
Abstract
Background
Although the tumour immune microenvironment is known to significantly influence immunotherapy outcomes, its association with changes in gene expression patterns in hepatocellular carcinoma (HCC) during immunotherapy and its effect on prognosis have not been clarified.
Methods
A total of 365 HCC samples from The Cancer Genome Atlas liver hepatocellular carcinoma (TCGA-LIHC) dataset were stratified into training datasets and verification datasets. In the training datasets, immune-related genes were analysed through univariate Cox regression analyses and least absolute shrinkage and selection operator (LASSO)-Cox analyses to build a prognostic model. The TCGA-LIHC, GSE14520, and Imvigor210 cohorts were subjected to time-dependent receiver operating characteristic (ROC) and Kaplan–Meier survival curve analyses to verify the reliability of the developed model. Finally, single-sample gene set enrichment analysis (ssGSEA) was used to study the underlying molecular mechanisms.
Results
Five immune-related genes (LDHA, PPAT, BFSP1, NR0B1, and PFKFB4) were identified and used to establish the prognostic model for patient response to HCC treatment. ROC curve analysis of the TCGA (training and validation sets) and GSE14520 cohorts confirmed the predictive ability of the five-gene-based model (AUC > 0.6). In addition, ROC and Kaplan–Meier analyses indicated that the model could stratify patients into a low-risk and a high-risk group, wherein the high-risk group exhibited worse prognosis and was less sensitive to immunotherapy than the low-risk group. Functional enrichment analysis predicted potential associations of the five genes with several metabolic processes and oncological signatures.
Conclusions
We established a novel five-gene-based prognostic model based on the tumour immune microenvironment that can predict immunotherapy efficacy in HCC patients.
Similar content being viewed by others
Background
Hepatocellular carcinoma (HCC) is a highly malignant cancer and ranks as the third leading cause of cancer-related deaths worldwide [1]. The 5‐year survival and overall survival (OS) rates are below 12%. Precursors of most HCC cases include liver cirrhosis, chronic hepatitis viral infections, alcohol-related liver disease, non-alcoholic fatty liver disease, and drug-induced hepatitis. As HCC is usually diagnosed at advanced stages [2], it can be difficult to treat. Thus, it is important to elucidate the molecular mechanisms underlying HCC progression and develop novel therapeutic targets to improve patient survival outcomes.
The immune microenvironment plays a critical role in tumorigenesis and is correlated with tumour progression and treatment efficacy [3, 4]. Systemic immune therapeutics have shown efficacy against HCC, especially for patients without an opportunity to undergo resection or liver transplantation [2, 5]. Common immunotherapy strategies include chimeric antigen receptor-engineered T cells (CAR-T cells), cancer vaccines, cytokine therapy, and immune checkpoint inhibitors (ICIs). Currently, ICIs are the most successful class of immune therapeutics, both as monotherapy and combination therapy [6]. For example, the efficacy of anti-programmed cell death 1 (anti-PD-1) and anti-programmed cell death ligand 1 (anti-PD-L1) in HCC has been investigated. PD-L1 expressed by T cells regulates immune responses at the initiation phase in lymph nodes and at the effector phase in tumour cells [7]. The restoration of function in “exhausted” T cells and the depletion of immunosuppressive regulatory T lymphocytes using monoclonal antibodies targeting these receptors have opened up new avenues for the treatment of several malignancies [8]. However, only approximately 25% of HCC patients with high infiltration of PD-1-expressing T cells respond to ICIs [9], and identification of patients who will respond well to ICIs is challenging.
Conventional strategies using tumour-node-metastasis (TNM) classification to predict HCC prognosis can help guide decisions in HCC clinical therapy [10]. However, their predictive efficacies are less than satisfactory. Notably, the use of genome-sequencing technologies coupled with bioinformatics analyses has improved tumour diagnosis and prognosis capabilities. Gene-based prognostic models have been established to identify differential mRNA expression patterns between cancer and normal tissues. Datasets reporting the expression profiles of long noncoding RNAs (lncRNAs) [11], genes that regulate epigenetic modifications [12], and immune-related genes [13] from The Cancer Genome Atlas (TCGA) and NCBI Gene Expression Omnibus (GEO) databases have been increasingly explored to study disease prognosis. However, there is no mature model that can stably predict patient response to HCC immunotherapy and treatment outcome. Therefore, we established a novel five-gene-based model pertaining to the immune microenvironment and conducted bioinformatics analyses to assess the ability of this model to predict HCC immunotherapy outcomes.
In the present study, we conducted univariate Cox regression analyses and least absolute shrinkage and selection operator (LASSO)-Cox regression analyses to build a risk model. Risk score, time-dependent receiver operating characteristic (ROC) AUC, and Kaplan–Meier survival analyses were used to assess the prognostic ability of the model. The results indicate that our model can effectively predict the efficacy of immunotherapy and that the five genes can serve as potential independent biomarkers in clinical applications. Functional enrichment analysis predicted the potential associations of the upregulated and downregulated genes identified in our model with relevant biological mechanisms. Conclusively, we established a five-gene-based model based on the influence of the tumour immune microenvironment that could potentially be applied to predict patient response to HCC immunotherapy.
