
Abstract
Background
Sjögren’s syndrome (SS) is an autoimmune disease mediated by lymphocytic infiltration into exocrine glands, resulting in progressive lacrimal and salivary destruction and dysfunctional glandular secretion. Metabolic syndrome influences the immune system. To investigate its relationship with metabolic abnormalities, we evaluated the pathogenesis of SS and the immune cell populations in non-obese diabetic NOD/ShiLtJ mice with type 1 diabetes (T1D).
Methods
To induce metabolic abnormalities, streptozotocin (STZ)—a glucosamine–nitrosourea compound that destroys pancreatic β cells, resulting in T1D—was injected into NOD/ShiLtJ mice. The blood glucose level was measured to evaluate induction of T1D. The severity of SS was assessed by determining the body weight, salivary flow rate, and histologic parameters. The expression levels of proinflammatory factors in the salivary glands, lacrimal gland, and spleen were quantified by real–time PCR. The populations of various T– and B–cell subtypes in the peripheral blood, spleen, and salivary glands were assessed by flow cytometry.
Results
Induction of T1D in NOD/ShiLtJ mice increased both the severity of SS and the levels of proinflammatory cytokines in the salivary glands compared to the controls. Furthermore, the number of interleukin-17–producing immune cells in the peripheral blood, spleen, and salivary glands was increased in STZ- compared to vehicle-treated NOD/ShiLtJ mice.
Conclusions
Metabolic abnormalities play an important role in the development of SS.
Similar content being viewed by others

Background
Sjögren’s syndrome (SS) is a systemic autoimmune disease characterized by infiltration of lymphocytes into the exocrine glands, inflammation, tissue damage, and dysfunctional glandular secretion. Destruction of the lacrimal and salivary glands, which typically occurs in patients with SS, results in ocular dryness (keratoconjunctivitis sicca) and oral dryness (xerostomia) [1]. Patients with SS often have extraglandular complications such as non-erosive polyarthritis, arthralgias, vasculitis, and chronic fatigue [2]. Furthermore, patients with SS have an increased incidence of progression to various non-Hodgkin lymphomas, which may influence the rate of morbidities [3].
The non-obese diabetic (NOD) mouse is not only a widely used model for diabetes mellitus type 1, but also recognized as an appropriate model to study autoimmune exocrinopathy prevalent in human SS patients. NOD mouse indicated the development of diabetes in 12-week-old NOD/ShiLtJ mice and an autoimmune exocrinopathy that shows significant similarities to SS becomes evident between 8 and 12 weeks [4]. “SS” in this model means cellular infiltration into the salivary gland or a change in salivary flow rate. In addition, the subsequent tissue specific immunological attack, antibody directed against the cell surface muscarinic/cholinergic receptors appears and increased cytokines in salivary gland [5,6,7,8].
The pathogenesis of SS is mediated by complex mechanisms involving infiltration by lymphocytes (mainly T and B cells) of target organs during a dysregulated adaptive immune response. In the T– and B–cell–containing ectopic lymphoid structures in the salivary and lacrimal glands, hyperactivated B cells produce autoantibodies; e.g., anti-SSA/Ro and -SSB/La, against small RNA molecules and rheumatoid factors [9, 10]. Activation of B cells by follicular helper T (Tfh) cells is crucial for the clonal selection and affinity maturation [11].
Immune cells are key mediators of chronic diseases associated with obesity or metabolic abnormalities. However, changes in metabolism also affect the immune system [12]. Metabolic abnormalities (cardiovascular risk factors, insulin resistance, and visceral obesity) lead to activation of immune cells in the adipose tissue, liver, and pancreas, and disrupt the co–ordination of the innate and adaptive immune responses [13,14,15]. In patients with SS, the prevalence and clinical significance of cardiovascular risk factors, which are associated with dyslipidemia, diabetes mellitus, and hyperuricemia, is higher than that in healthy control subjects [16, 17]. Furthermore, SS patients with metabolic syndrome have increased serum levels of leptin and interleukin (IL)-1β compared to those without metabolic syndrome [18]. However, little is known about the effects of metabolic abnormalities on the T and B lymphocytes that mediate the pathogenesis of SS.
