
Abstract
Background and objectives
In hepatocellular carcinoma (HCC) patients, microvascular invasion (MVI) is associated with worse outcomes regardless of treatment. No single reliable preoperative factor exists to predict MVI. The aim of the work described here was to develop a new MVI− based mRNA biomarker to differentiate between high and low risk patients.
Methods
Using The Cancer Genome Atlas (TCGA) database, we collected data from 315 HCC patients, including mRNA expression and complete clinical data. We generated a seven-mRNA signature to predict patient outcomes. The mRNA signature was validated using the GSE36376 cohort. Finally, we tested the formula in our own 53 HCC patients using qPCR for the seven mRNAs and analyzing the computed tomography (CT) features.
Results
This seven‐mRNA signature significantly correlated with length of recurrence-free survival (RFS) and overall survival (OS) for both the training and validation groups. RFS and OS were briefer in high risk versus low risk patients. A Kaplan–Meier analysis also indicated that survival time was significantly shortened in the high risk group versus the low risk group. Time-dependent receiver operating characteristic analysis demonstrated good predictive performance for the seven-mRNA signature. The mRNA signature also acts as an independent factor according to a Multivariate analysis. Our results are consistent with the seven-mRNA formula risk score.
Conclusion
Our research showed a novel seven-mRNA biomarker based on MVI predicting RFS and OS in HCC patients. This mRNA signature can stratify patients into subgroups based on their risk of recurrence to help guide individualized treatment and precision management in HCC.
Similar content being viewed by others

Background
Liver cancer, a highly aggressive form of cancer, is a leading cause of cancer-related deaths worldwide, including China [1]. Hepatocellular carcinoma (HCC) makes up 90% of liver cancer cases. Although developments in medical oncology and surgery have revolutionized the treatment of HCC, the prognosis remains poor, with high recurrence. Recurrence occurs in a quarter of HCC patients after liver transplantation and in more than two-thirds of patients after hepatic resection within 5 years post-remission [2]. Currently, no approved treatment reduces the risk of recurrence, progression, or death. Thus, methods that can predict patient prognosis are urgently needed, so that effective therapeutic and management strategies can be designed for distinct subsets of HCC patients.
The high recurrence of HCC is partly due to microvascular invasion (MVI). MVI is characterized by tumor emboli in vessels, including veins, capillaries, and lymphatic spaces. Tumor cell dissemination is associated with poor outcomes. All the evidence and diagnosis of MVI is post-surgery, based on the histopathology of the specimen. Thus, prior to surgery, very little is known about the diagnosis.
The prognostic scoring systems currently available to predict survival in HCC patients before resection or transplantation involve variables such as the tumor size and number, serum alpha-fetoprotein (AFP), and underlying liver disease [3,4,5,6]. These scoring systems make use of clinical information or international criterion, while ignoring the underlying conditions and internal changes. Patients who develop HCC usually have inflammation associated with fibrotic and cirrhotic livers, which is often accompanied by widespread lymphocyte infiltration in patients with chronic hepatitis. Thus, the surrounding tissue microenvironment likely has an important influence on HCC metastatic tendency. These scoring systems may help us understand MVI but the prognostic value of these systems is limited. Other systems incorporate morphology features to the advanced examinations, which is an improvement [7]. These systems also make use of qualified image analysis or Radionics to determine which patients have a higher risk for recurrence. With the advancement of microarray and high-throughput technology, several studies have shown a significant relationship between gene expression profiles and signatures with survival and outcome of cancer patients, including HCC [8, 9]. Several research groups have focused on relating gene expression to imaging. The results of these studies offer more clues for predicting MVI, but are not very clinically applicable. Therefore, more research is needed to improve and highlight the function of biomarkers.
The aim of this study was to establish the differential mRNA expression in MVI + HCC patients compared to MVI− patients. This differential mRNA expression pattern can help physicians make more accurate diagnoses. Better diagnoses can lead to optimization of medical resources, such as limited organ supplies for transplantation, and improved treatment for the individual.
