; https://github.com/lh3/bwa/tree/master/bwakit) [22]. Genome Analysis Toolkit (GATK 3.4.0; https://software.broadinstitute.org/gatk/) was employed to perform local realignment around the insertions/deletions (INDELs) and base quality score recalibration. Picard was used to remove PCR duplicates. VarScan2 was applied to detect single-nucleotide variations (SNVs) and INDELs. SNVs were filtered out if the mutant allele frequency (MAF) was less than 1% for tumor tissue.
Tumor mutation burden calculation
Tumor mutation burden (TMB) was calculated as the number of non-synonymous somatic mutations, including missense, nonsense, splice-site, in-frame and frameshift mutations.
mIHC and multispectral imaging
Immune cell subsets in the TIME were identified by mIHC and multispectral imaging. Of the cohort of ninety cases, sixty-four patients had available results of both NGS and mIHC staining. Multiplexed immunofluorescence staining was performed using PANO 7-plex IHC kit (Panovue, Beijing, China), according to the manufacturer’s instructions. T cells were identified using the CD8 marker. NK cells were identified using the CD56 marker and were divided into two categories according to the intensity of membrane staining for the CD56 protein: CD56dim (weak staining) and CD56bright (strong staining). TAMs were identified by CD68 and HLA-DR and were divided into two categories: subtype M1 (CD68+ and HLA-DR+) and subtype M2 (CD68+ and HLA-DR−). Different primary antibodies were sequentially applied, including anti-CD8 (CST70306, Cell Signaling Technology, USA), anti-CD56 (CST3576), anti-panCK (CST4545), anti-CD68 (BX50031, Biolynx, China), anti-HLA-DR (ab92511), and anti-S100 (ab52642). S100 staining was used to define the invasive margin and tumor parenchyma [12, 23]. The Mantra System (PerkinElmer, Waltham, Massachusetts, US) was used to scan the stained slides and subsequently build a single stack image. The inForm image analysis software (PerkinElmer, Waltham, Massachusetts, US) was used for the reconstruction of images of sections with autofluorescence removal, based on a spectral library for multispectral unmixing. The positive rate was defined as the ratio of the number of positive cells in total number of cells in the TC or IM, excluding necrotic cells and tissues.
Statistical methods
Fisher’s exact tests were used to test the categorical variables between groups. Kaplan–Meier curves were used to analyze overall survival of various patient groups, and the statistical difference was analyzed using the log-rank test. A two-sided p value of no more than 0.05 was considered significant for all tests unless indicated otherwise (*p ≤ 0.05). All statistical analyses in this study were performed using the R Project for Statistical Computing (version 3.4.0).
Results
Patient overview and prognostic values of clinicopathological features
The study included 90 Chinese patients with primary AM who underwent surgery at our hospital. The clinicopathological features of the cohort were summarized in Table 1. The median age of diagnosis was 62 years (38–86 years), and 55 (61.1%) patients were females. The majority of patients presented with stage II (41/90, 45.6%) and stage III (35/90, 38.9%) disease. A high proportion of patients presented Breslow thickness > 4.0 mm (41.1%). Of the patients with known histology, ALM was the most common histological subtype, which accounted for 44.4% (40/90) of the entire cohort. Other subtypes included nodular melanoma (NM) (30, 33.3%), superficial spreading melanoma (10, 11.1%) and one case of lentigo maligna melanoma. Most lesions were located in the feet (79, 87.8%) and a few in the hands (11, 12.2%). The incidence of ulcers at the primary sites was 62.2%. None of the patients had received any anti-tumor treatment prior to surgery. Following surgical resection, sixty-six (73.3%) patients received treatments, including interferon, interferon combined with other drugs, chemotherapy, and anti-PD-1 therapy. The median follow-up time was 34 months (range: 5–114 months). The median disease-free survival (mDFS) was 21.3 months and estimated median overall survival (mOS) was 60 months. At the last follow-up, 36 patients had died.
Through multivariate analysis, we found several clinical features independently associated with clinical outcome (Table 2). As expected, patients with more advanced stage diseases (stages III and IV) had worse survival compared with those with earlier stages (stages I and II)(HR = 2.57, 95% CI 1.25–5.29, p = 0.01). Also, most patients had derived clinical benefit from post-surgical treatments compared with those who only underwent surgery (HR = 0.36, 95% CI 0.17–0.76, p = 0.01). In addition, we also found that patients with Breslow thickness > 4.0 mm had worse survival than those with Breslow thickness ≤ 4.0 mm (HR = 3.43, 95% CI 1.51–7.82, p < 0.01). Patients with older ages also had poorer survival compared with younger patients (HR = 2.77, 95% CI 1.22–6.28, p = 0.02).
