
Abstract
Background
Detecting trans-ethnic common associated genetic loci can offer important insights into shared genetic components underlying complex diseases/traits across diverse continental populations. However, effective statistical methods for such a goal are currently lacking.
Methods
By leveraging summary statistics available from global-scale genome-wide association studies, we herein proposed a novel genetic overlap detection method called CONTO (COmposite Null hypothesis test for Trans-ethnic genetic Overlap) from the perspective of high-dimensional composite null hypothesis testing. Unlike previous studies which generally analyzed individual genetic variants, CONTO is a gene-centric method which focuses on a set of genetic variants located within a gene simultaneously and assesses their joint significance with the trait of interest. By borrowing the similar principle of joint significance test (JST), CONTO takes the maximum P value of multiple associations as the significance measurement.
Results
Compared to JST which is often overly conservative, CONTO is improved in two aspects, including the construction of three-component mixture null distribution and the adjustment of trans-ethnic genetic correlation. Consequently, CONTO corrects the conservativeness of JST with well-calibrated P values and is much more powerful validated by extensive simulation studies. We applied CONTO to discover common associated genes for 31 complex diseases/traits between the East Asian and European populations, and identified many shared trait-associated genes that had otherwise been missed by JST. We further revealed that population-common genes were generally more evolutionarily conserved than population-specific or null ones.
Conclusion
Overall, CONTO represents a powerful method for detecting common associated genes across diverse ancestral groups; our results provide important implications on the transferability of GWAS discoveries in one population to others.
Similar content being viewed by others

Background
The past decade has witnessed great success of global-scale genome-wide association studies (GWASs) in discovering a large number of single nucleotide polymorphisms (SNPs) associated with many diseases and traits in diverse ancestries (e.g., blood lipids [1], prostate cancer [2], blood pressure [3, 4], type II diabetes [5, 6], glycemic traits [7], and schizophrenia [8]). These discoveries dramatically revolutionize our biological knowledge regarding genetic basis underlying complex phenotypes [9,10,11], and pave the way toward novel molecular therapeutic strategies for complex diseases and disorders [12,13,14,15,16]. One of the important findings of trans-ethnic GWASs is that a few of association signals identified in one population are found to be replicated in others in the sense that trait-associated genes exhibit consistently statistical association and effect direction in multiple populations [12, 14, 17,18,19,20,21], indicating that the same trait shares similar genetic components across diverse continental populations [22,23,24,25,26,27,28].
Indeed, current GWASs have sought to identify trait-associated genetic loci in the hope that discoveries in one population are likely transferred to others. However, given the population genetic differentiation among various ancestral groups worldwide [14, 29,30,31], the extent to which discovered associations can be generalized across populations is not completely known. We herein aim to formally investigate a central problem in population genetics using widespread summary statistics data publicly available from large-scale GWASs in different populations: are significant genes associated with a complex trait in one population also statistically related to the same trait in another population?
Understanding of shared genetic foundation for traits across diverse populations has profound implications from both statistical and practical perspectives. First, it helps improve power for trans-ethnic analysis by integrating multiple studies from various ethnicities [11, 32,33,34,35], increase accuracy of trans-ethnic genetic risk prediction in racial and ethnic minorities [28, 36], and enhance resolution in fine-mapping causal genes in various populations [37, 38]. Second, it offers additional insight into biological mechanism underlying complex diseases and helps measure the extent of interaction between genetic and environmental influences on disease risk [14]. Third, it holds the key to examine the transferability of associations discovered from current medical genomics researches which focus primarily on European (EUR) descent to other populations; that is, it is greatly of interest to examine whether the identified associations also hold in other populations and even discover more trait-related loci with higher power by leveraging genetic similarity across populations [39,40,41]. Fourth, biologically, replicating trait-associated genes across various ancestral groups is crucial for identifying truly causal genes as genetic loci that are simultaneously related to the trait in various populations much more likely contain important causal variants.
To assess the common genetic component underlying traits across distinct populations, novel trans-ethnic genetic correlation has been proposed using only summary statistics data [26, 28, 42]. Conceptually, trans-ethnic genetic correlation quantifies the extent to which a set of SNPs exert the same or similar effects on phenotypic variation in various ancestral groups. Although such correlation provides an overall insight into genetic foundation of the trait shared between populations, it cannot characterize detailed association pattern for individual genetic loci or genes. Moreover, a near-zero estimate of trans-ethnic genetic correlation does not necessarily indicate the absence of genetic overlap because mixed trans-ethnic genetic correlations in both positive and negative directions might dilute the overall estimate. The standard GWAS conventionally considers only significant genetic loci to examine whether they could be consistently discovered across ethnicities [19], which however ignores many significantly insignificant ones but with weak effects and thus might lead to biased conclusions. In addition, trans-ethnic meta-analysis is widely used [11, 32,33,34,35], whereas the determined associations might be present only in a single population. To our knowledge, only few statistical methods have been currently developed to identify trans-ethnic trait-associated genes shared across the entire genome.
To fill this knowledge gap, in the present study we propose a novel gene-centric genetic overlap detection method called CONTO. Unlike previous studies which analyzed individual SNPs [19, 28, 43], we instead focus on a set of SNPs located within a gene simultaneously and assess their joint significance with the trait of interest. From a statistical perspective, we observe that the identification of population-common genes across the whole genome can be effectively handled under the high-dimensional framework of composite null hypothesis testing by borrowing the idea of joint significance test (JST). Methodologically, JST employs the maximum P value of multiple associations as the significance measurement [44] and can be equivalently expressed as a combination of three disjoint component null hypotheses [45]. However, JST is often overly conservative because it depends on the 0–1 uniform distribution as its null distribution, which fails to consider the nature of composite null hypothesis test [46].