Materials and methods
Patients
After approval of the study by the Institutional Ethical Committee of the First Affiliated Hospital of Zhengzhou University, China, 53 patients were enrolled and written informed consent was obtained for all patients. Liver cancer samples were collected from untreated patients between September 2017 and April 2018. A three-phase contrast-enhanced computed tomography examination was conducted in all patients. Under double-blind conditions, two hepatobiliary radiologists with more than 10 years’ experience reviewed and analyzed the CT image features independently. Discrepancies about image features were resolved by consensus review.
Data research
Level 3 data from The Cancer Genome Atlas (TCGA) for 315 patients with HCC were procured from the Cancer Genomics Browser from University of California Santa Cruz, including mRNA expression profiles and clinical information associated with these samples. Patients who satisfied the following criteria were used for the model development: (a) histologic diagnosis of HCC; (b) available microvascular invasion information, mRNA expression data, and comprehensive clinic pathological and follow-up information; and (c) recurrence-free survival (RFS) between 30 and 3000 days. Publicly available data were collected from TCGA. Thus, further authorization by the institutional ethics committee was unnecessary. GSE36376 was procured from Gene Expression Omnibus (GEO) for the validation group.
Construction of a risk-score formula
Using the TCGA training set, a univariate Cox proportional regression model comparing MVI+ and MVI− groups differential gene expression was used to identify potential mRNAs. The relationship of these identified mRNAs to RFS was then analyzed. A good method for regression analysis of high-dimensional data, the least absolute shrinkage and selection operator (LASSO) Cox regression algorithm, was implemented for the identification of prognostic genes. In addition, the log2 fold change and average expression levels were utilized for ranking potential candidates. A linear combination of selected genes weighted by their respective coefficients was designed to develop a patient risk score formula. Risk score = (expr Gene1 * coif Gene1) + (expr Gene2 * coif Gene2) + … + (expr Gene5 * coif Gene5).
Identical Ƀ-values were tested in the validation set. Survival curves of the risk scores were generated in the training and validation sets with a Kaplan–Meier survival analysis and a two-tailed log-rank test. Prognostic performance was determined using y calculated from the area under the curve (AUC) from the time-dependent receiver operating characteristic (ROC) analysis. Risk score accuracy for predicting RFS at 1, 3, and 5 years was determined. The independence of the predictive mRNA signature was determined using a multivariable Cox proportional hazards regression analysis, where the dependent variable was RFS and the mRNA signature and other clinical factors were covariables.
Statistical analysis
R language (Version 3.3.3) was utilized for all statistical analyses. The ‘survminer’, ‘survival’, and ‘survival ROC’ packages were utilized for the creation of survival curves and ROC curves. Differences in clinic pathological features were assessed with the Student’s t test or Chi squared test. Statistical tests were two-tailed. P less than 0.05 was considered significant.
Results
Determining the prognostic mRNA signature in the discovery group
The model was constructed from the TCGA data of 315 HCC patients. Of the 315 patients, 209 were MVI− and 106 MVI+. The mRNA expression data were compared. The logFC value was > 0.5, P < 0.01. A total of 341 different genes were differentially expressed (Fig. 1a). A univariable Cox proportional hazards regression analysis of the mRNA profiling data from the discovery group was conducted resulting in 59 mRNAs with P-values less than 0.05. A LASSO Cox regression model was utilized to identify the most prognostic genes. We obtained two constitute one is one standard error (SE) of the minimum criteria and minimum criteria [10]. Based on the simplified criteria, the lse model was used (Fig. 1b, c), resulting in 12 candidate genes. Genes were further filtered with a stepwise regression using a multivariable Cox proportional hazards regression model. Seven genes (IM2A, STAG3, ADH1C, NEIL3, GULP1, PPAP2C, CKMT1B) were identified. Correlations among the seven genes in the TCGA cohorts were evaluated using a heatmap. The risk score was calculated using the expression levels of the seven genes and the corresponding Coif. The resulting risk-score formula was: risk score = + (CKMT1B*−0.15788) + (PPAP2C*−0.06074) + (GULP1*0.111848) + (NEIL3*0.114841)) + (ADH1C *−0.10131)) + (STAG3*−0.19945) + (ITM2A*−0.3719).