Methods
Data collection
Data from a total of 365 HCC patient samples from the TCGA-liver hepatocellular carcinoma (LIHC) dataset were retrieved and used for the analysis of prognostic gene expression signatures and the development of a prognostic model. Genes were excluded if corresponding patient sample data were lacking. Random sampling with arrangement was performed, wherein the 365 samples (including the training set [n = 219] and validation set [n = 146]) were randomly sampled 100 times with replacement. There was no significant difference in TNM stage, grade, OS, sex, or age between the training set and validation set (p > 0.05). The clinical data and mRNA expression data of the GSE14520 dataset (n = 221) were retrieved from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/), and an immunotherapy dataset (Imvigor210) was obtained from published study [14]. All patient data that were used in the present study had complete clinical information, including TNM stage, grade, survival time, sex, age, and immune-related gene expression.
Establishment of the five-gene model based on immune-related genes
First, we screened for immune-related genes associated with HCC prognosis. The immune-related genes involved in HCC pathogenesis were collected from previous studies [15,16,17]. In the TCGA-LIHC training set, immune-related gene and survival data were analysed by univariate Cox regression analysis using the R package “coxph”. The criterion of p < 0.001 was selected as the filtering threshold. We screened the immune-related genes associated with HCC prognosis. Second, a LASSO-Cox regression analysis was used to further filter the prognostic genes[18]. This method enables the simultaneous selection of variables and parameter estimation and can better solve the multicollinearity problem in regression analyses [18]. A prognostic gene signature was established based on the LASSO-Cox regression model coefficients (β values) multiplied by the mRNA expression level. The risk score = [0.307 × mRNA expression level of LDHA] + [0.268 × mRNA expression level of PPAT] + [0.455 × mRNA expression level of BFSP1] + [0.234 × mRNA expression level of NR0B1] + [0.109 × mRNA expression level of PFKFB4].
Assessment of the five-gene-based model by tissue microarray (TMA) analysis
To assess the prognostic ability of the five-gene-based model, we constructed a TMA comprised of 90 carcinoma tissues from HCC patients (Shanghai Outdo Biotech Co. Ltd., Shanghai, China) according to a reference method that was described previously [19]. Subsequently, immunohistochemistry (IHC) and integrated optical density (IOD) analyses were performed as described previously [20]. The primary antibodies used are shown in Additional file 1: Table S1. The IHC scores were obtained from independent assessments by three senior pathologists without any prior knowledge of patient characteristics. The IHC score of each patient was calculated using the following formula: IHC score = [0.307 × protein expression level of LDHA] + [0.268 × protein expression level of PPAT] + [0.455 × protein expression level of BFSP1] + [0.234 × protein expression level of NR0B1] + [0.109 × protein expression level of PFKFB4]. A Kaplan–Meier log-rank analysis was used to evaluate the difference in survival rates between the groups with a high IHC score and low IHC score.
Validation of the performance and prognostic ability of the five-gene-based model
Time-dependent ROC analyses and Kaplan–Meier log-rank tests were used to evaluate the performance and prognostic ability of the model using verification datasets from the TCGA-LIHC dataset, the entire TCGA-LIHC dataset, the GSE14520 cohort, and the immunotherapy dataset (Imvigor210).
Functional enrichment analyses
To explore the underlying molecular mechanisms of the five-gene-based model, single-sample gene set enrichment analysis (ssGSEA) of the gene expression profiles was used to identify the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways predicted to be correlated with the risk score [21, 22]. Clustering analyses were performed by gene set variation analysis (GSVA). A p < 0.05 and false discovery rate (FDR) < 0.05 were considered statistically significant.
Statistical analysis
Statistical analyses were performed using SPSS v25 (IBM, Chicago, IL, USA), GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA), and R software (version 3.5.1). Student's t-test was used for statistical comparisons, the Kaplan–Meier method was used to estimate OS, and p < 0.05 was considered statistically significant.
Results
Identification of HCC prognostic gene expression signatures to construct the HCC prognostic model
A flowchart of the analysis workflow is illustrated in Fig. 1a. Using the TCGA-LIHC training set, univariate Cox regression analysis of the screening results, including 4,227 immune-related genes, led to the identification of 245 immune-related genesas potential prognostic indicators of HCC OS.

Overall analysis workflow and LASSO model profile plots for the potential prognostic indicators of HCC. a. Schematic flowchart of the workflow performed to build and validate the HCC prognostic model. b. LASSO coefficient profile plots of the 245 prognostic genes showing that the variations in the size of the coefficients of parameters shrink with an increasing value of the k penalty. c. Penalty plot for the LASSO model for the 245 prognostic genes with error bars denoting the standard errors. LASSO least absolute shrinkage and selection operator, HCC hepatocellular carcinoma, OS overall survival
Construction of the five-gene-based HCC prognostic model
After primary filtering, a LASSO-penalized Cox analysis was performed to further narrow down the mRNA expression profiles (Fig. 1b and c). Five genes were identified: lactate dehydrogenase A (LDHA), phosphoribosyl pyrophosphate amidotransferase (PPAT), beaded filament structural protein 1 (BFSP1), nuclear receptor subfamily 0 group B member 1 (NR0B1), and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4). A stepwise multivariate Cox regression analysis was then performed to establish the prognostic model. The risk score was calculated by summing the weighted gene expression level of each of the five genes multiplied by their respective LASSO coefficients: risk score = [0.307 × mRNA expression level of LDHA] + [0.268 × mRNA expression level of PPAT] + [0.455 × mRNA expression level of BFSP1] + [0.234 × mRNA expression level of NR0B1] + [0.109 × mRNA expression level of PFKFB4]. All five genes showed positive LASSO coefficients in the multivariate Cox regression analysis. Next, the five-gene-based model was further evaluated for stability and reliability.