We make two key improvements of CONTO relative to JST. First, it constructs three-component mixture null distribution by taking the nature of composite null hypothesis test into account [47]. Second, it generates decorrelated test statistics to explain the trans-ethnic genetic correlation, which ultimately leads to well-calibrated P value for significance evaluation. Consequently, CONTO corrects the conservativeness of JST and is expected to more powerful than JST, which is validated through a wide range of simulation scenarios. We finally applied CONTO to detect population-common genes for 31 complex traits between the East Asian (EAS) and EUR populations. We identified many shared trait-associated genes that had otherwise been missed by JST. We also revealed that population-common genes were generally more evolutionarily conserved than population-specific ones.
Methods
Gene-set association method
Let the marginal Z score and P value of a gene for the analyzed trait in the EAS and EUR populations to be Z1 and Z2, and P1 and P2, respectively. These gene-level summary statistics can be easily to obtain with SNP-level association results publicly released by GWASs [48,49,50,51]. Therefore, we first aggregate multiple association signals at the SNP level into a single association signal at the gene level [52]. To this aim, we employ a powerful gene-set based association method called MAGMA [53], which is efficiently conducted via user-friendly software. Afterwards, the P value for each gene is obtained in both populations, which is immediately converted into Z score. The direction of Z score is determined by the sign of the summation of the product of effect sizes and MAFs across all SNPs of that gene [54, 55]. These gene-level summary statistics would be taken as inputs to measure the evidence of gene association with the trait in the two populations.
Trans-ethnic genetic overlap test under the composite null hypothesis framework
Our primary objective is to examine whether a particular trait-associated gene identified in one population is also significant in another population throughout the entire genome. The trans-ethnic genetic overlap can be defined in terms of distinct types of summary statistics. For example, the alternative hypothesis implies that both |Z1| and |Z2| are larger than a pre-assigned threshold value or that both P1 and P2 are less than a given significance level [say α; in our analysis we sought to control false discovery rate (FDR)]. This alternative hypothesis corresponds to three null hypotheses: (i) H00: the gene is not associated with the trait in either population; (ii) H10: the gene is associated with the trait in the first population but not the second; (iii) H01: the gene is associated with the trait in the second population but not the first. Formally, if defining the hypothesis test according to P values, we have
Under this framework we intend to identify shared trait-associated genes in both populations from the viewpoint of composite null hypothesis testing.
CONTO: composite null hypothesis test for trans-ethnic genetic overlap
Like JST, we take Pmax = max (P1, P2) as our test statistic for the detection of trans-ethnic genetic overlap. However, in contrast to JST which uses the 0–1 uniform distribution as its null distribution, we directly build the null distribution of Pmax to correct the conservativeness of JST by borrowing the idea given in [47], which was proposed under the context of high-dimensional epigenetic mediation analysis [45]. Specifically, we estimate the proportions of the three sub-null hypotheses and fit a mixture null distribution for Pmax
where p01 is the power of rejecting P2 ≤ α under H01 and p10 is the power of rejecting P1 ≤ α under H10, both of which are estimated via the Grenander method [56]; λ01, λ10, and λ00 are the proportions for the three sub-null null hypotheses, all of which are calculated with well-established methods for estimating FDR [57, 58]; see more details in Additional file 1.
It needs to highlight that the proposed method above implicitly assumes that the two P values are uncorrelated with each other. Although this condition is guaranteed by the sequential negligibility assumption in the mediation analysis [45, 59, 60], such independence does not necessarily hold in trans-ethnic genetic overlap test because of pervasive cross-population genetic correlation [26, 28, 42], which could cause inflated false discoveries than expected if not properly handled. Therefore, when implementing our method, we first decorrelate test statistics for each gene across populations by multiplying Z scores by the inverse of a correlation matrix. The cross-population correlation coefficient is calculated with Z scores of null genes (e.g., those with P1 > 0.05 and P2 > 0.05) [61, 62]. The uncorrelated Z scores can be in turn transformed into two-sided P values based on the normal approximation. Theoretically, this decorrelation strategy maximizes the transformed test statistics and the original ones [63]; therefore, it has the minimal influence on identifying shared associations. We refer to the above method as CONTO. The code for implementing CONTO is freely available at https://github.com/biostatpzeng/CONTO.
Simulation studies and real data applications
Simulation settings
We here implemented simulation studies to evaluate the performance of CONTO. Because it is conducted with only summary-level data, we thus directly sampled two sets of Z scores from a given multivariate normal (MVN) distribution under various scenarios. Specifically, for a gene in the first population, we generated its Z scores randomly from MVN((0, 0), Λ) under H00 with a probability λ00, or from MVN((τ10, 0), Λ) under H10 with a probability λ10, or from MVN((τ01, 0), Λ) under H01 with a probability λ01, or from MVN((τ11, 0), Λ) under H11 with a probability λ11. For the same gene in the second population, we drew its Z scores at random from MVN((0, 0), Λ) under H00 with a probability π00, or from MVN((0, 0), Λ) under H10 with a probability π10, or from MVN((0, τ01), Λ) under H01 with a probability λ01, or from MVN((0, τ11), Λ) under H11 with a probability λ11. The magnitude of τ10 (or τ01 and τ11) measures the strength of association, with larger value indicating stronger association signal. For simplicity, we set τ10 = τ01 = τ11 = 2, 3 or 4, and the total number of genes to 10000, 15000, or 20000.