The different mRNA expression between MVI+ and MVI− in TCGA discovery group. a Univariate Cox regression analysis shows 341 genes significantly related to RFS. b A tenfold cross-validation for tuning parameter selection in the LASSO model. c The LASSO coefficient profiles for the associated genes
Patients in the training cohort were categorized as high risk or low risk based on the optimum cutoff(−4.52432). The training cohort consisted of 87 high risk patients with 54 recurrences and 184 low risk patients with 53 recurrences. High risk patients had worse outcomes compared to low risk patients (P < 0.0001; Fig. 2a), as determined by the Kaplan–Meier analysis. Notably, if the mRNA expression value was weighted by a negative coefficient in this formula, the gene was protective with low risk for recurrence; a positive coefficient signified that the gene was harmful to the prognosis. In the discovery group, patients with low risk scores had a longer RFS time (median 572 days) compared to high risk patients (median 239 days). According to the heat map, the protective five mRNAs were increased in the low risk group, while the other two genes (NEIL3, GULP1) were downregulated (Fig. 2b). Thus, the risk-score formula stratified the HCC patients based on the RFS.

Kaplan–Meier analysis of RFS of the formula in different groups. a Risk formula analysis in the TCGA set: The recurrence time in high and low risk groups were applied using the Kaplan–Meier analysis. b The distribution of the risk formula, patients’ status, and the expression heat map for the discovery set. c Risk formula analysis in the GEO set: Recurrence time in high and low risk groups based on the Kaplan–Meier analysis. d Distribution of the risk formula, patients’ status, and the expression heat map for the discovery set. e Risk formula analysis in the 53 patient set: Recurrence time in high and low risk groups based on the Kaplan–Meier analysis. f The distribution of the risk formula, patients’ status, and the expression heat map for the discovery set
The prognostic performance of the seven mRNA signature was assessed with a time-dependent ROC curve analysis. AUC values for the RNA signature were 0.782, 0.793, and 0.749 for predicting recurrence in the TCGA set at 1, 3, and 5 years (Additional file 1: Figure S1). AUC values exceeded 0.7 [11], indicating that our selection mRNA formula was suitable for predicting recurrence in HCC patients.
Prognostic value of the seven mRNA formula in the validation groups
To validate the seven mRNA risk score formula, we used GSE36376 containing 401 patients. Similar to the discovery group, high risk patients had shorter recurrence times and worse outcomes than low risk patients (P = 0.00024). There were 97 high risk patients with 61 recurrences, whereas there were 304 low risk patients with 184 recurrences. The RFS time in low risk patients was longer compared to high risk patients (Fig. 2c, d). In addition, the five protective mRNAs increased in low risk patients; the opposite occurred in high risk patients. These results are consistent with the data from the discovery group. Thus, these seven mRNAs are risk factors for recurrence in HCC patients, supporting the risk prediction model.
The relationship between risk scores and clinical pathological factors
We conducted a set of predefined stratified analyses to identify whether the seven mRNAs have an independent predictive ability in the discovery and validation sets separately. A Cox multivariate regression analysis was performed with clinical characteristics, including age, gender, tumor stage, and WHO grade. The RFS probabilities for high and low risk groups, visualized with Kaplan–Meier plots (Additional file 1: Figure S2 and Figure S3), indicate that the mRNA signature is an independent factor for predicting RFS in HCC patients, both in the discovery and validated sets (Table 1).