We investigated the role of metabolic abnormalities induced by streptozotocin (STZ)—a glucosamine–nitrosourea compound that destroys pancreatic β cells, resulting in a type 1 diabetes (T1D) phenotype [19] —in the pathogenesis of SS in NOD/ShiLtJ mice. Induction of T1D in NOD/ShiLtJ mice increased the severity of SS, the levels of proinflammatory cytokines, and the number of IL–17–producing immune cells in the peripheral blood, spleen, and salivary glands. Also, STZ-induced metabolic abnormalities promoted the infiltration of IL–17–producing cells into the salivary glands.
Methods
Animals
Seven-week-old female NOD/ShiLtJ mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were housed under specific pathogen-free conditions at the Catholic Research Institute of Medical Science, Catholic University of Korea. The mice were fed standard mouse chow and water. The procedures were approved by the Animal Research Ethics Committee of the Catholic University of Korea and conformed to the National Institutes of Health of the United States guidelines (Permit Number: CUMS- 2019-0001-01). All animals were treated and euthanized in accordance with the Guidelines on the Use and Care of Animals of the Catholic University of Korea. Surgery was performed under anesthesia with isoflurane, and every effort was made to minimize suffering. At the end of the study, the mice were euthanized in a CO2 chamber for sample collection.
Injection of agents
To induce T1D, 10-week-old female NOD/ShiLtJ mice were fasted for 24 h and intraperitoneally injected with 180 mg/kg STZ (Sigma-Aldrich, St. Louis, MO) in 0.1 M Na-citrate buffer (pH 4.5), or with only 0.1 M citrate buffer. The blood glucose levels of the mice were determined using an Accu-Check™ Compact Glucometer on days 0, 3, 7, and 11 after STZ injection (Roche Diagnostics, Indianapolis, IN).
Collection of saliva from NOD/ShiLtJ mice
Mice were anesthetized by inhalation of isoflurane (2%) and secretion of saliva was stimulated by intraperitoneal injection of pilocarpine (100 μg/mouse; Sigma-Aldrich). Ninety seconds later, saliva was collected using a micropipette from the oral cavity for 7 min. The volume of saliva collected was determined gravimetrically (μL/g/min). Saliva was stored at −70 °C until analysis.
Histological assessment of salivary-gland inflammation
Parotid glands were fixed in 4% paraformaldehyde and embedded in paraffin; next, Sects. (5 μm thickness) were prepared and stained with hematoxylin and eosin. The degree of inflammation was quantified as described previously [20]. The sections were incubated with primary antibodies against IL-6, tumor necrosis factor-α (TNF-α), and IL-17 (Abcam, Cambridge, UK) overnight at 4 °C, and with a biotinylated secondary antibody (REAL™ EnVision™/horseradish peroxidase; Dako, Glostrup, Denmark) for 1 h at 4 °C. The color was developed using the chromogen diaminobenzidine (Thermo Scientific, Rockford, IL) and the sections were examined under a light microscope (Olympus, Tokyo, Japan). Cells positive for IL-6, TNF-α, or IL-17 were enumerated visually by four individuals on high-magnification images projected onto a screen; mean values are presented.