After obtaining Z scores, we transformed them into P values based on the normal approximation. In our simulation, we set Λ to be a two-dimensional identify matrix. We considered three various probability settings with λ11 ≠ 0 to evaluate FDR control and power: (i) λ00 = 0.40, λ10 = 0.20, λ01 = 0.20, and λ11 = 0.20, constructing a highly polygenic but less overlapped genetic architecture, in which 40% genes were related to the trait in each population. and approximately 33.3% of associated genes were shared across populations; (ii) λ00 = 0.80, λ10 = 0.05, λ01 = 0.05, and λ11 = 0.10, building a less polygenic and moderately overlapped genetic architecture, in which 15% genes were related to the trait in each population and approximately 50% of associated genes were shared across populations; (iii) λ00 = 0.90, λ10 = 0.01, λ01 = 0.01, and λ11 = 0.08, generating a sparse but highly overlapped genetic architecture, in which 9% genes were related to the trait in each population, but approximately 80% of associated genes were shared by the trait across populations. Note that, to a great extent, these simulation parameters were selected based on our results of real data applications (see below).
Besides CONTO, for comparison we also carried out three other composite null methods (Additional file 1), including JST [44], joint significance composite-null test (JT-comp) [64], and divide-aggregate composite-null test (DACT) [65]. We repeated 103 times for each simulation setting and displayed the average across these replicates for these methods.
Summary statistics of 31 complex diseases from the EAS and EUR populations
We applied these methods to 31 complex traits of EAS-only or EUR-only individuals available from distinct GWAS consortia (Table 1 and Additional file 1: Table S1). These traits were analyzed in our previous work and more detailed descriptions regarding them can be found therein and in respective original paper [28, 62]. We downloaded summary statistics of these traits and performed stringent quality control in both populations for each trait: (i) removed SNPs without rs label; (ii) filtered out non-biallelic SNPs and those with strand-ambiguous alleles; (iii) deleted SNPs whose alleles did not match with those in the 1000 Genomes Project [66]; (iv) excluded duplicated SNPs and those with inconsistent alleles between EAS and EUR populations; (v) kept only common SNPs (MAF > 1%) which were shared in the two populations; (vi) removed SNPs located within the major histocompatibility complex region because of its complicated LD structure.
After quality control, we implemented MAGMA with genotypes of 504 EAS or 503 EUR individuals from the 1000 Genomes Project as the reference panel. We defined the set of cis-SNPs for a specific gene in terms of the annotation file provided by VIGAS [67]. The P value and Z score for each gene of traits were thus available. To handle possible residual influence of population stratification, family structures and cryptic relatedness [68,69,70,71], we further conducted genomic control for the gene-based association results of MAGMA if an inflation in these gene-level test statistics was observed (indicated by the inflation factor > 1.05). We took the resulting P values or Z scores as input to implement JST, JT-comp, DACT and CONTO for detecting trait-associated genes shared across the EAS and EUR populations.
Afterwards, for each trait we could classify these genes into three groups: (i) null genes which were not associated with the trait in either population; (ii) population-specific genes that were related to the trait in the EAS or EUR population; (iii) population-common genes that were shared across the two populations. To understand the characteristics of these genes in distinct groups, we used several conservation scores to examine the extent to which a particular gene varied across populations, which included phyloP score [72], phastCons score [73], and dN/dS ratio [74]. Specifically, higher phyloP or phastCons score indicates more conservativeness, while smaller dN/dS ratio represents higher conservativeness. We obtained these scores from [75], and compared the average scores across all genes of these traits in the three groups described above using the Friedman F test method.
Results
Estimated false discovery rate and statistical power
We first assessed whether these methods could correctly control FDR at a given level. Here, we primarily focused on the results obtained under the setting that the number of genes was set to 15000 (Fig. 1). First, it is shown that JS-com could lead to inflated control of FDR under the polygenic and less overlapped case regardless of the magnitude of association signals (Fig. 1A). JS-com could maintain an efficient FDR control if the genetic architecture was less polygenic or sparse, but moderately or highly overlapped, especially when the association evidence was weak (e.g., τ10 = τ01 = τ11 = 2) (Fig. 1B–C). However, as the increase of association evidence the estimated FDR of JS-com became inflated, which was particularly evident when the trait had a less polygenic but highly overlapped genetic architecture (Fig. 1B–C). This finding was consistent with that observed in previous mediation analysis literature because the assumption of weak association signal was violated in JS-com as the increase in τ10, τ01 and τ11 [64, 65].

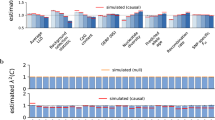
Estimated false discovery rate under the simulation settings: A λ00 = 0.40, λ10 = 0.20, λ01 = 0.20, and λ11 = 0.2; B λ00 = 0.80, λ10 = 0.05, λ01 = 0.05, and λ11 = 0.10, and C λ00 = 0.90, λ10 = 0.01, λ01 = 0.01, and λ11 = 0.08. Here, the number of genes was set to 15000, and the false discovery rate was calculated as the proportion of non-overlapped associated genes among all identified ones
Second, DACT always resulted in overestimated FDR in our simulation scenarios; however, the inflation seemed to be less obvious as the genetic impact became strong if the genetic architecture of the trait was less polygenic or sparse but highly overlapped (Fig. 1B–C). Third, in contrast to DACT and JS-com, JST was overly conservative under all our simulation settings, which was in line with prior observations [44]. Fourth, CONTO effectively controlled FDR at the nominal level across our simulation cases; however, it was slightly conservative when the association evidence was relatively weak (e.g., τ10 = τ01 = τ11 = 2) under the genetic architecture which was highly polygenic but less overlapped (Fig. 1A), or less polygenic but moderately overlapped (Fig. 1B).