Evaluation of the formula with our 53 patient set
Fifty-three fresh liver cancer tissues were collected from patients (Table 2) without any treatment. Total mRNA was isolated using TRIzol reagent (Invitrogen, Waltham, MA). A PrimeScript RT Reagent Kit was used for reverse transcription of the isolated RNA (Takara Bio, Otsu, Shiga, Japan). Gene expression levels were quantified using qRT-PCR. In the 53 HCC patients, RFS was shorter in the 19 high risk patients versus the 34 low risk patients. In the high risk group, 18 patients had recurrences, while 15 low risk patients had recurrences. The low risk patients had longer RFS than high risk patients (617.5 days VS 413 days). In this patient group, the seven mRNA formula differentiated the high risk from low risk HCC patients, with respect to RFS (Fig. 2e, f).
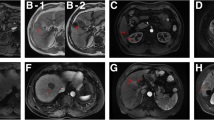
Imaging examination is widely used for assessing tumors. Therefore, for our patient group, we evaluated the association between imaging features and the mRNA signature. In previous research, we found that tumor dimension, non-smooth tumor margins, peritumoral enhancement, and TTPVI have the best ability to predict MVI in HCC [12]. Non-smooth tumor margins, peritumoral enhancement, and TTPVI are related to tumor size [13]. Therefore, we did not collect information about tumor size. After comparing the CT images, we found more “worrisome” features in high risk patients versus low risk patients (Fig. 3). Patients had high similarities between the imaging features and the mRNA formula (Fig. 4). In other words, the mRNA formula was consistent with our radiology consensus.

The CT features between the high and low risk group in the 53 patients set: The red column displays the high risk group, while the green column displays the low risk group. a The nonsmooth tumor margins, b the peritumoral enhancement, and c TTPVI

Computed tomography scan revealed the patients with different RFS. a peritumoral enhancement: recurrence 3.2 months, external portion enhancing in the arterial phase (arrows). b nonsmooth tumor margin positive: recurrence 4.5 months, focal extranodular extension (arrows) in the venous phase. c The two-trait algorithm predictive of MVI (TTPVI) positive: recurrence 2.8 months, blue arrows show internal artery and lower image means noncontinuous hypoattenuating halos
Cancer-related death is the most important index for patients. Thus, we assessed the association of risk score with overall survival (OS) time. Data from the discovery group indicated that patients with low risk scores had longer OS times than patients with high risk scores. Mortality rate in high risk patients was significantly elevated compared to low risk patients (Fig. 5), indicating that the seven-mRNA formula might play an important role in identifying patients with more risk of recurrence and worse outcomes. This formula showed the same effect in our 53 patient set. When patients were stratified according to tumor grade, the seven-mRNA formula could predict the recurrence based on the risks. In other words, the formula has the ability to divide the HCC patients with respect to OS and RFS based on the MVI.

Kaplan–Meier analysis of OS of the formula in different groups and stages. a Risk formula analysis in the TCGA set: The OS time in high and low risk groups based on the Kaplan–Meier analysis, b Kaplan–Meier curves obtained for TCGA set patient stages I and II. c Kaplan–Meier curves obtained for TCGA set patient stages III and IV. d Risk formula analysis in the GEO set: The OS time in high and low risk groups based on the Kaplan–Meier analysis. e Kaplan–Meier curves obtained for GEO set patient stages I and II. f Kaplan–Meier curves obtained for GEO set patient stages III and IV. g Risk formula analysis in the 53 patient set: The OS time in high and low risk groups based on the Kaplan-Meier analysis, h Kaplan–Meier curves obtained for 53 patient stages I and II, i Kaplan–Meier curves obtained for 53 patient stages III and IV
Discussion
Although diagnosis and management of HCC have advanced, the median patient survival time is less than 8 months; HCC is still a highly malignant cancer [14]. Even if patients choose curative treatment, such as surgical resection, liver transplantation, and local ablation, the frequent recurrence impedes successful outcome. For personalized patient management and elucidation of the biological features of recurrence, the development of a feasible and reliable signature that can predict high risk of recurrence is necessary. In our study, we developed and validated an mRNA formula which predicts recurrence. This is the first research based on mRNA to predict recurrence.