Mutational landscape of AM patients
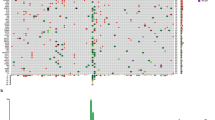
The top frequently altered genes in the 83 patients who had evaluable NGS results were illustrated in Fig. 1. Overall, a low TMB was observed, with a median TMB of 2.4 muts/Mb (range 0.0–15.90 muts/Mb). BRAF, NRAS, and KIT were most commonly altered, with respective alteration frequencies of 14.5%, 12.0%, and 16.9%. In particular, BRAF p.V600E and NRAS p.Q61K/R were the most common driver mutations, found in eight and seven patients, respectively. Missense mutations accounted for 77% of these driver mutations, and 9 (17%) copy number variations (CNV) were found in KIT (n = 7), BRAF (n = 1) and NRAS (n = 1). Other variant types found in these driver genes included three indel mutations (two KIT p.P573_D579dup, and one BRAF p.T599_V600delinsRE) and one fusion gene (TRB-BRAF). In addition, mutations were also commonly found in NF1 (10.8%), APC (7.2%), and ARID2 (6%). While TMB was low, the genomic landscape of AM was characterized by a high level of CNV, which was commonly found in CCND1 (19.3%), CDK4 (19.3%), MDM2 (14.5%) and FGF19 (12%). Comparing the different histological subtypes, we found that NRAS mutations were more commonly found in the ALM subgroup (p = 0.04) while CCND1 CNVs were more enriched in the NM subgroup (p = 0.04) (Additional file 4: Table S1). No significant difference in TMB was observed between the ALM and NM subgroups.

Genomic landscape of AM patients in the study (N = 83). Distributions of individual gene mutations and copy number variations in the study cohort as assessed by next-generation sequencing. Each column represents one patient. Genetic alterations were indicated in different colors according to type and clinical characteristics of each patient were shown at the top. ALM, acral lentiginous melanoma; AM, acral melanoma; NM, nodular melanoma
Associations of overall survival with genetic aberrations
To identify putative prognostic genetic biomarkers, we performed univariate analysis of OS on genes with alteration frequencies of ≥ 3% in the study cohort. Alterations in six genes (NSD1, KDM5A, MAP3K1, ERBB3, CDK4, TERT) were significantly associated with OS (p ≤ 0.05) (Additional file 5: Table S2). Among these, TERT CNV was not suitable for subsequent multivariate analysis as all patients harboring TERT CNV were still alive at last follow-up. Multivariate analysis adjusting for clinicopathological factors on the remaining five genes revealed strong negative associations of survival with CDK4 CNV (HR = 3.61, 95% CI 1.38–9.46, p = 0.01) (Additional file 6: Table S3). Patients with CDK4 CNV exhibited markedly shorted OS compared with those without CDK4 CNV (mOS = 28.6 m vs. not reached, p = 0.0044) (Fig. 2). By contrast, patients carrying mutations in the common AM driver genes, including BRAF, NRAS, and KIT, displayed comparable survival outcomes to the wild-type patients. In addition, there was no significant survival difference comparing TMB-high and TMB-low patients (Additional file 1: Fig. S1).

Kaplan Meier estimates of overall survival in AM patients. Survival plot comparing patients with and without CDK4 copy number variations (CDK4 CNV) (mOS: 28.6 m vs. not reached)
Quantitative analysis of tumor microenvironment
To further explore potential markers of prognosis, we analyzed 69 primary AMs for the compositions of various immune cells, including CD8+ T cells, M1 macrophages, M2 macrophages, CD56 bright NK cells, and CD56 dim NK cells both in the tumor center (TC) and the invasive margin (IM) by using mIHC. Overall, AM demonstrated low levels of immune infiltration (Fig. 3A). In particular, very low levels of CD56 bright cells were detected in both the TC and IM (Fig. 3B). Comparing the immune compositions in the TC and IM, we found that CD8+ T cells (p < 0.001) and M1 macrophages (p = 0.05) were more highly enriched in the IM. No significant difference between TC and IM was found in the spatial distributions of other immune cell types, including M2 macrophages, CD56 bright NK, and CD56 dim NK cells (Fig. 3B). Significant lower levels of CD56 dim NK cells in the IM were seen in patients with primary lesions located in the hands (p = 0.05) compared with those with primary lesions located in the feet. Patients with driver mutations displayed higher levels of M2 macrophages both in the TC (p < 0.01) and IM (p < 0.01). Compared with other pathological types, M2 macrophages (p < 0.01) and CD56 bright NK cells (p < 0.05) were highly enriched in the patients with NM and ALM pathological type (Additional file 2: Fig. S2).