Evaluation of the predictive efficacy of the five-gene-based model using the TCGA-LIHC training set
To determine the association between the gene expression signatures of these five genes and HCC patient survival outcome, risk scores (AUC values) were calculated with the five-gene-based model for each sample separately, and the optimal cut-off for the risk score was defined using the TCGA-LIHC training set (Fig. 2a). Higher AUC values indicated better classification performance of the five-gene-based HCC prognostic model. AUC values of 0.80, 0.77 and 0.73 were obtained for the 1-year, 3-year and 5-year survival rates, respectively. Kaplan–Meier survival analysis revealed that patients in the high-risk group showed worse prognosis than patients in the low-risk group (p < 0.0001; Fig. 2a). Taken together, these results indicate good performance of the established five-gene-based model for predicting HCC survival outcomes.

Output profiles from the analyses of the TCGA and GSE dataset based on the prognostic model. Risk score (top left), patient status (top right), mRNA expression heatmap (bottom left), and time-dependent ROC and Kaplan–Meier curves (bottom right) of the five-gene-based model for the a. TCGA-LIHC training set and b. GSE14520 dataset. TCGA The Cancer Genome Atlas, ROC receiver operating characteristic, PPAT phosphoribosyl pyrophosphate amidotransferase, BFSP1 beaded filament structural protein 1, LDHA lactate dehydrogenase A, NR0B1 nuclear receptor subfamily 0 group B member 1, PFKFB4 6-phosphofructo- 2-kinase/fructose-2,6-bisphosphatase 4
Validation of the five-gene-based HCC prognostic model using the GSE14520 dataset, TCGA-LIHC testing set, and whole TCGA-LIHC dataset
To validate the stability of the five-gene-based model, a similar workflow was employed for the training set, wherein three datasets (the GSE14520 dataset, TCGA-LIHC testing set, and whole TCGA-LIHC dataset) were analysed. Risk scores for the five-gene-based model were obtained by calculating the AUC values of the respective ROC curves. AUC values of 0.75, 0.73 and 0.67 were obtained for 1-year, 3-year and 5-year survival rates, respectively, for the GSE14520 validation dataset. The results indicated that patients in the high-risk group showed significantly worse survival rates than patients in the low-risk group (p < 0.001; Fig. 2b). Consistent with the results of the GSE14520 dataset, patients in the high-risk group showed poorer OS than patients in the low-risk group (all p < 0.05) and the AUC values were above 0.6 for the whole TCGA-LIHC validation dataset and the TCGA-LIHC dataset (Additional file 2: Figure S1a and b). The results collectively showed that the five-gene-based model could predict patient survival duration based on gene expression levels.
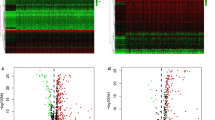
Upregulated genes (LDHA, PPAT, BFSP1, NR0B1, and PFKFB4) identified from the IHC TMA analysis predict poor prognosis
Next, the protein expression levels of the five genes LDHA, PPAT, BFSP1, NR0B1 and PFKFB4 were determined with an HCC TMA using IHC. Notably, we found that the protein levels of the five genes were significantly different in HCC tissues (Fig. 3a). The IHC score was calculated using the following formula: IHC score = [0.307 × protein expression level of LDHA] + [0.268 × protein expression level of PPAT] + [0.455 × protein expression level of BFSP1] + [0.234 × protein expression level of NROB1] + [0.109 × protein expression level of PFKFB4]. The group with high IHC scores was associated with higher protein expression levels and poorer disease prognosis than the group with low IHC scores (Fig. 3b). Variables that showed statistical significance in the univariate analysis were included in the Cox multivariate survival regression analysis. The results showed that the five-gene-based model score, grade and TNM stage were statistically significant (p < 0.05; Table 1). Thus, the results suggest that high protein expression of the five genes predicts poor disease prognosis.

The expression levels of the 5 genes in tumour tissues linked to prognosis of HCC. a. IHC images of the expression of proteins included in the five-gene-based model in HCC tumour tissues in the tissue microarray. b. Kaplan–Meier analysis of the five-gene-based model for patients with low- or high-risk scores. PPAT phosphoribosyl pyrophosphate amidotransferase, BFSP1 beaded filament structural protein 1, LDHA lactate dehydrogenase A, NR0B1 nuclear receptor subfamily 0 group B member 1, PFKFB4 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4, IHC immunohistochemistry, HCC hepatocellular carcinoma
Assessment of the ability of the five-gene-based model to predict immunotherapy efficacy
Biomarkers that can effectively predict the efficacy of immunotherapy drugs are currently lacking. Thus, identifying new predictive markers is necessary to further improve precision immunotherapy. A transcriptome dataset (Imvigor210) of the treatment response data of patients who underwent anti-PD-L1 immunotherapy was retrieved to assess the ability of the five-gene-based model to predict immunotherapy efficacy. Kaplan–Meier analysis showed that a high-risk score was associated with a poorer survival rate than a low risk score (Fig. 4a). ROC curve analysis showed that the combined consideration of neoantigen (NEO) burden, tumour mutational burden (TMB) and risk score output a higher AUC value (AUC = 0.91) than NEO burden (AUC = 0.7), TMB (AUC = 0.65), or risk score (AUC = 0.54) alone (Fig. 4b). The risk score was not correlated with the immunotherapy efficacy subgroup (p > 0.05; Fig. 4c) but was correlated with the immune cell and tumour cell subgroups (p < 0.05; Fig. 4d and e). These results suggest that considering the five-gene-based model score together with NEO burden and TMB could enhance the assessment of immunotherapy efficacy and identify patients who will respond to immunotherapy.