As shown above, because only JST and CONTO could maintain FDR at or below the given nominal level, we thus mainly considered these two methods in our following analyses. When assessing power, it was obviously observed that CONTO was consistently more powerful compared to JST across our simulation scenarios (Fig. 2). For instance, under the polygenic but less overlapped case, CONTO had a 26.6% higher power compared to JST when the association evidence was strong (τ10 = τ01 = τ11 = 4) and the number of tested genes was 15000 (Fig. 2A). The advantage of CONTO over JST became more remarkable under other two simulation scenarios (Fig. 2B–C). Finally, it was found that the similar patterns of FDR control and power behaviors were consistently observed when the number of tested genes was 10000 or 20000 (Additional file 1: Figure S1–S4).

Estimated statistical power under the simulation settings: A λ00 = 0.40, λ10 = 0.20, λ01 = 0.20, and λ11 = 0.2; B λ00 = 0.80, λ10 = 0.05, λ01 = 0.05, and λ11 = 0.10, and C λ00 = 0.90, λ10 = 0.01, λ01 = 0.01, and λ11 = 0.08. Here, the number of genes was set to 15000, and the power was calculated as the proportion of truly overlapped associated genes among all identified ones
Associated genes discovered by MAGMA and CONTO
Based on the association results of MAGMA, we identified a set of trait-associated genes (FDR < 0.01) shared across the EAS and EUR populations using JST and CONTO (Table 1). Consistent with the results shown in the simulation studies above, we found that CONTO had higher power and thus discovered approximately two-fold more associated genes compared to JST across all the traits, with the average number of shared associated genes increased from 109 detected by JST to 291 discovered by CONTO. Moreover, for every trait all genes identified by JST were also simultaneously detected by CONTO. With regards to CONTO, the number of detected genes related to the trait in both populations ranged from 53 for AOA to 1,544 for Height.
As expected, the number of common traits-associated genes was highly correlated with the trans-ethnic genetic overlap (Spearman correlation = 0.593, P = 8.75 × 10−4) (Fig. 3A). Conceptually, greater trans-ethnic genetic correlation implies higher degree of common genetic foundation underlying the trait between two diverse populations [26, 28, 42]. For example, the trans-ethnic genetic correlation was 0.93 (se = 0.04) (calculated with the popcorn method [26]) and the number of population-common associated genes was 824 for T2D; while the trans-ethnic genetic correlation was 0.53 (se = 0.11) and the number of population-common associated genes was 53 for adult-onset asthma.

A Relationship between the number of population-common genes identified by CONTO and the trans-ethnic genetic correlation calculated with the popcorn method; B Relationship between the number of associated genes identified by MAGMA and the sample size of each trait in the EAS population; and C Relationship between the number of associated genes identified by MAGMA and the sample size of each trait in the EUR population
Besides population-common genes, we could also identify many population-specific genes (FDR < 0.01) (Table 1). It is evident that more population-specific genes were detected for traits in the EUR population because of higher power resulting from larger sample size (Fig. 3B–C). Consequently, the majority (an average of 92.4% across all traits) of trait-associated genes identified in the EAS population were also discovered in the EUR population, while only 56.1% of trait-associated genes identified in the EUR population were detected in the EAS population.
Conservation of associated genes
In terms of conservation score analysis, we observed that these population-common genes were often more evolutionarily conserved compared to unique associated genes identified in a single population, which were also more evolutionarily conserved than null genes. The increased conservation pattern in population-common genes was reflected by each of the three conservation scores including dN/dS ratio (Fig. 4A), phyloP score (Fig. 4B), and phastCons score (Fig. 4C). More specifically, the average phyloP scores were 0.209, 0.188, and 0.153 for common trait-associated genes, population-specific trait-associated genes, and null genes (P = 1.14 × 10−11), respectively; the corresponding average phastCons scores were 0.132, 0.127, and 0.116 (P = 9.36 × 10−14), and the corresponding average dN/dS ratio were 0.091, 0.096, and 0.097 (P = 6.86 × 10−10).

Average dN/dS ratio score A, phyloP score B, and phastCons score C across genes for all traits in distinct gene groups
Discussion
As large-scale medical genomics studies have become increasingly diverse, understanding similarity and diversity of associated genes for complex diseases/traits and consequently the transferability of genetic discoveries across various populations is essential [76]. In the present study, we proposed a novel gene-centric method called CONTO to identify common associated genes shared across distinct populations by leveraging GWAS summary statistics. As a result, we detected many population-common association signals for complex diseases and traits. From a biological perspective, the existence of population-common genes is to some extent widespread because shared genetic loci are often believed to be of ancient origin and thus shared among diverse ancestral groups [77].
We also discovered a great deal of population-specific associated genes, indicating that genetic association discoveries identified in one population were not always generalized to other ancestral groups although there indeed shows substantial trans-ethnic genetic overlap underlying diseases/traits between distinct populations. These population-specific discoveries also imply the urgent requirement of including sufficient individuals from underrepresented populations in trans-ethnic GWASs so that more ethnic groups can benefit from current medical genomics researches [18, 40, 78]. The genetic diversity can be explained by the difference in clinical definitions and phenotype measurements, gene-gene and gene-environment interaction [79, 80]. As another interpretation, the genetic inconsistency across various populations might result from various sample sizes and therefore different statistical power of studies. Furthermore, we revealed that population-common genes were in general more evolutionarily conserved compared to these population-specific or null ones.
Our work distinguishes itself from previous studies in three aspects. First, unlike prior studies which often focused only on a single trait [22, 81, 82], we considered a lot of diseases/traits, which could offer more unbiased insights into the extent of trans-ethnic genetic foundation shared across different populations. Second, in contrast to prior studies which performed single-marker association analysis, we here implemented a gene-centric trans-ethnic genetic overlap identification which jointly analyzed the association of a set of SNPs with the trait. Gene-set analysis is a popular complementary strategy in association studies and often much powerful as it aggregates multiple weak, sparse association signals at the SNP level into a strong association signal at the gene level and effectively reduces the burden of multiple tests [53]. In addition, correlation among SNPs in a gene was also considered in our gene-set association analysis; consequently, the trans-ethnic difference of LD structures was naturally explained by CONTO. Third, methodologically, we considered the trans-ethnic genetic overlap identification from a perspective of composite null hypothesis testing, which effectively takes the nature of trans-ethnic genetic overlap test into account and is thus much powerful compared to competitive methods.