A risk score formula that can estimate HCC recurrence and prognosis has value in guiding the management of HCC. A risk score may help doctors identify candidates for liver transplantation and guide the design of clinical therapies. For example, patients who have high risk scores may not be the best candidates for liver transplantation compared to low risk patients, but may be candidates for more aggressive treatments.
Cancer gene expression can reveal the etiology, prognosis, and treatment response [15, 16]. With gene sequencing advancements, a large number of messenger RNAs (mRNA) have been explored in relation to cancer. Our study revealed that a seven mRNA signature (ITM2A, STAG3, ADH1C, NEIL3, GULP1, PPAP2C, CKMT1B) is associated with HCC recurrence. Patients with high mRNA signature based risk scores have shorter recurrence and OS times. The mRNAs in the predictive seven mRNA risk score shown below were also reported in other cancers. Risk score = + (CKMT1B*−0.15788) +(PPAP2C*−0.06074)) + (GULP1*0.111848) +(NEIL3*0.114841)) + (ADH1C *−0.10131)) + (STAG3*−0.19945) + (ITM2A *−0.3719). CKMT1B is involved in breast cancer. However, in contrast to our study, high CKMT1B expression was associated with poor outcomes [17,18,19,20]. CKMT1B encodes for a protein responsible for transferring high energy phosphate from mitochondria to cytosolic creatine. Thus, the upregulated expression of CKMT1B may result in more energy for the growing tumor tissue. This gene is also a porin of the mitochondrial membrane pore and impacts apoptosis. More research is necessary to elucidate the role of CKMT1B in HCC. PPAP2C belongs to the phosphatidic acid phosphatase enzyme family responsible for regulating dephosphorylation of lipid phosphates [21, 22]. This gene is overexpressed in many cancers, including ovarian carcinoma, lung cancer, and bladder, prostate cancers, as well as sarcomas. PPAP2C can regulate cell proliferation and may be an anticancer drug. This is consistent with our finding that this PPAP2C plays a protective role in HCC patients. GULP1 expression is significantly decreased in cancer tissue versus normal tissues and highly hypermethylated. Consistent with our data, GULP1 expression is associated with outcomes in ovarian cancer patients [23, 24]. Elevated expression of NEIL3 occurs in many human cancer cell types and tissues and is associated with primary malignant melanomas and metastasis [25,26,27]. NEIL3 may be a latent tumor suppressor gene for hepatocellular carcinoma [28]. This is consistent with our risk formula. The ADH1C gene has two alleles (ADH1C*1 and ADH1C*2), which code for the γ1 and γ2 enzyme subunits with different in vitro kinetic properties. ADH1C is associated with susceptibility to oral cancer, and may have anti-oncogenic, proapoptotic effects. In addition, ADHIC may be involved in rapid metabolism of ethanol and accumulation of acetaldehyde in tissue, resulting in increased cancer risk. We found that ADH1C may be protective in HCC. HCC is often associated with alcohol consumption; thus, ADH1C may have the same mechanism in HCC patients [29,30,31]. STAG3 has a tumor suppression function in ovarian cancer. This is consistent with our finding. STAG3 is also associated with lymphoma, and colorectal and testicular cancers [32, 33]. ITM2A is GATA3-related gene and has been reported as a downstream target in T-cell lymphoma. ITM2A also acts as a tumor suppressor of ovarian cancer via G2/M cell cycle arrest. According to our study, this gene is also protective in HCC [34,35,36].
This seven-mRNA risk formula was validated in three independent cohorts, including our fifty-three HCC patient set. Because imaging examination is popular in the clinical practice, we also compared our finding and the accepted MVI imaging feature; the results from this comparison are consistent concerning recurrence and OS. All the results showed that this mRNA signature is worthy to apply in clinical practice.