The five-gene-based model can predict immunotherapy efficacy for the Imvigor210 dataset. a Kaplan–Meier analysis of the five-gene-based model. b ROC curves of TMB, NEO burden, risk score, and the combination (NEO burden, TMB and risk score). The risk score was grouped by c immunotherapy efficacy, d immune cell subgroups, and e. tumour cell subgroups. ROC receiver operating characteristic, NEO neoantigens, TMB tumour mutational burden, CR complete response, PR partial response, SD stable disease, PD progressive disease, IC immune cell, TC tumour cell
The five-gene-based model can predict different clinical characteristics
After confirming the efficacy of the five-gene-based model in predicting patient response to immunotherapy, whether the five-gene-based model could be applied to determine the survival outcomes in subgroups with different clinical characteristics was investigated. The results indicated that the five-gene-based model could be used to predict different clinical characteristics (p < 0.05; Fig. 5).

Kaplan–Meier analyses of the five-gene-based model in subgroups with different clinical characteristics. Kaplan–Meier survival plot analyses to assess the efficacy of the five-gene-based model in predicting the survival outcomes of subgroups with different clinical characteristics (age, sex, T1 + T2, T3 + T4, N0, M0, stage I + II, stage III + IV, G1 + G2, and G3 + G4). Kaplan–Meier survival curves for a age > 60, b age < 60, c male, d female, e T1 + T2, f T3 + T4, g N0, h M0, i stage I + II, j stage III + IV, k G1 + G2, and l G3 + G4 for the low-risk and high-risk groups. T tumour, N node, M metastasis, G grade
Risk model and prognostic analyses of different gene mutation subtypes
To determine the clinical efficacy of the model in predicting treatment response in HCC patients with somatic mutations, Kaplan–Meier survival analyses were performed. First, 11 genes that are commonly mutated in HCC (tumour protein p53 [TP53], phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha [PIK3CA], retinoblastoma protein [RB1], cyclin-dependent kinase inhibitor 2A [CDKN2A], tuberous sclerosis-2 [TSC2], β-catenin [CTNNB1], AT-rich interactive domain-containing protein 2 [ARID2], axin 1 [AXIN1], ribosomal protein S6 kinase A3 [RPS6KA3], AT-rich interactive domain-containing protein 1A [ARID1A], and lysine methyltransferase 2D [KMT2D]) were selected. TP53, CTNNB1, AXIN1, and ARID1A showed the highest mutation frequencies (28%, 24%, 8% and 7%, respectively) and were therefore used for the Kaplan–Meier analysis (Additional file 3: Figure S2). There was a significant difference in OS between patients in the high-risk and low-risk groups (Additional file 3: Figure S2a with TP53 mutation; Figure S2b without TP53 mutation; Figure S2c with CTNNB1 mutation; and Figure S2d without CTNNB1 mutation [p < 0.05; Additional file 4: Figure S3]). Patients with AXIN1 or ARID1A mutations showed no significant difference in OS between the high-risk and low-risk groups, but a significant difference was observed for patients without AXIN1 or ARID1A mutations between the high-risk and low-risk groups. The results suggest that the five-gene-based model can also be used to predict the survival outcomes of HCC patients with genetic mutations.
The five-gene-based model can serve as an independent biomarker in clinical applications
Next, we evaluated whether the five-gene-based model could serve as an independent biomarker with clinical implications. Univariate and multivariate Cox regression analyses of the clinicopathological parameters (including age, sex, T stage, stage, and tumour grade) of 365 patients (Table 2) revealed that the HR of the risk model was approximately 1.7 (p < 0.001), and the HR of the T stage model was approximately 1.3 (p < 0.001; Fig. 6a). Multivariate Cox regression analysis showed that T stage and the prognostic model were independent risk factors associated with OS.

The forest plot and comparison of the five-gene-based model and other models. a Forrest plot of the clinicopathological parameters age, T stage, stage, grade, and risk score of 365 HCC patients. Time-dependent ROC analysis and Kaplan–Meier analysis for b, e. the five-gene signature, c, f. the HCC prognostic evaluation model, and d, g. the six-gene model. ROC receiver operating characteristic, HCC hepatocellular carcinoma, T tumour, AUC area under the curve, HR hazard ratio, CI confidence interval
Comparison of the five-gene-based model and other models
Next, the ability of our established model to determine HCC prognosis was compared with those of three other prognostic models: the four-gene signature [23], the HCC prognostic evaluation model [24] and the six-gene signature [25]. We calculated the risk score of the corresponding genes in these three models for the TCGA-LIHC dataset using a method similar to that used for the establishment of our five-gene-based model (described above). The four-gene-based model had lower AUC values for 1-, 3- and 5-year survival rates than our model; the HCC prognostic evaluation model and the six-gene-based model had slightly higher AUC values for the 1-year survival rate than our model but had lower AUC values for 3- and 5-year survival rates (Fig. 6b-d). These results indicated that our model performed better at predicting the long-term survival (3-year and 5-year survival) than the short-term survival (1-year) of HCC patients. Similar to our model, these three models could also predict the OS of the high- and low-risk groups (log-rank p < 0.001; Fig. 6e–g).