Distribution and compositions of immune cells in melanoma environment (N = 69). A Heatmap_Distribution of immune cell compositions in the tumor center (TC) and invasive margin (IM) in AM patients. B Positive rate of immune cells in TC and IM of acral melanoma patients. Wilcoxon test was used to compare the immune compositions between TC and IM
Associations of overall survival with immune infiltration
Using median positive rate of each immune cell type as the cutoff, we performed univariate analysis examining their associations with patient survival. We found that consistent with their roles on the immune system, higher levels of M2 macrophages in the TC was associated with poor prognosis (HR = 2.19, 95% CI 0.95–5.05, p = 0.06) (Additional file 7: Table S4), while an enrichment of M1 macrophages in the IM was correlated with favorable outcome (mOS = not reached vs. 40.1 m, HR = 0.43, 95% CI 0.20–0.95, p = 0.03) (Fig. 4 and Additional file 7: Table S4). Following adjusting for clinicopathological factors in multivariate analysis, high M1 macrophage infiltration in the IM remained significantly correlated with prolonged survival (HR = 0.42, 95% CI 0.81–1.01, p = 0.05) (Additional file 8: Table S5).

Survival analysis of M1 macrophage infiltration in the IM. Kaplan Meier estimates of overall survival in acral melanoma patients with high and low positive rate of M1 macrophages in the invasive margin (IM). mOS: not reached vs. 40.1 m
Genetic aberrations and immune infiltration
Finally, we sought to determine whether there was a correlation between genetic aberrations and immune cell infiltrations in AM. Association analysis of immune cell compositions with genes (with ≥ 5% alteration frequencies), as well as TMB, revealed significant positive correlations between TMB and M1 macrophage infiltration in the TC, and CD56 dim NK cell infiltration in the IM (p < 0.05, p < 0.05) (Additional file 3: Fig. S3). We further examined the correlation of the prognosis-related genes (p < 0.1) with the infiltration of M1 macrophages in the IM. Interestingly, we found that AM patients with CDK4 amplification tended to be less infiltrated with M1 macrophages in the IM (p = 0.06) (Fig. 5).

Correlation of prognosis-related genes with infiltration of M1 macrophages in the IM. A Influence of nine putative prognostic genetic markers on the positive rate of M1 macrophages in the invasive margin (IM). The degree of co-occurrence (green) or mutual exclusivity (pink) among factors were indicated by the color gradient. p value was calculated by log-rank test. B The correlation of CDK4 copy number variations (CDK4 CNV) and the positive rate (PR) of M1 macrophages in the IM. The number and proportion of patients were indicated on the top of the bar chart. p value was calculated by Chi-square test
Discussion
To the best of our knowledge, this is the first study to systematically explore the genetic aberrations and TIME characteristics in Chinese AM patients. Not surprisingly, patient’s age, clinical stage, Breslow thickness and postoperative treatment status had significant impact on prognosis. Interestingly, our study suggests that CDK4 amplification may serve as a prognostic factor of AM. CDK4 encodes a cyclin-dependent protein kinase that mediates cell cycle progression and proliferation [24]. Although previous studies have indicated a potential prognostic value of CDK4 amplification or overexpression in several cancer types, including liposarcoma and osteosarcoma, as well as acral and mucosal melanoma [25,26,27,28], it has not been proven as an independent risk factor for melanoma prognosis [27, 28]. In our study, by adjusting for key clinicopathological features, we found that CDK4 amplification was independently associated with unfavorable AM prognosis. Notably, no difference in survival was observed between patients with driver gene mutations (BRAF, NRAS, KIT) and otherwise wild-type patients. However, given the limited sample size and small number of patients with CDK4 amplification, the prognostic role of CDK4 in AM needs to be further validated in large prospective cohorts.