Conclusion
This seven-mRNA risk formula is an independent factor from other clinical traits for micro-vessel invasion. Therefore, the mRNA risk formula can be a predictor of HCC recurrence. In summary, our finding has tremendous value in the diagnosis and treatment in HCC patients, especially in predicting the recurrence of MVI.
Availability of data and materials
All data generated or analyzed during this study are included in this article.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- MVI:
-
Microvascular invasion
- TCGA:
-
The Cancer Genome Atlas
- CT:
-
Computed tomography
- RFS:
-
Recurrence-free survival
- OS:
-
Overall survival
- LASSO:
-
Least absolute shrinkage and selection operator
- AUC:
-
Area under the curve
- ROC:
-
Time-dependent receiver operating characteristic
- TTPVI:
-
Two-trait predictor of venous invasion: Internal arteries and hypodense halos
- coif:
-
Coefficient
References
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32.
Fisher RA, Kulik LM, Freise CE, Lok ASF, Shearon TH, Brown RS, Ghobrial RM, Fair JH, Olthoff KM, Kam I, Berg CL. Hepatocellular carcinoma recurrence and death following living and deceased donor liver transplantation. Am J Transplant. 2007;7:1601–8.
Hanazaki K, Kajikawa S, Koide N, Adachi W, Amano J. Prognostic factors after hepatic resection for hepatocellular carcinoma with hepatitis C viral infection: univariate and multivariate analysis. Am J Gastroenterol. 2001;96:1243–50.
Sasaki Y, Yamada T, Tanaka H, Ohigashi H, Eguchi H, Yano M, Ishikawa O, Imaoka S. Risk of recurrence in a long-term follow-up after surgery in 417 patients with hepatitis B- or hepatitis C-related hepatocellular carcinoma. Ann Surg. 2006;244:771–80.
Pandey D, Lee KH, Wai CT, Wagholikar G, Tan KC. Long term outcome and prognostic factors for large hepatocellular carcinoma (10 cm or more) after surgical resection. Ann Surg Oncol. 2007;14:2817–23.
Sun HC, Zhuang PY, Qin LX, Ye QH, Wang L, Ren N, Zhang JB, Qian YB, Lu L, Fan J, Tang ZY. Incidence and prognostic values of lymph node metastasis in operable hepatocellular carcinoma and evaluation of routine complete lymphadenectomy. J Surg Oncol. 2007;96:37–45.
Xu X, Zhang HL, Liu QP, Sun SW, Zhang J, Zhu FP, Yang G, Yan X, Zhang YD, Liu XS. Radiomic analysis of contrast-enhanced CT predicts microvascular invasion and outcome in hepatocellular carcinoma. J Hepatol. 2019;70:1133–44.
Bai F, Zhou H, Ma M, Guan C, Lyu J, Meng QH. A novel RNA sequencing-based miRNA signature predicts with recurrence and outcome of hepatocellular carcinoma. Mol Oncol. 2018;12:1125–37.
Lin T, Fu Y, Zhang X, Gu J, Ma X, Miao R, Xiang X, Niu W, Qu K, Liu C, Wu Q. A seven-long noncoding RNA signature predicts overall survival for patients with early stage non-small cell lung cancer. Aging (Albany NY). 2018;10:2356–66.
Qian Z, Li Y, Fan X, Zhang C, Wang Y, Jiang T, Liu X. Molecular and clinical characterization of IDH associated immune signature in lower-grade gliomas. Oncoimmunology. 2018;7:e1434466.
Zhang C, Zhang Z, Zhang G, Zhang Z, Luo Y, Wang F, Wang S, Che Y, Zeng Q, Sun N, He J. Clinical significance and inflammatory landscapes of a novel recurrence-associated immune signature in early-stage lung adenocarcinoma. Cancer Lett. 2020;479:31–41.
Renzulli M, Brocchi S, Cucchetti A, Mazzotti F, Mosconi C, Sportoletti C, Brandi G, Pinna AD, Golfieri R. Can current preoperative imaging be used to detect microvascular invasion of hepatocellular carcinoma? Radiology. 2016;279:432–42.