Relationship between risk score and KEGG pathways
We performed ssGSEA to identify potential KEGG pathways (with a correlation coefficient > 0.45) associated with the risk score. A total of 21 KEGG pathways were identified (Fig. 7a). Analysis of the relationship between gene sets and the risk score revealed that the KEGG pathways positively correlated with the risk score included DNA replication, mismatch repair, cell cycle, homologous recombination, spliceosome, oocyte meiosis, progesterone-mediated oocyte maturation, and pathogenic Escherichia coli infection. There were also downregulated pathway terms that negatively correlated with the risk score in HCC, including butanoate metabolism, peroxisome, fatty acid, linoleic acid, tryptophan and tyrosine metabolism (Fig. 7b). The enrichment analysis revealed potential critical pathways implicated in HCC pathogenesis.

Relationships between the risk score and KEGG pathways. a. Twenty-one KEGG pathways were found to be correlated with the risk score by ssGSEA. b. The ssGSEA score of the KEGG pathway changes as the risk score increases. Samples (rows) are ranked from low to high risk scores. The colour scale indicates upregulation (red) or downregulation (blue) of gene expression. ssGSEA single-sample gene set enrichment analysis, KEGG Kyoto Encyclopedia of Genes and Genomes
Discussion
Immune subsets were found to be significant effectors of immune defence [26,27,28]. The use of gene expression signatures to determine the outcomes of treatments has been reported by several groups. We showed that the correlation between gene signatures and the tumour immune microenvironment can be used to predict HCC immunotherapy outcome. Similarly, He et al. [13] established a model based on the expression of immune-related genes to predict treatment outcome, Shen et al. [29] identified a clinical-immune signature to estimate OS in ovarian cancer, and Tekpli et al. [30] discovered a novel independent prognostic factor based on the tumour immune microenvironment in breast cancer. Recently, integrative analysis of multi-omics data revealed epidermal growth factor receptor (EGFR) as a critical node in the gene regulatory network that is related to immune phenotype, and the inclusion of therapeutic EGFR inhibition enhanced head and neck squamous cell carcinoma patient response to ICIs [31]. This finding suggests that critical genes associated with the tumour immune microenvironment may serve as prognostic signatures and be useful for clinical immunotherapy.
HCC is a highly heterogeneous tumour at the molecular level and is pathological [32]. The most frequent mutations were identified in telomerase reverse transcriptase (TERT), ARID1A, TP53, and CTNNB1 [33,34,35,36,37]. These gene mutations were also associated with cell differentiation, proliferation, and clinical features [32, 38]. In our study, the five identified genes had a low frequency of mutations, and they were conducive to constructing a stable prognostic model. In addition, the five-gene-based model established in the present study could predict HCC treatment outcomes for patients with or without tumour-specific mutations in the TP53, CTNNB1, AXIN1 or ARID1A genes. Thus, the five-gene-based prognostic model was a useful classification tool for patients with various genetic mutation backgrounds.
TMB is an emerging biomarker of sensitivity to immunotherapy [39, 40]. Cai et al. [41] showed that high TMB in liver cancer patients with radical resection was significantly correlated with poor prognosis. Stenzinger et al. [42] reported that high TMB correlated with increased patient response rates and survival benefits from immune checkpoint inhibitors. Tumour-specific NEO that form as a consequence of mutations are thought to be another important biomarker for the therapeutic efficacy of cancer immunotherapies [43, 44]. In our study, ROC curve analysis showed that the combined consideration of NEO burden, TMB and risk score output a higher AUC value than NEO burden, TMB, or risk score alone. Combining the five-gene-based model with NEO burden and TMB could enhance the assessment of immunotherapy efficacy and identify patients who will respond to immunotherapy. In the present study, we identified five genes (LDHA, PPAT, BFSP1, NR0B1, and PFKFB4) associated with the tumour immune microenvironment in HCC patient cohorts that may be applied as potential biomarkers of HCC immunotherapy outcome. The correlation between the risk score and HCC prognosis was determined by analysing the mRNA expression and protein expression of the identified genes with an HCC TMA by qPCR and IHC, respectively. We showed for the first time that BFSP1 is correlated with HCC outcomes. PPAT catalyses the first committed step of de novo purine nucleotide biosynthesis [45, 46], suggesting that targeting PPAT might be a promising cancer strategy [47]. PPAT was also identified as a prognostic biomarker in HCC [48]. Many studies have reported that upregulated LDHA promotes tumour metastasis and is correlated with poor prognosis in several cancers, including lung adenocarcinoma [49], breast cancer [50], HCC [51], gallbladder carcinoma [52], and renal cell carcinoma [53]. Since high LDHA expression can reduce the oxygen dependency of tumour cells by promoting efficient anaerobic/glycolytic metabolism, targeting LDHA is a potential anti-tumour strategy. Epigenetic modification of NR0B1 leads to its ectopic activation in Ewing's sarcoma and lung cancer, enabling it to promote cancer cell proliferation [54,55,56,57]. The overexpression of PFKFB4 was found to be associated with a poor prognosis in gastric cancer [58], bladder cancer [59], colon cancer [60], acute monocytic leukaemia [61], glioblastoma [62], thyroid cancer [63], and breast cancers [64,65,66]. A better understanding of the molecular mechanisms underlying the ability of the five-gene signature to predict HCC pathogenesis as well as prospective studies to validate its utility in clinical applications are needed for the further development of new therapeutic and prognostication strategies (Additional file 5: Table S2).