There are two directions that need to further explore for CONTO in the future. First, when implementing CONTO, it requires us to first generate gene-level P values using gene-set association analysis methods. Certainly, the used gene-set based method would play a critical role in CONTO. Intuitively, the performance of a gene-set association analysis method is determined by how well its modeling assumption matches the true genetic architecture of a group of SNPs [83, 84], which however is generally unknown a priori and varies from one gene to another. Consequently, it is difficult to choose a consistently optimal gene-set based method for all genes across the whole genome [52, 85, 86]. In the present study, we applied MAGMA to simultaneously examine the association evidence of multiple SNPs, which was in nature a variance-component based score test for multilocus genetic association studies built based on random-effects models [53, 87]. Although MAGMA might be not the optimal method for every gene, it exhibited excellent performance in statistical power compared to many existing gene-set based methods and was widely used in gene-centric integrative analysis in post-GWAS era [52,53,54,55].
Nevertheless, the use of robust and powerful gene-set based methods in CONTO is of importance. Prior studies have demonstrated that aggregating association evidence available from diverse sources is an effective strategy for boosting power; such as integrating multiple gene expression prediction models in transcriptome-wide association studies using the harmonic mean P-value combining method [88], and combining a group of methods into a single powerful omnibus test using the minimum P-value method [86] or the aggregated Cauchy combination method [89, 90]. Therefore, leveraging these similar aggregation strategies to CONTO is an interesting direction that warrants further explorations.
Second, to a great degree, CONTO should be viewed as a qualitative trans-ethnic genetic overlap identification method because it can only offer the determination whether a particular gene is associated with the trait in both populations, but is difficult to give accurate evidence regarding the consistence of trans-ethnic genetic effects across populations. Understanding the effect difference could provide more in-depth insight into the diversity and similarity of genetic architecture underlying the same trait across distinct ancestral groups. To examine the genetic influence of a gene on complex traits in GWAS, prior studies attempted to employ the polygenic risk score calculated based on SNP effect sizes and genotypes of individuals available from external reference panels [2, 30, 54, 55]. Thus, such score may be also useful for the evaluation of trans-ethnic genetic effect of a gene in CONTO.
Conclusion
In summary, CONTO stands for a powerful method for detecting trans-ethnic genetic overlap across diverse ancestral groups; our results provide important implications on the transferability of GWAS discoveries in one population to others.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information file.
Abbreviations
- CONTO:
-
Composite null hypothesis test for trans-ethnic genetic overlap
- GWASs:
-
Genome-wide association studies
- SNPs:
-
Single nucleotide polymorphisms
- EUR:
-
European
- JST:
-
Joint significance test
- EAS:
-
East Asian
- FDR:
-
False discovery rate
- MVN:
-
Multivariate normal
- JT-comp:
-
Joint significance composite-null test
- DACT:
-
Divide-aggregate composite-null test
References
Graham SE, Clarke SL, Wu K-HH, Kanoni S, Zajac GJM, Ramdas S, Surakka I, Ntalla I, Vedantam S, Winkler TW, et al. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600:675–9.
Conti DV, Darst BF, Moss LC, Saunders EJ, Sheng X, Chou A, Schumacher FR, Olama AAA, Benlloch S, Dadaev T, et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet. 2021;53:65–75.
Giri A, Hellwege JN, Keaton JM, Park J, Qiu C, Warren HR, Torstenson ES, Kovesdy CP, Sun YV, Wilson OD, et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat Genet. 2019;51:51–62.
Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang WH, Kelly TN, Saleheen D, Lehne B, Leach IM, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet. 2015;47:1282–93.
Spracklen CN, Horikoshi M, Kim YJ, Lin K, Bragg F, Moon S, Suzuki K, Tam CHT, Tabara Y, Kwak S-H, et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature. 2020;582:240–5.
Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, Huffman JE, Assimes TL, Lorenz K, Zhu X, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020;52:680–91.
Sun S, Zhu J, Zhou X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat Methods. 2020;17:193–200.
Lam M, Chen C-Y, Li Z, Martin AR, Bryois J, Ma X, Gaspar H, Ikeda M, Benyamin B, Brown BC, et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat Genet. 2019;51:1670–8.
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J. 10 Years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22.
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20:467–84.
Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, Highland HM, Patel YM, Sorokin EP, Avery CL, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–8.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci. 2009;106:9362–7.
Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017;169:1177–86.
Gurdasani D, Barroso I, Zeggini E, Sandhu MS. Genomics of disease risk in globally diverse populations. Nat Rev Genet. 2019;20:520–35.
Chen J, Spracklen CN, Marenne G, Varshney A, Corbin LJ, Ja Luan, Willems SM, Wu Y, Zhang X, Horikoshi M, et al. The trans-ancestral genomic architecture of glycemic traits. Nat Genet. 2021;53:840–60.
Breen G, Li QQ, Roth BL, O’Donnell P, Didriksen M, Dolmetsch R, O’Reilly PF, Gaspar HA, Manji H, Huebel C, et al. Translating genome-wide association findings into new therapeutics for psychiatry. Nat Neurosci. 2016;19:1392–6.