Intracellular staining and flow cytometry
Cells were isolated from the spleen, salivary glands, and peripheral blood and stimulated with 25 ng/mL phorbol myristate acetate (Sigma-Aldrich) and 250 ng/mL ionomycin in the presence of GolgiStop (BD Biosciences, San Jose, CA) for 4 h. The cells were stained with anti-mouse CD4 peridin chlorophyll protein (PerCP) (clone RM4-5), anti-mouse C–X-C chemokine receptor type 5 (CXCR5)- PerCP-eFluor710 (clone SPRCL5) (eBioscience, San Diego, CA) and/or anti-mouse CD19 Phycoerythrin-Cy7 (clone 1D3) (BD Pharmingen, San Jose, CA) antibodies. Surface-labeled cells were permeabilized with Cytofix/Cytoperm solution (BD Pharmingen) and intracellular staining for IL-17 was performed using an anti-mouse IL-17–fluorescein isothiocyanate (clone eBio17B7) antibody (eBioscience, San Diego, CA). The samples were subjected to flow cytometry using a FACSCalibur (BD Pharmingen) and the data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Real-time polymerase chain reaction
A LightCycler 2.0 instrument (software version 4.0; Roche Diagnostics) and SensiFAST SYBR® Hi–ROX (Bioline USA Inc., Taunton, MA) were used for polymerase chain reaction (PCR) amplification according to the manufacturer’s instructions. The following primers were used: IL-6, 5′-AAC GAT GCA CTT GCA GAA A-3′ (sense) and 5′-TCT GAA GGA CTC TGG CTT TGT C-3′ (antisense); RORγt, 5′–TGT CCT GGG CTA CCC TAC TG -3′ (sense), 5′- GTG CAG GAG TAG GCC ACA TT-3′ (antisense); IL-17, 5′- CCT CAA AGC TCA GCG TGT CC-3′ (sense), 5′- GAG CTC ACT TTT GCG CCA AG -3′ (antisense); TNF-α, 5′-ATG AGC ACA GAA AGC ATG ATC-3′ (sense) and 5′–TAC AGG CTT GTC ACT CGA ATT-3′ (antisense); and β-actin, 5′-GTA CGA CCA GAG GCA TAC AGG-3′ (sense) and 5′-GAT GAC GAT ATC GCT GCG CTG-3′ (antisense). The mRNA levels were normalized to that of β-actin.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (version 5 for Windows; GraphPad Software, San Diego, CA). Normally distributed continuous data were analyzed by parametric Student’s t test. Differences in mean values among groups were subjected to analysis of variance. Values are presented as means ± SD. Values of P < 0.05 (two-tailed) were considered indicative of significance.
Results
Induction of T1D in NOD/ShiLtJ mice increases the severity of SS
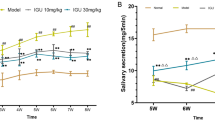
To evaluate the role of metabolic abnormalities in SS in vivo, 10-week-old female NOD/ShiLtJ mice were fasted for 24 h and STZ was administered intraperitoneally. Injection of STZ to mice and rats is associated with diabetes and weight loss [21,22,23]. Injection of STZ led to a rapid increase in the blood glucose level on day 3 and a decrease in body weight on day 7 compared to vehicle-treated NOD/ShiLtJ mice (Fig. 1a, b). Also, the salivary flow rate was markedly reduced in STZ-treated NOD/ShiLtJ mice compared to vehicle-treated NOD/ShiLtJ mice (P < 0.001) (Fig. 1c). Furthermore, infiltration of inflammatory cells into the salivary glands was exacerbated in STZ-treated NOD/ShiLtJ mice compared to vehicle-treated NOD/ShiLtJ mice (P < 0.001) (Fig. 1d). Therefore, metabolic abnormalities may contribute to the development of SS in NOD/ShiLtJ mice.