In this work, we established a prognostic signature for HCC OS prediction that also effectively predicts the efficacy of immunotherapy through combined analysis of gene expression datasets from GEO and TCGA. The model was based on gene mRNA expression but not gene mutations or epigenetic modifications of these five genes. Therefore, it has good clinical feasibility, as it does not require whole-genome sequencing for all patients. Moreover, the methods used in this study might also be suitable for other types of malignancies. In further studies, we plan to detect the expression of these five genes in circulating tumour cells. However, there are several limitations in our study. First, the prognostic role of the five genes at the protein level warrants further research. Second, the model was established with tumour tissues, so it can only predict the prognosis of HCC patients after surgery and cannot detect and diagnose tumours at the early stage. Third, further functional experiments are needed, and the underlying mechanism of the five genes needs to be clarified (Additional file 6).
Conclusions
In this study, we established a novel five-gene-based prognostic model based on the tumour immune microenvironment. Importantly, our model could effectively predict the efficacy of immunotherapy and might serve as a potential independent biomarker in clinical applications.
Availability of data and materials
All data in our study are available upon request.
Abbreviations
- LASSO:
-
Least absolute shrinkage and selection operator
- OS:
-
Overall survival
- TCGA-LIHC:
-
The Cancer Genome Atlas liver hepatocellular carcinoma
- HCC:
-
Hepatocellular carcinoma
- PFKFB4:
-
6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4
- PPAT:
-
Phosphoribosyl pyrophosphate amidotransferase
- BFSP1:
-
Beaded filament structural protein 1
- LDHA:
-
Lactate dehydrogenase A
- NR0B1:
-
Nuclear receptor subfamily 0 group B member 1
- IHC:
-
Immunohistochemistry
- ROC:
-
Receiver operating characteristic
- NEO:
-
Neoantigens
- TMB:
-
Tumour mutational burden
- CR:
-
Complete response
- PR:
-
Partial response
- SD:
-
Stable disease
- PD:
-
Progressive disease
- IC:
-
Immune cell
- TC:
-
Tumour cell
- T:
-
Tumour
- N:
-
Node
- M:
-
Metastasis
- G:
-
Grade
- ssGSEA:
-
Single-sample gene set enrichment analysis
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Colagrande S, Inghilesi AL, Aburas S, Taliani GG, Nardi C, Marra F. Challenges of advanced hepatocellular carcinoma. World J Gastroenterol. 2016;22:7645–59.
Zhao Y, Shuen TWH, Toh TB, Chan XY, Liu M, Tan SY, et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut. 2018;67:1845–54.
Nishida N, Kudo M. Immunological microenvironment of hepatocellular carcinoma and its clinical implication. Oncology. 2017;92:40–9.
Wang L, Wang FS. Clinical immunology and immunotherapy for hepatocellular carcinoma: current progress and challenges. Hepatol Int. 2019;13:521–33.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30.
Mizukoshi E, Kaneko S. Immune cell therapy for hepatocellular carcinoma. J Hematol Oncol. 2019;12:52.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, de Moura MC, et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology. 2017;153:812–26.
Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology. 2016;150:835–53.
Xue C, Zhao Y, Jiang J, Li L. Expression levels of lncRNAs are prognostic for hepatocellular carcinoma overall survival. Am J Transl Res. 2020;12:1873–83.
He Y, Yu X, Li J, Zhang Q, Zheng Q, Guo W. Role of m(5)C-related regulatory genes in the diagnosis and prognosis of hepatocellular carcinoma. Am J Transl Res. 2020;12:912–22.
He Y, Dang Q, Li J, Zhang Q, Yu X, Xue M, et al. Prediction of hepatocellular carcinoma prognosis based on expression of an immune-related gene set. Aging (Albany NY). 2020;12:965–77.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–8.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61.
Chifman J, Pullikuth A, Chou JW, Bedognetti D, Miller LD. Conservation of immune gene signatures in solid tumors and prognostic implications. BMC Cancer. 2016;16:911.
Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218.
Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16:385–95.
Cai MY, Zhang B, He WP, Yang GF, Rao HL, Rao ZY, et al. Decreased expression of PinX1 protein is correlated with tumor development and is a new independent poor prognostic factor in ovarian carcinoma. Cancer Sci. 2010;101:1543–9.
Jin H, Zhang Y, You H, Tao X, Wang C, Jin G, et al. Prognostic significance of kynurenine 3-monooxygenase and effects on proliferation, migration, and invasion of human hepatocellular carcinoma. Sci Rep. 2015;5:10466.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50.