In line with previous reports, TMB was low and copy number aberrations were frequent in our AM cohort [29, 30]. Consistent with a low TMB level, which can serve a surrogate for neoantigens, we found that the AM tumors were only moderately infiltrated with immune cells. Such a unique immune profile might account for the poor response to ICIs treatment in AM patients [31]. The infiltration of CD8+ T cells and M1 macrophages were markedly lower in the TC than in the IM, consistent with the result reported by Gartrell et al. [11]. Their study also found a positive correlation between CD8+ T cells and the disease specific survival (DSS), but a negative correlation between M2 macrophages and DSS in primary melanoma. While our study demonstrated positive prognostic value of M1 macrophages in the IM in AM patients, and CD8+ T cell infiltration had no association with outcome, which may be due to an overall low level of CD8+ T cell infiltration, especially in the TC.
The impact of the genomic landscape of the tumor on the TIME has been studied in other tumor types [32, 33]. In our study, CDK4 amplification was associated with a reduced level of M1 macrophage infiltration in the IM. Previous study has shown that selective inhibitors of CDK4/6 induce tumor cell cycle arrest and promote anti-tumor immunity [34]. Whether CDK4 inhibition could promote M1 macrophage infiltration in AM tumors remains unknown. In the future, more in vivo and clinical studies are needed to verify the linkage between CDK4 and M1 macrophage as well as their roles in AM.
However, our study has several limitations. First, this is a single-center study with a relatively small sample size. Second, our study did not account for the spatio-temporal heterogeneity of immune infiltration in the tumor tissues, as composition of the TIME may undergo drastic changes during tumor evolution, which was not captured by the mIHC analysis. Finally, our study examined a small number of immune cell types, which may not reflect the complex TIME in the tumor that is generally characterized by manifold markers. Future studies are necessary to fully depict the immune cell profiles and their dynamic changes for a comprehensive understanding of the TIME.
Despite these limitations, our study provides a comprehensive analysis of AM through systematic characterization of the clinicopathological features, genetic aberrations and TIME. This is also the first study to provide clinical evidence for the positive prognostic role of M1 macrophages in AM, and to provide possible mechanistic explanations for the poor prognosis in patients with CDK4-amplified AM. Future studies should focus on the infiltration of M1 macrophages and explore ways to improve AM patient prognosis, likely through enhancing M1 infiltration and preventing M2 transformation in the TIME.
Conclusions
This study systematically explores the genetic aberrations and TIME characteristics in Chinese AM patients. Our study demonstrates the negative prognostic value of CDK4 amplification. The infiltration of M1 macrophages in the IM was also associated with poor prognostic in AM. Especially, in CDK4-amplified patients, there tended to be low M1 macrophage infiltration in the IM. Based on these prognostic factors, further studies should focus on enhancing M1 macrophage polarization, especially in CDK4-amplified AM patients.
Data availability
The datasets generated and/or analysed during the current study are not publicly available due [REASON WHY DATA ARE NOT PUBLIC] but are available from the corresponding author on reasonable request.
Abbreviations
- ALM:
-
Acral lentiginous melanoma
- AM:
-
Acral melanoma
- CNV:
-
Copy number variations
- DFS:
-
Disease-free survival
- DSS:
-
Disease-specific survival
- FFPE:
-
Formalin-fixed paraffin-embedded
- ICIs:
-
Immune checkpoint inhibitors
- IM:
-
Invasive margin
- mIHC:
-
Multiplexed immunohistochemistry
- NGS:
-
Next-generation sequencing
- NM:
-
Nodular melanoma
- OS:
-
Overall survival
- TAM:
-
Tumor-associated macrophage
- TC:
-
Tumor center
- TIME:
-
Tumor immune microenvironment
- TMB:
-
Tumor mutation burden
References
Li Z, Fang Y, Chen H, et al. Spatiotemporal trends of the global burden of melanoma in 204 countries and territories from 1990 to 2019: results from the 2019 global burden of disease study. Neoplasia. 2022;24(1):12–21. https://doi.org/10.1016/j.neo.2021.11.013.
Bai X, Kong Y, Chi Z, et al. MAPK pathway and TERT promoter gene mutation pattern and its prognostic value in melanoma patients: a retrospective study of 2,793 cases. Clin Cancer Res. 2017;23(20):6120–7. https://doi.org/10.1158/1078-0432.CCR-17-0980.
Chi Z, Li S, Sheng X, et al. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: a study of 522 consecutive cases. BMC Cancer. 2011;11:85. https://doi.org/10.1186/1471-2407-11-85.
Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017;14(8):463–82. https://doi.org/10.1038/nrclinonc.2017.43.
Lv J, Dai B, Kong Y, Shen X, Kong J. Acral melanoma in Chinese: a clinicopathological and prognostic study of 142 cases. Sci Rep. 2016;6:31432. https://doi.org/10.1038/srep31432.