Carlson CS, Matise TC, North KE, Haiman CA, Fesinmeyer MD, Buyske S, Schumacher FR, Peters U, Franceschini N, Ritchie MD, et al. Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLoS Biol. 2013;11:e1001661.
Waters KM, Stram DO, Hassanein MT, Le Marchand L, Wilkens LR, Maskarinec G, Monroe KR, Kolonel LN, Altshuler D, Henderson BE, Haiman CA. Consistent association of type 2 diabetes risk variants found in europeans in diverse racial and ethnic groups. PLoS Genet. 2010;6:e1001078.
Marigorta UM, Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 2013;9:e1003566.
Kraft P, Zeggini E, Ioannidis JP. Replication in genome-wide association studies. Stat sci. 2009;24:561.
Li Y, Lan L, Wang Y, Yang C, Tang W, Cui G, Luo S, Cheng Y, Liu Y, Liu J, Jin Y. Extremely cold and hot temperatures increase the risk of diabetes mortality in metropolitan areas of two Chinese cities. Environ Res. 2014;134:91–7.
Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–86.
Ikeda M, Takahashi A, Kamatani Y, Okahisa Y, Kunugi H, Mori N, Sasaki T, Ohmori T, Okamoto Y, Kawasaki H. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol Psychiatry. 2018;23:639–47.
Bigdeli TB, Ripke S, Peterson RE, Trzaskowski M, Bacanu S-A, Abdellaoui A, Andlauer T, Beekman A, Berger K, Blackwood DH. Genetic effects influencing risk for major depressive disorder in China and Europe. Transl Psychiatry. 2017;7:e1074–e1074.
Guo J, Bakshi A, Wang Y, Jiang L, Yengo L, Goddard ME, Visscher PM, Yang J. Quantifying genetic heterogeneity between continental populations for human height and body mass index. Sci Rep. 2021;11:1–9.
Brown BC, Ye CJ, Price AL, Zaitlen N. Consortium AGENTD: transethnic genetic-correlation estimates from summary statistics. Am J Hum Genet. 2016;99:76–88.
Veturi Y, de Campos G, Yi N, Huang W, Vazquez AI, Kühnel B. Modeling heterogeneity in the genetic architecture of ethnically diverse groups using random effect interaction models. Genetics. 2019;211:1395–407.
Lu H, Wang T, Zhang J, Zhang S, Huang S, Zeng P. Evaluating marginal genetic correlation of associated loci for complex diseases and traits between European and East Asian populations. Hum Genet. 2021;140:1285–97.
Kim MS, Patel KP, Teng AK, Berens AJ, Lachance J. Genetic disease risks can be misestimated across global populations. Genome Biol. 2018;19:179.
Guo J, Wu Y, Zhu Z, Zheng Z, Trzaskowski M, Zeng J, Robinson MR, Visscher PM, Yang J. Global genetic differentiation of complex traits shaped by natural selection in humans. Nat Commun. 1865;2018:9.
Robinson MR, Hemani G, Medina-Gomez C, Mezzavilla M, Esko- T, Shakhbazov K, Powell JE, Vinkhuyzen A, Berndt SI, Gustafsson S, et al. Population genetic differentiation of height and body mass index across Europe. Nat Genet. 2015;47:1357–62.
Li YR, Keating BJ. Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med. 2014;6:91.
Zaitlen N, Paşaniuc B, Gur T, Ziv E, Halperin E. Leveraging genetic variability across populations for the identification of causal variants. Am J Hum Genet. 2010;86:23–33.
Morris AP. Transethnic meta-analysis of genomewide association studies. Genet Epidemiol. 2011;35:809–22.
Fu J, Festen EAM, Wijmenga C. Multi-ethnic studies in complex traits. Hum Mol Genet. 2011;20:R206-13.
Coram MA, Fang H, Candille SI, Assimes TL, Tang H. Leveraging multi-ethnic evidence for risk assessment of quantitative traits in minority populations. Am J Hum Genet. 2017;101:218–26.
Coram MA, Candille SI, Duan Q, Chan KHK, Li Y, Kooperberg C, Reiner AP, Tang H. Leveraging multi-ethnic evidence for mapping complex traits in minority populations: an empirical Bayes approach. Am J Hum Genet. 2015;96:740–52.
Spracklen CN, Chen P, Kim YJ, Wang X, Cai H, Li S, Long J, Wu Y, Wang YX, Takeuchi F. Association analyses of East Asian individuals and trans-ancestry analyses with European individuals reveal new loci associated with cholesterol and triglyceride levels. Hum Mol Genet. 2017;26:1770–84.
Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11:356–66.
Bustamante CD, Burchard EG, De la Vega FM. Genomics for the world. Nature. 2011;475:163–5.
Teo Y-Y, Small KS, Kwiatkowski DP. Methodological challenges of genome-wide association analysis in Africa. Nat Rev Genet. 2010;11:149–60.
Galinsky KJ, Reshef YA, Finucane HK, Loh P-R, Zaitlen N, Patterson NJ, Brown BC, Price AL. Estimating cross-population genetic correlations of causal effect sizes. Genet Epidemiol. 2019;43:180–8.
Ongen H, Brown AA, Delaneau O, Panousis NI, Nica AC, Dermitzakis ET, Consortium GT. Estimating the causal tissues for complex traits and diseases. Nat Genet. 2017;49:1676–83.
Barfield R, Shen J, Just AC, Vokonas PS, Schwartz J, Baccarelli AA, VanderWeele TJ, Lin X. Testing for the indirect effect under the null for genome-wide mediation analyses. Genet Epidemiol. 2017;41:824–33.
Zeng P, Shao Z, Zhou X. Statistical methods for mediation analysis in the era of high-throughput genomics: current successes and future challenges. Comput Struct Biotechnol J. 2021;19:3209–24.