Streptozotocin (STZ) increases the severity of Sjögren’s syndrome (SS) in NOD/ShiLtJ mice. a After intraperitoneal injection of STZ (180 mg/kg) or vehicle into 10-week-old female NOD/ShiLtJ mice (n = 3 per group), the blood glucose level was determined using a glucometer. b Changes in body weight. c Salivary flow rates at 0, 7, and 11 days after injection of STZ. d Sections of parotid glands obtained 11 days after administration of STZ were stained with hematoxylin and eosin. Representative histological features and histologic grades are shown. ***P < 0.001 vs. vehicle–treated group. Data are mean ± SD
Induction of T1D in NOD/ShiLtJ mice upregulates the levels of proinflammatory cytokines
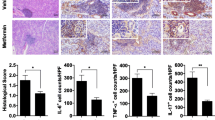
To examine whether metabolic abnormalities affect the levels of proinflammatory cytokines, salivary-gland sections were immunohistochemically stained for IL-6, IL-17, and TNF-α. The number of IL-6 + and IL-17 + cells was significantly increased in the salivary glands of STZ-treated compared to vehicle-treated mice (P < 0.01 and P < 0.05, respectively) (Fig. 2a). The number of TNF-α + cells was non-significantly increased in the STZ-injected mice (Fig. 2). In addition, the mRNA levels of IL-6, RORγt, IL-17, IFN-γ, and TNF-α in cells from the salivary gland, lacrimal gland, and spleen were increased in STZ-treated compared to vehicle-treated mice (Fig. 3). Also, in STZ–treated mice, the mRNA levels of these proinflammatory cytokines were higher in the salivary glands and lacrimal glands than in the spleen. Therefore, induction of T1D in NOD/ShiLtJ mice promoted the secretion of proinflammatory cytokines.

STZ increases the levels of proinflammatory cytokines in NOD/ShiLtJ mice. STZ (180 mg/kg) or vehicle was injected intraperitoneally into 10-week-old female NOD/ShiLtJ mice (n = 3 per group). Sections of parotid glands obtained 11 days after administration of STZ were stained with antibodies against interleukin (IL)-6, IL-17, and tumor necrosis factor-α (TNF-α). Representative histological features and numbers of antibody-positive cells are shown. Scale bar = 100 µm. *P < 0.05 and **P < 0.01 vs. vehicle-treated group. Data are mean ± SD

STZ increases the expression of proinflammatory cytokines in NOD/ShiLtJ mice. STZ (180 mg/kg) or vehicle was injected intraperitoneally into 10-week-old female NOD/ShiLtJ mice (n = 3 per group). RNA was extracted from the salivary glands (a), lacrimal glands (b), and spleens (c) of STZ– or vehicle-treated mice obtained 11 days after administration of STZ, and the mRNA levels of IL-6, RORγt, IL-17, IFN-γ, and TNF-α were analyzed by real-time polymerase chain reaction (PCR). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle–treated group. Data are mean ± SD
Metabolic abnormalities augment the number of IL–17–producing cells in NOD/ShiLtJ mice
To investigate the cell types related to the increased severity of SS in STZ-treated NOD/ShiLtJ mice, we analyzed the populations of T– and B–cell subtypes in peripheral blood and splenocytes. The numbers of Th17 cells and IL–17–producing B cells were significantly increased in the peripheral blood (Fig. 4a) and splenocytes (Fig. 4c) of STZ-treated NOD/ShiLtJ mice compared to those of vehicle-treated mice (P < 0.05). The number of CD4 + CXCR5 + IL-17 + Tfh17 cells in the peripheral blood was also significantly increased in NOD/ShiLtJ mice with T1D compared to vehicle-treated mice (P < 0.05) (Fig. 4b). However, there was no difference in the number of Tfh17 cells in splenocytes between the STZ- and vehicle-treated mice (Fig. 4d). Therefore, metabolic abnormalities may promote differentiation towards IL-17–producing cells.