Segal E, Sirlin CB, Ooi C, Adler AS, Gollub J, Chen X, Chan BK, Matcuk GR, Barry CT, Chang HY, Kuo MD. Decoding global gene expression programs in liver cancer by noninvasive imaging. Nat Biotechnol. 2007;25:675–80.
Banerjee S, Wang DS, Kim HJ, Sirlin CB, Chan MG, Korn RL, Rutman AM, Siripongsakun S, Lu D, Imanbayev G, Kuo MD. A computed tomography radiogenomic biomarker predicts microvascular invasion and clinical outcomes in hepatocellular carcinoma. Hepatology. 2015;62:792–800.
Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, Cheng CL, Wang CH, Terng HJ, Kao SF, et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med. 2007;356:11–20.
Lu Y, Wang L, Liu P, Yang P, You M. Gene-expression signature predicts postoperative recurrence in stage I non-small cell lung cancer patients. PLoS ONE. 2012;7:e30880.
Redondo M, Villar E, Torres-Munoz J, Tellez T, Morell M, Petito CK. Overexpression of clusterin in human breast carcinoma. Am J Pathol. 2000;157:393–9.
Folgueira MA, Brentani H, Katayama ML, Patrao DF, Carraro DM, Mourao Netto M, Barbosa EM, Caldeira JR, Abreu AP, Lyra EC, et al. Gene expression profiling of clinical stages II and III breast cancer. Braz J Med Biol Res. 2006;39:1101–13.
Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006;98:262–72.
Cimino D, Fuso L, Sfiligoi C, Biglia N, Ponzone R, Maggiorotto F, Russo G, Cicatiello L, Weisz A, Taverna D, et al. Identification of new genes associated with breast cancer progression by gene expression analysis of predefined sets of neoplastic tissues. Int J Cancer. 2008;123:1327–38.
Tanyi JL, Morris AJ, Wolf JK, Fang X, Hasegawa Y, Lapushin R, Auersperg N, Sigal YJ, Newman RA, Felix EA, et al. The human lipid phosphate phosphatase-3 decreases the growth, survival, and tumorigenesis of ovarian cancer cells: validation of the lysophosphatidic acid signaling cascade as a target for therapy in ovarian cancer. Cancer Res. 2003;63:1073–82.
Brindley DN, Pilquil C. Lipid phosphate phosphatases and signaling. J Lipid Res. 2009;50(Suppl):S225–30.
Ma CI, Martin C, Ma Z, Hafiane A, Dai M, Lebrun JJ, Kiss RS. Engulfment protein GULP is regulator of transforming growth factor-beta response in ovarian cells. J Biol Chem. 2012;287:20636–51.
Maldonado L, Brait M, Izumchenko E, Begum S, Chatterjee A, Sen T, Loyo M, Barbosa A, Poeta ML, Makarev E, et al. Integrated transcriptomic and epigenomic analysis of ovarian cancer reveals epigenetically silenced GULP1. Cancer Lett. 2018;433:242–51.
Kauffmann A, Rosselli F, Lazar V, Winnepenninckx V, Mansuet-Lupo A, Dessen P, van den Oord JJ, Spatz A, Sarasin A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene. 2008;27:565–73.
Krishnamurthy N, Zhao X, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–46.
Liu M, Doublie S, Wallace SS. Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat Res. 2013;743–744:4–11.
Zhang H, Ma H, Wang Q, Chen M, Weng D, Wang H, Zhou J, Li Y, Sun J, Chen Y, et al. Analysis of loss of heterozygosity on chromosome 4q in hepatocellular carcinoma using high-throughput SNP array. Oncol Rep. 2010;23:445–55.
Bouchardy C, Hirvonen A, Coutelle C, Ward PJ, Dayer P, Benhamou S. Role of alcohol dehydrogenase 3 and cytochrome P-4502E1 genotypes in susceptibility to cancers of the upper aerodigestive tract. Int J Cancer. 2000;87:734–40.