Shao Z, Wang T, Zhang M, Jiang Z, Huang S, Zeng P. IUSMMT: survival mediation analysis of gene expression with multiple DNA methylation exposures and its application to cancers of TCGA. PLoS Comput Biol. 2021;17:e1009250.
Dai JY, Stanford JL, LeBlanc M. A multiple-testing procedure for high-dimensional mediation hypotheses. J Am Stat Assoc. 2022;117:198–213. https://doi.org/10.1080/01621459.2020.1765785.
Eicher JD, Landowski C, Stackhouse B, Sloan A, Chen W, Jensen N, Lien J-P, Leslie R, Johnson AD. GRASP v2. 0: an update on the genome-wide repository of associations between SNPs and phenotypes. Nucleic Acids Res. 2015;43:D799–804.
Li MJ, Liu Z, Wang P, Wong MP, Nelson MR, Kocher J-PA, Yeager M, Sham PC, Chanock SJ, Xia Z, Wang J. GWASdb: v2 an update database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2016;44:D869–76.
McMahon A, Malangone C, Suveges D, Sollis E, Cunningham F, Riat HS, MacArthur JAL, Hayhurst J, Morales J, Guillen JA, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–12.
Wang J, Huang D, Zhou Y, Yao H, Liu H, Zhai S, Wu C, Zheng Z, Zhao K, Wang Z, et al. CAUSALdb: a database for disease/trait causal variants identified using summary statistics of genome-wide association studies. Nucleic Acids Res. 2019;48:D807–16.
Shao Z, Wang T, Qiao J, Zhang Y, Huang S, Zeng P. A comprehensive comparison of multilocus association methods with summary statistics in genome-wide association studies. BMC Bioinformatics. 2022;23:359.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219.
Lu H, Qiao J, Shao Z, Wang T, Huang S, Zeng P. A comprehensive gene-centric pleiotropic association analysis for 14 psychiatric disorders with GWAS summary statistics. BMC Med. 2021;19:314.
Wang T, Lu H, Zeng P. Identifying pleiotropic genes for complex phenotypes with summary statistics from a perspective of composite null hypothesis testing. Brief Bioinform. 2022;23:bbab389.
Langaas M, Lindqvist BH, Ferkingstad E. Estimating the proportion of true null hypotheses, with application to DNA microarray data. J R Stat Soc Ser B. 2005;67:555–72.
Jin J, Cai TT. Estimating the null and the proportion of nonnull effects in large-scale multiple comparisons. J Am Stat Assoc. 2007;102:495–506.
Efron B. Large-scale simultaneous hypothesis testing. J Am Stat Assoc. 2004;99:96–104.
Imai K, Keele L, Yamamoto T. Identification, inference and sensitivity analysis for causal mediation effects. Stat Sci. 2010;25:51–71.
VanderWeele T. Explanation in causal inference methods for mediation and interaction. Oxford: Oxford University Press; 2015.
Lin D-Y, Sullivan PF. Meta-analysis of genome-wide association studies with overlapping subjects. Am J Hum Genet. 2009;85:862–72.
Zeng P, Hao X, Zhou X. Pleiotropic mapping and annotation selection in genome-wide association studies with penalized Gaussian mixture models. Bioinformatics. 2018;34:2797–807.
LeBlanc M, Zuber V, Thompson WK, Andreassen OA, Frigessi A, Andreassen BK, Psychiat GC. A correction for sample overlap in genome-wide association studies in a polygenic pleiotropy-informed framework. BMC Genomics. 2018;19:494.
Huang Y-T. Genome-wide analyses of sparse mediation effects under composite null hypotheses. Ann Appl Stat. 2019;13:60–84.
Liu Z, Shen J, Barfield R, Schwartz J, Baccarelli AA, Lin X. Large-scale hypothesis testing for causal mediation effects with applications in genome-wide epigenetic studies. J Am Stat Assoc. 2022;117:67–81. https://doi.org/10.1080/01621459.2021.1914634.
Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, Hayward NK, Montgomery GW, Visscher PM, Martin NG, Macgregor S. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87:139–45.
Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361:598–604.
Jiang Y, Epstein MP, Conneely KN. Assessing the impact of population stratification on association studies of rare variation. Hum Hered. 2013;76:28–35.
van den Berg S, Vandenplas J, van Eeuwijk FA, Lopes MS, Veerkamp RF. Significance testing and genomic inflation factor using high-density genotypes or whole-genome sequence data. J Anim Breed Genet. 2019;136:418–29.
Zeng P, Zhao Y, Qian C, Zhang L, Zhang R, Gou J, Liu J, Liu L, Chen F. Statistical analysis for genome-wide association study. J Biomed Res. 2015;29:285–97.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–21.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50.
Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304.
Shang L, Smith JA, Zhao W, Kho M, Turner ST, Mosley TH, Kardia SLR, Zhou X. Genetic architecture of gene expression in European and African Americans: an eQTL mapping study in GENOA. Am J Hum Genet. 2020;106:496–512.
Coram Marc A, Duan Q, Hoffmann Thomas J, Thornton T, Knowles Joshua W, Johnson Nicholas A, Ochs-Balcom Heather M, Donlon Timothy A, Martin Lisa W, Eaton Charles B, et al. Genome-wide characterization of shared and distinct genetic components that influence blood lipid levels in ethnically diverse human populations. Am J Hum Genet. 2013;92:904–16.
Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639.
Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538:161–4.
Baye TM, Abebe T, Wilke RA. Genotype-environment interactions and their translational implications. Per Med. 2011;8:59–70.
Shi H, Gazal S, Kanai M, Koch EM, Schoech AP, Siewert KM, Kim SS, Luo Y, Amariuta T, Huang H, et al. Population-specific causal disease effect sizes in functionally important regions impacted by selection. Nat Commun. 2021;12:1098–1098.