Induction of type 1 diabetes (T1D) increases the number of IL–17–producing cells in NOD/ShiLtJ mice. STZ (180 mg/kg) or vehicle was injected intraperitoneally into 10-week-old female NOD/ShiLtJ mice (n = 3 per group). a, c Peripheral blood cells (a) and splenocytes (c) obtained 11 days after administration of STZ were stimulated with PMA, ionomycin, and GolgiStop for 4 h and stained with antibodies against CD4+IL-17+ (Th17) and CD19+IL-17+ (B17) cells. b, d Peripheral blood cells (b) and splenocytes (d) obtained 11 days after administration of STZ were stimulated with PMA, ionomycin, and GolgiStop for 4 h and stained with antibodies against CD4+CXCR5+IL-17+ (Tfh17) cells, followed by analysis by flow cytometry. *P < 0.05 vs. vehicle-treated group. Data are mean ± SD
STZ-induced metabolic abnormalities increase infiltration of IL–17–producing cells in the salivary glands
We investigated whether the metabolic abnormalities induced by STZ promote infiltration of IL–17–producing cells into the salivary glands. Compared to vehicle-treated mice, the number of IL–17–producing T and B cells in the salivary glands of STZ-treated mice was dramatically increased (Fig. 5). Therefore, increased infiltration of IL–17–producing cells into the salivary glands results in destruction of salivary-gland tissue and an increase in the severity of SS.

Induction of T1D promotes infiltration of IL–17–producing cells into the salivary glands of NOD/ShiLtJ mice. STZ (180 mg/kg) or vehicle was injected intraperitoneally into 10-week-old female NOD/ShiLtJ mice (n = 3 per group). Cells from salivary-gland tissue obtained 11 days after administration of STZ were stimulated with PMA, ionomycin, and GolgiStop for 4 h and stained with antibodies against CD4+IL-17+ (Th17) and CD19+IL-17+ (B17) cells, followed by analysis by flow cytometry. *P < 0.05 vs. vehicle-treated group. Data are mean ± SD
Discussion
We investigated the effect of metabolic abnormalities on T and B lymphocytes in NOD/ShiLtJ mice with SS and STZ-induced T1D. STZ resulted in a decrease in body weight and salivary flow rate, as well as in an increase in the infiltration of inflammatory cells into the salivary glands in NOD/ShiLtJ mice compared to vehicle-treated NOD/ShiLtJ mice. The number of IL–17–producing Th17, Tfh17, and B cells was increased in the peripheral blood and splenocytes from STZ- compared to vehicle-treated NOD/ShiLtJ mice. Furthermore, infiltration of IL–17–producing T and B cells into the salivary glands was increased in STZ-treated NOD/ShiLtJ mice. Therefore, metabolic abnormalities may contribute to the development of SS by increasing the population of IL–17–producing immune cells, as well as the infiltration of these cells into the salivary glands.
Metabolic syndrome, which comprises hypertension, diabetes, and obesity, is closely related to autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus [24,25,26,27]. However, the clinical significance of metabolic abnormalities in patients with SS is unclear. Dyslipidemia is closely related to hyperuricemia and the clinical features of SS [16], and patients with SS have an increased prevalence of diabetes mellitus and hypertriglyceridemia compared to healthy control subjects [17]. Furthermore, patients with SS who also have metabolic syndrome have increased serum levels of leptin and IL-1β [18]. In this study, we evaluated the effect of metabolic abnormalities on the development of SS. Induction of T1D in NOD/ShiLtJ mice greatly decreased the salivary flow rate and increased the degree of inflammation in the salivary glands. Also, the number of IL–17–producing cells in peripheral blood and splenocytes was increased in STZ- compared to vehicle-treated NOD/ShiLtJ mice. In particular, the increase in the population of IL-17–producing B cells can be used as an indicator of lymphoma in SS; thus, it is an important guide for SS treatment strategies [3]. These results clarify the effect of metabolic abnormalities on the immune cells involved in the development of SS. Further studies should aim to identify the molecular mechanism by which metabolic abnormalities increase IL-17 production by T and B cells.