Si L, Kong Y, Xu X, et al. Prevalence of BRAF V600E mutation in Chinese melanoma patients: large scale analysis of BRAF and NRAS mutations in a 432-case cohort. Eur J Cancer. 2012;48(1):94–100. https://doi.org/10.1016/j.ejca.2011.06.056.
Kong Y, Si L, Zhu Y, et al. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin Cancer Res. 2011;17(7):1684–91. https://doi.org/10.1158/1078-0432.CCR-10-2346.
Jakob JA, Bassett RL, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118(16):4014–23. https://doi.org/10.1002/cncr.26724.
Hong JW, Lee S, Kim DC, Kim KH, Song KH. Prognostic and clinicopathologic associations of BRAF mutation in primary acral lentiginous melanoma in Korean patients: a preliminary study. Ann Dermatol. 2014;26(2):195–202. https://doi.org/10.5021/ad.2014.26.2.195.
Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. https://doi.org/10.1038/nrc3245.
Gartrell RD, Marks DK, Hart TD, et al. Quantitative analysis of immune infiltrates in primary melanoma. Cancer Immunol Res. 2018;6(4):481–93. https://doi.org/10.1158/2326-6066.CIR-17-0360.
Cabrita R, Lauss M, Sanna A, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–5. https://doi.org/10.1038/s41586-019-1914-8.
Wong PF, Wei W, Smithy JW, et al. Multiplex quantitative analysis of tumor-infiltrating lymphocytes and immunotherapy outcome in metastatic melanoma. Clin Cancer Res. 2019;25(8):2442–9. https://doi.org/10.1158/1078-0432.CCR-18-2652.
Tremble LF, McCabe M, Walker SP, et al. Differential association of CD68+ and CD163+ macrophages with macrophage enzymes, whole tumour gene expression and overall survival in advanced melanoma. Br J Cancer. 2020;123(10):1553–61. https://doi.org/10.1038/s41416-020-01037-7.
Piras F, Colombari R, Minerba L, et al. The predictive value of CD8, CD4, CD68, and human leukocyte antigen-D-related cells in the prognosis of cutaneous malignant melanoma with vertical growth phase. Cancer. 2005;104(6):1246–54. https://doi.org/10.1002/cncr.21283.
Jensen TO, Schmidt H, Møller HJ, et al. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J Clin Oncol. 2009;27(20):3330–7. https://doi.org/10.1200/JCO.2008.19.9919.
Salmi S, Siiskonen H, Sironen R, et al. The number and localization of CD68+ and CD163+ macrophages in different stages of cutaneous melanoma. Melanoma Res. 2019;29(3):237–47. https://doi.org/10.1097/CMR.0000000000000522.
Yang Z, Yang N, Ou Q, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin Cancer Res. 2018;24(13):3097–107. https://doi.org/10.1158/1078-0432.CCR-17-2310.
Shu Y, Wu X, Tong X, et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep. 2017;7(1):583. https://doi.org/10.1038/s41598-017-00520-1.
Fang W, Ma Y, Yin JC, et al. Comprehensive genomic profiling identifies novel genetic predictors of response to anti-PD-(L)1 therapies in non-small cell lung cancer. Clin Cancer Res. 2019;25(16):5015–26. https://doi.org/10.1158/1078-0432.CCR-19-0585.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–60. https://doi.org/10.1093/bioinformatics/btp324.
Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. https://doi.org/10.1038/nature13954.
Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122. https://doi.org/10.1186/gb4184.
Lee SE, Kim YJ, Kwon MJ, et al. High level of CDK4 amplification is a poor prognostic factor in well-differentiated and dedifferentiated liposarcoma. Histol Histopathol. 2014;29(1):127–38. https://doi.org/10.14670/HH-29.127.
Zhou Y, Shen JK, Yu Z, Hornicek FJ, Kan Q, Duan Z. Expression and therapeutic implications of cyclin-dependent kinase 4 (CDK4) in osteosarcoma. Biochim Biophys Acta Mol. 2018;1864(5 Pt A):1573–82. https://doi.org/10.1016/j.bbadis.2018.02.004.
Wang F, Chen G, Quinn MJ, et al. Increased CDK4 protein expression predicts a poor prognosis in mucosal melanoma associated with the p16INK4a-CDK4-pRb pathway. Int J Clin Exp Pathol. 2019;12(8):2819–25.