Zheng Y, Liu Y, Zhao S, Zheng Z, Shen C, An L, et al. Large-scale analysis reveals a novel risk score to predict overall survival in hepatocellular carcinoma. Cancer Manag Res. 2018;10:6079–96.
Ke K, Chen G, Cai Z, Huang Y, Zhao B, Wang Y, et al. Evaluation and prediction of hepatocellular carcinoma prognosis based on molecular classification. Cancer Manag Res. 2018;10:5291–302.
Liu GM, Zeng HD, Zhang CY, Xu JW. Identification of a six-gene signature predicting overall survival for hepatocellular carcinoma. Cancer Cell Int. 2019;19:138.
Miao YR, Zhang Q, Lei Q, Luo M, Xie GY, Wang H, et al. ImmuCellAI: a unique method for comprehensive T-cell subsets abundance prediction and its application in cancer immunotherapy. Adv Sci. 2020;7:1902880.
Jurmeister P, von Laffert M, Jöhrens K. Dissecting the spatial heterogeneity of different immune cell subsets in non-small cell lung cancer. Pathol Res Pract. 2020;216:152904.
Zhou R, Zhang J, Zeng D, Sun H, Rong X, Shi M, et al. Immune cell infiltration as a biomarker for the diagnosis and prognosis of stage I-III colon cancer. Cancer Immunol Immunother. 2019;68:433–42.
Shen S, Wang G, Zhang R, Zhao Y, Yu H, Wei Y, et al. Development and validation of an immune gene-set based prognostic signature in ovarian cancer. EBioMedicine. 2019;40:318–26.
Tekpli X, Lien T, Røssevold AH, Nebdal D, Borgen E, Ohnstad HO, et al. An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment. Nat Commun. 2019;10:5499.
Feng B, Shen Y, Pastor Hostench X, Bieg M, Plath M, Ishaque N, et al. Integrative analysis of multi-omics data identified EGFR and PTGS2 as key nodes in a gene regulatory network related to immune phenotypes in head and neck cancer. Clin Cancer Res. 2020;26:3616–28.
Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouzé E, Blanc JF, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol. 2017;67:727–38.
Nault JC, Martin Y, Caruso S, Hirsch TZ, Bayard Q, Calderaro J, et al. Clinical impact of genomic diversity from early to advanced hepatocellular carcinoma. Hepatology. 2020;71:164–82.
Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–8.
Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–11.
Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–73.
Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, et al. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. 2014;60:1983–92.
Rao CV, Asch AS, Yamada HY. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis. 2017;38:2–11.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–1.
Cai H, Zhang Y, Zhang H, Cui C, Li C, Lu S. Prognostic role of tumor mutation burden in hepatocellular carcinoma after radical hepatectomy. J Surg Oncol. 2020;121:1007–14.
Stenzinger A, Allen JD, Maas J, Stewart MD, Merino DM, Wempe MM, et al. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer. 2019;58:578–88.
Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21:81–5.
van den Bulk J, Verdegaal EM, de Miranda NF. Cancer immunotherapy: broadening the scope of targetable tumours. Open Biol. 2018;8:180037.
Iwahana H, Oka J, Mizusawa N, Kudo E, Ii S, Yoshimoto K, et al. Molecular cloning of human amidophosphoribosyltransferase. Biochem Biophys Res Commun. 1993;190:192–200.
Yamaoka T, Kondo M, Honda S, Iwahana H, Moritani M, Ii S, et al. Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. J Biol Chem. 1997;272:17719–25.
Bibi N, Parveen Z, Nawaz MS, Kamal MA. In silico structure modeling and molecular docking analysis of phosphoribosyl pyrophosphate amidotransferase (PPAT) with antifolate inhibitors. Curr Cancer Drug Targets. 2019;19:408–16.
Hu X, Bao M, Huang J, Zhou L, Zheng S. Identification and validation of novel biomarkers for diagnosis and prognosis of hepatocellular carcinoma. Front Oncol. 2020;10:541479.
Yu C, Hou L, Cui H, Zhang L, Tan X, Leng X, et al. LDHA upregulation independently predicts poor survival in lung adenocarcinoma, but not in lung squamous cell carcinoma. Future Oncol. 2018;14:2483–92.
Guddeti RK, Bali P, Karyala P, Pakala SB. MTA1 coregulator regulates LDHA expression and function in breast cancer. Biochem Biophys Res Commun. 2019;520:54–9.
Hua S, Liu C, Liu L, Wu D. miR-142-3p inhibits aerobic glycolysis and cell proliferation in hepatocellular carcinoma via targeting LDHA. Biochem Biophys Res Commun. 2018;496:947–54.
He Y, Chen X, Yu Y, Li J, Hu Q, Xue C, et al. LDHA is a direct target of miR-30d-5p and contributes to aggressive progression of gallbladder carcinoma. Mol Carcinog. 2018;57:772–83.
Zhao J, Huang X, Xu Z, Dai J, He H, Zhu Y, et al. LDHA promotes tumor metastasis by facilitating epithelial-mesenchymal transition in renal cell carcinoma. Mol Med Rep. 2017;16:8335–44.