Weiss JR, Baer MR, Ambrosone CB, Blanco JG, Hutson A, Ford LA, Moysich KB. Concordance of pharmacogenetic polymorphisms in tumor and germ line DNA in adult patients with acute myeloid leukemia. Cancer Epidemiol Biomarkers Prev. 2007;16:1038–41.
Brocic M, Supic G, Zeljic K, Jovic N, Kozomara R, Zagorac S, Zlatkovic M, Magic Z. Genetic polymorphisms of ADH1C and CYP2E1 and risk of oral squamous cell carcinoma. Otolaryngol Head Neck Surg. 2011;145:586–93.
Song H, Ramus SJ, Tyrer J, Bolton KL, Gentry-Maharaj A, Wozniak E, Anton-Culver H, Chang-Claude J, Cramer DW, DiCioccio R, et al. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet. 2009;41:996–1000.
Notaridou M, Quaye L, Dafou D, Jones C, Song H, Hogdall E, Kjaer SK, Christensen L, Hogdall C, Blaakaer J, et al. Common alleles in candidate susceptibility genes associated with risk and development of epithelial ovarian cancer. Int J Cancer. 2011;128:2063–74.
Barber TD, Barber MC, Tomescu O, Barr FG, Ruben S, Friedman TB. Identification of target genes regulated by PAX3 and PAX3-FKHR in embryogenesis and alveolar rhabdomyosarcoma. Genomics. 2002;79:278–84.
Van den Plas D, Merregaert J. Constitutive overexpression of the integral membrane protein Itm2A enhances myogenic differentiation of C2C12 cells. Cell Biol Int. 2004;28:199–207.
Nguyen TM, Shin IW, Lee TJ, Park J, Kim JH, Park MS, Lee EJ. Loss of ITM2A, a novel tumor suppressor of ovarian cancer through G2/M cell cycle arrest, is a poor prognostic factor of epithelial ovarian cancer. Gynecol Oncol. 2016;140:545–53.
Acknowledgements
I am grateful to all of those who help me during this research work. I especially need to say thank you to my family members.
Funding
This research was supported by Henan Province Health Planning Technology Talents Overseas Research Project (formerly 5451 Project). Grant Numbers: 2018010 and HWYX2019045.
Author information
Authors and Affiliations
Contributions
YZ, BD and FW contributed to the study design. YZ, BD and FW, BJ, and ZW contributed to data collection, performed statistical analysis and interpretation. BD and FW drafted the manuscript. All authors contributed to critical revision of the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Experimental procedures were approved by the Care Committee of the first affiliated Hospital of ZhengZhou University.
Consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Time‐dependent survival ROC analysis of the formula signature in the TCGA set. AUC: area under the curve. Figure S2. Supplement. Kaplan-Meier analysis of RFS of the formula in different groups and stages. Risk formula analysis in the TCGA set: The RFS time in high and low risk groups based on the Kaplan-Meier analysis, Kaplan–Meier curves obtained for TCGA set patient stage I (a), stage II (b), stage III & IV (c). Risk formula analysis in the GEO set: The RFS time in high and low risk groups based on the Kaplan-Meier analysis, Kaplan–Meier curves obtained for GEO set patient stages I (d), II (e), stage III & IV (f). Figure S3. Kaplan-Meier analysis of RFS of the formula in 53 patients group.Risk formula analysis in the 53 patients group set: The RFS time in high and low risk groups based on the Kaplan-Meier analysis, Kaplan–Meier curves obtained for patient stages I & II (a), Kaplan–Meier curves obtained for patient stages III & IV (b).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, B., Wang, F., Jarad, B. et al. A novel signature based on microvascular invasion predicts the recurrence of HCC. J Transl Med 18, 272 (2020). https://doi.org/10.1186/s12967-020-02432-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02432-7