Kong Y, Sheng X, Wu X, et al. Frequent genetic aberrations in the CDK4 pathway in acral melanoma indicate the potential for CDK4/6 inhibitors in targeted therapy. Clin Cancer Res. 2017;23(22):6946–57. https://doi.org/10.1158/1078-0432.CCR-17-0070.
Hayward NK, Wilmott JS, Waddell N, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545(7653):175–80. https://doi.org/10.1038/nature22071.
Zou Z, Ou Q, Ren Y, et al. Distinct genomic traits of acral and mucosal melanomas revealed by targeted mutational profiling. Pigment Cell Melanoma Res. 2020;33(4):601–11. https://doi.org/10.1111/pcmr.12865.
van Not OJ, de Meza MM, van den Eertwegh AJM, et al. Response to immune checkpoint inhibitors in acral melanoma: a nationwide cohort study. Eur J Cancer. 1990;2022(167):70–80. https://doi.org/10.1016/j.ejca.2022.02.026.
Hiltbrunner S, Mannarino L, Kirschner MB, et al. Tumor immune microenvironment and genetic alterations in mesothelioma. Front Oncol. 2021;11:660039. https://doi.org/10.3389/fonc.2021.660039.
Feng B, Hess J. Immune-related mutational landscape and gene signatures: prognostic value and therapeutic impact for head and neck cancer. Cancers. 2021;13(5):1162. https://doi.org/10.3390/cancers13051162.
Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–5. https://doi.org/10.1038/nature23465.
Acknowledgements
None.
Funding
This work was supported by the Social Development Fund of Jiangsu Province (No.BE2019605) and the National Natural Science Foundation of China (No.81872484, No.82073365).
Author information
Authors and Affiliations
Contributions
ZYZ, RH and YR conceived the study concept and design. RH, KLZ, JYW, YR, LQQ, GYZ, MKZ, XYS and LQL contributed to clinical information collection and clinical follow-up. RH, GGS and FFW performed statistical analysis. RH drafted the article. ZYZ, YS and BRL contributed to the revision. YS and JCY polished the manuscript. All authors significantly contributed to the critical revision of the article, data interpretation and approved the submission. The work reported in this study was performed by the authors, unless clearly specified in the text. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Reviewer Board the Affiliated Drum Tower Hospital, Medical School of Nanjing University (Approval No. 2022-170-02) and performed according to the Declaration of Helsinki Principle. Informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
GGS, YS, JCY, FFW, and YS are employees or shareholders of Nanjing Geneseeq Technology Inc., China. The remaining authors have no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Additional file 1: Figure S1.
Survival analysis of TMB. Kaplan Meier overall survival curve of acral melanoma patients with high tumor mutation burden (TMB-High) (≥ 3.5 muts/Mb) and low tumor mutation burden (TMB-Low) (< 3.5 muts/Mb). mOS: 60.0 m vs. 58.1 m.

Additional file 2: Figure S2
. Correlation of clinicopathological features with the positive rate of immune cells. (A) Correlation of primary lesions with the positive rate of immune cells in the invasive margin (IM). (B) Correlation of driver mutations with the positive rate of immune cells in the tumor center (TC) and IM. (C) Correlation of pathological types with the positive rate of immune cells in the IM. P value was calculated by Wilcoxon test. ALM, acral lentiginous melanoma; NM, nodular melanoma.
Additional file 3: Figure S3.
Correlation of common genetic aberrations with the positive rate of immune cells. (A, B) Correlation of genetic features with the positive rate (PR) of immune cells in the tumor center (TC) (A) and invasive margin (IM) (B). Genes with ≥ 5% alteration frequencies are included. The degree of co-occurrence (green) or mutual exclusivity (pink) are indicated by the color gradient. *: P < 0.05. P value was calculated by log-rank test.
Additional file 4: Table S1.
Molecular differences between ALM and NM pathological subtypes.
Additional file 5: Table S2.
Univariate analysis of genetic aberrations associated with overall survival.
Additional file 6: Table S3.
Multivariate analysis of genetic aberrations associated with overall survival.
Additional file 7: Table S4.
Univariate analysis of the positive rate of immune cells associated with overall survival.
Additional file 8: Table S5.
Multivariate analysis of positive rate of immune cells associated with overall survival.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Huang, R., Shen, G., Ren, Y. et al. Prognostic value of genetic aberrations and tumor immune microenvironment in primary acral melanoma. J Transl Med 21, 78 (2023). https://doi.org/10.1186/s12967-022-03856-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03856-z