Lu Y, Liu Y, Liao S, Tu W, Shen Y, Yan Y, et al. Epigenetic modifications promote the expression of the orphan nuclear receptor NR0B1 in human lung adenocarcinoma cells. Oncotarget. 2016;7:43162–76.
Döpke W, Schwertner E, Wagner J, Flöther F. Resolution of racemates of the beta-receptor blocker propranolol. Pharmazie. 1990;45:673–5.
Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res. 2009;69:9047–55.
Kinsey M, Smith R, Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol Cancer Res. 2006;4:851–9.
Minchenko O, Tsuchihara K, Minchenko D, Bikfalvi A, Esumi H. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J Gastroenterol. 2014;20:13705–17.
Yun SJ, Jo SW, Ha YS, Lee OJ, Kim WT, Kim YJ, et al. PFKFB4 as a prognostic marker in non-muscle-invasive bladder cancer. Urol Oncol. 2012;30:893–9.
Minchenko O, Ochiai A, Opentanova I, Ogura T, Minchenko D, Caro J, et al. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie. 2005;87:1005–10.
Wang G, Li S, Xue K, Dong S. PFKFB4 is critical for the survival of acute monocytic leukemia cells. Biochem Biophys Res Commun. 2020;526:978–85.
Goidts V, Bageritz J, Puccio L, Nakata S, Zapatka M, Barbus S, et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene. 2012;31:3235–43.
Lu H, Chen S, You Z, Xie C, Huang S, Hu X. PFKFB4 negatively regulated the expression of histone acetyltransferase GCN5 to mediate the tumorigenesis of thyroid cancer. Dev Growth Differ. 2020;62:129–38.
Yao L, Wang L, Cao Z, Hu X, Shao Z. High expression of metabolic enzyme PFKFB4 is associated with poor prognosis of operable breast cancer. Cancer Cell Int. 2019;19:165.
Gao R, Liu Y, Li D, Xun J, Zhou W, Wang P, et al. PFKFB4 promotes breast cancer metastasis via induction of hyaluronan production in a p38-dependent manner. Cell Physiol Biochem. 2018;50:2108–23.
Dasgupta S, Rajapakshe K, Zhu B, Nikolai BC, Yi P, Putluri N, et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature. 2018;556:249–54.
Acknowledgements
Not applicable.
Funding
This study was supported by grants awarded by the National Science and Technology Major Project of China (No. 2018ZX10302206), the Science and Technology Major Projects of Zhejiang Province (No. 2018C04016), and the Science and Technology Major Projects of Ningbo (No. 2016C51008).
Author information
Authors and Affiliations
Contributions
XG and HZ conceived the study design and wrote the manuscript. JG, QZ and JX performed the statistical analyses and data interpretation. QY, CH, GW, CC, and HZ collected the data and conducted the literature search. HZ and ZC reviewed the manuscript and made critical corrections. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the institutional ethical committee, and all patients signed informed consent forms.
Consent for publication
Consent for publication was obtained from all authors.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1. Primary antibody information.
Additional file 2
: Figure S1. Output profiles from the analyses of the TCGA validation set and entire TCGA dataset based on the five-gene-based HCC prognostic model. Risk score (top left), patient status (top right), mRNA expression heatmap (bottom left), and time-dependent ROC and Kaplan-Meier curves (bottom right) of the five-gene-based model for S1a. the TCGA-LIHC validation set and S1b. the entire TCGA dataset. Abbreviations: TCGA, The Cancer Genome Atlas; HCC, hepatocellular carcinoma; ROC, receiver operating characteristic; PPAT, phosphoribosyl pyrophosphate amidotransferase; BFSP1, beaded filament structural protein 1; LDHA, lactate dehydrogenase A; NR0B1, nuclear receptor subfamily 0 group B member 1; PFKFB4, 6-phosphofructo- 2-kinase/fructose-2,6-bisphosphatase 4.
Additional file 3
: Figure S2. Genes showing the highest mutation frequency in the TCGA-LIHC dataset. Abbreviations: TP53, tumour protein p53; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; RB1, retinoblastoma protein; CDKN2A, cyclin-dependent kinase inhibitor 2A; TSC2, tuberous sclerosis-2; CTNNB1, β-catenin; ARID2, AT-rich interactive domain-containing protein 2; AXIN1, axin 1; RPS6KA3, ribosomal protein S6 kinase A3; ARID1A, AT-rich interactive domain-containing protein 1A; KMT2D, lysine methyltransferase 2D.
Additional file 4
: Figure S3. Kaplan-Meier survival analysis to assess the ability of the five-gene-based model to predict HCC therapy outcome for patient groups a. with TP53 mutation, b. without TP53 mutation, c. with CTNNB1 mutation, and d. without CTNNB1 mutation. Abbreviations: HCC, hepatocellular carcinoma; TP53, tumour protein p53; CTNNB1, β-catenin; AXIN1, axin 1; ARID1A, AT-rich interactive domain-containing protein 1A.
Additional file 5: Table S2.
The sample information of TCGA training set and validation set.
Additional file 6.
Additional material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gu, X., Guan, J., Xu, J. et al. Model based on five tumour immune microenvironment-related genes for predicting hepatocellular carcinoma immunotherapy outcomes. J Transl Med 19, 26 (2021). https://doi.org/10.1186/s12967-020-02691-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02691-4