Wheeler E, Leong A, Liu CT, Hivert MF, Strawbridge RJ, Podmore C, Li M, Yao J, Sim X, Hong J, et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med. 2017;14:e1002383.
Dastani Z, Johnson T, Kronenberg F, Nelson CP, Assimes TL, März W, Richards JB, Consortium C, Consortium A. The shared allelic architecture of adiponectin levels and coronary artery disease. Atherosclerosis. 2013;229:145–8.
Zeng P, Zhao Y, Liu J, Liu L, Zhang L, Wang T, Huang S, Chen F. Likelihood ratio tests in rare variant detection for continuous phenotypes. Ann Hum Genet. 2014;78:320–32.
Zeng P, Zhou X. Non-parametric genetic prediction of complex traits with latent Dirichlet process regression models. Nat Commun. 2017;8:456.
Sun R, Hui S, Bader GD, Lin X, Kraft P. Powerful gene set analysis in GWAS with the generalized Berk-Jones statistic. PLoS Genet. 2019;15:e1007530.
Sun R, Lin X. Genetic variant set-based tests using the generalized berk-jones statistic with application to a genome-wide association study of breast cancer. J Am Stat Assoc. 2020;115:1079–91.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence Kernel association test. Am J Hum Genet. 2011;89:82–93.
Zeng P, Dai J, Jin S, Zhou X. Aggregating multiple expression prediction models improves the power of transcriptome-wide association studies. Hum Mol Genet. 2021;30:939–51.
Liu Y, Xie J. Cauchy combination test: a powerful test with analytic p-value calculation under arbitrary dependency structures. J Am Stat Assoc. 2020;115:393–402.
Liu Y, Chen S, Li Z, Morrison AC, Boerwinkle E, Lin X. ACAT: a fast and powerful p value combination method for rare-variant analysis in sequencing studies. Am J Hum Genet. 2019;104:410–21.
Acknowledgements
We thank all the GWAS consortia for making summary statistics publicly available for us and are grateful to all the investigators and participants contributed to those studies. The data analyses in the present study were carried out with the high-performance computing cluster that was supported by the special central finance project of local universities for Xuzhou Medical University. We are very grateful to the Editor and two Reviewers for their constructive comments which substantially improved our manuscript.
Funding
The research of Ping Zeng was supported in part by the National Natural Science Foundation of China (82173630 and 81402765), the Youth Foundation of Humanity and Social Science funded by Ministry of Education of China (18YJC910002), the Natural Science Foundation of Jiangsu Province of China (BK20181472), the China Postdoctoral Science Foundation (2018M630607 and 2019T120465), the QingLan Research Project of Jiangsu Province for Young and Middle-aged Academic Leaders, the Six-Talent Peaks Project in Jiangsu Province of China (WSN-087), the Training Project for Youth Teams of Science and Technology Innovation at Xuzhou Medical University (TD202008), the Postdoctoral Science Foundation of Xuzhou Medical University, and the Statistical Science Research Project from National Bureau of Statistics of China (2014LY112). The research of Ting Wang was supported in part by the Social Development Project of Xuzhou City (KC20062).
Author information
Authors and Affiliations
Contributions
PZ conceived the idea for the study. PZ, JQ, ZS and TW obtained and cleared the datasets; JQ and ZS performed the simulations and data analyses. PZ, JQ, YW, and TW interpreted the results of the data analyses. ZP, JQ and TW wrote the manuscript with the help from other authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors agreed that this manuscript be submitted to the Journal of Translational Medicine for publication.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1. Complex traits available from the European and East Asian analyzed in the present study. Figure S1. Estimated false discovery rate under the simulation settings: (A) λ00=0.40, λ10=0.20, λ01=0.20, and λ11=0.2; (B) λ00=0.80, λ10=0.05, λ01=0.05, and λ11=0.10, and (C) λ00=0.90, λ10=0.01, λ01=0.01, and λ11=0.08. Here, the number of genes was set to 10000, and the false discovery rate was calculated as the proportion of non-overlapped associated genes among all identified ones. Figure S2. Estimated statistical power under the simulation settings: (A) λ00=0.40, λ10=0.20, λ01=0.20, and λ11=0.2; (B) λ00=0.80, λ10=0.05, λ01=0.05, and λ11=0.10, and (C) λ00=0.90, λ10=0.01, λ01=0.01, and λ11=0.08. Here, the number of genes was set to 10000, and the power was calculated as the proportion of truly overlapped associated genes among all identified ones. Figure S3. Estimated false discovery rate under the simulation settings: (A) λ00=0.40, λ10=0.20, λ01=0.20, and λ11=0.2; (B) λ00=0.80, λ10=0.05, λ01=0.05, and λ11=0.10, and (C) λ00=0.90, λ10=0.01, λ01=0.01, and λ11=0.08. Here, the number of genes was set to 20000, and the false discovery rate was calculated as the proportion of non-overlapped associated genes among all identified ones. Figure S4. Estimated statistical power under the simulation settings: (A) λ00=0.40, λ10=0.20, λ01=0.20, and λ11=0.2; (B) λ00=0.80, λ10=0.05, λ01=0.05, and λ11=0.10, and (C) λ00=0.90, λ10=0.01, λ01=0.01, and λ11=0.08. Here, the number of genes was set to 20000, and the power was calculated as the proportion of truly overlapped associated genes among all identified ones.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qiao, J., Shao, Z., Wu, Y. et al. Detecting associated genes for complex traits shared across East Asian and European populations under the framework of composite null hypothesis testing. J Transl Med 20, 424 (2022). https://doi.org/10.1186/s12967-022-03637-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03637-8