IL-17 is a proinflammatory cytokine involved in the pathogenesis of various autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus, and psoriasis [28]. IL-17 triggers the production of proinflammatory factors, including IL-21, IL-6, CXCL8/IL-8, and CCL2/MIP–3a, which promotes lymphocyte recruitment, activation, and migration to target tissues [29,30,31]. Although IL-17 is mainly secreted by Th17 cells, it is also produced by Tfh and B cells [32,33,34]. The expression level of IL-17 is high in salivary gland tissues from C57BL/6.NOD-Aec1Aec2 mice and SS patients [35], and IL-17–expressing CD4 + T cells are present in the salivary glands of SS patients [36]. In addition, IL-17–deficient salivary gland protein-immunized SS mice exhibit reduced susceptibility to SS, but adoptive transfer of Th17 cells into IL-17–knockout mice induces SS by mimicking immunization with salivary gland proteins [37]. Diabetic children have higher levels of IL-17 and salivary mediators in saliva than nondiabetic children [38]. Therefore, IL-17 is related to the severity of SS. Metabolic syndrome reportedly exacerbates IL-17–mediated inflammatory reactions. The level of IL-17 in the periapical, hepatic, and renal regions was increased in apical periodontitis with STZ-induced diabetes compared to apical periodontitis [39]. Furthermore, administration of an IL-17 blocker reduces the pathogenicity of the oral microbiota in diabetic mice [40], and STZ-treated IL-17–knockout mice show decreased hyperglycemia and insulitis compared to control mice [41]. Adiponectin secreted from white adipose tissue promotes the development of arthritis by increasing the number of Th17 cells and the expression of receptor activator of nuclear factor-κB ligand in the joint tissues of mice with collagen-induced arthritis [42]. In addition, obesity promotes the differentiation of Th17 cells by increasing the expression of lipid kinase in CD4 T cells [43]. These previous reports support our hypothesis that metabolic abnormalities promote the development of IL-17–mediated SS. Further study of the mechanism(s) by which metabolic abnormalities promote the pathogenesis of SS is needed.
Conclusion
To our knowledge, this is the first report showing that metabolic syndrome exacerbates SS by increasing both the number of IL–17–producing immune cells and their infiltration into the salivary glands. Our findings suggest that metabolic disorders play an important role in the development of SS.
Availability of data and materials
All data are available in the manuscript or upon request to the authors.
Abbreviations
- SS:
-
Sjögren’s syndrome
- Tfh:
-
Follicular helper T
- IL:
-
Interleukin
- STZ:
-
Streptozotocin
- T1D:
-
Type 1 diabetes
- TNF-α:
-
Tumor necrosis factor-α
- CXCR5:
-
C X-C chemokine receptor type 5
- FITC:
-
Fluorescein isothiocyanate
- PCR:
-
Polymerase chain reaction
References
Brito-Zeron P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, Sivils K, Theander E, Tzioufas A, Ramos-Casals M. Sjogren syndrome. Nat Rev Dis Primers. 2016;2:16047.
Ramos-Casals M, Brito-Zeron P, Font J. The overlap of Sjogren’s syndrome with other systemic autoimmune diseases. Semin Arthritis Rheum. 2007;36:246–55.
Voulgarelis M, Dafni UG, Isenberg DA, Moutsopoulos HM. Malignant lymphoma in primary Sjogren’s syndrome: a multicenter, retrospective, clinical study by the European Concerted Action on Sjogren’s syndrome. Arthritis Rheum. 1999;42:1765–72.
Karnell JL, Mahmoud TI, Herbst R, Ettinger R. Discerning the kinetics of autoimmune manifestations in a model of Sjogren’s syndrome. Mol Immunol. 2014;62:277–82.
Cha S, Peck AB, Humphreys-Beher MG. Progress in understanding autoimmune exocrinopathy using the non-obese diabetic mouse: an update. Crit Rev Oral Biol Med. 2002;13:5–16.
Humphreys-Beher MG, Peck AB. New concepts for the development of autoimmune exocrinopathy derived from studies with the NOD mouse model. Arch Oral Biol. 1999;44(Suppl 1):S21–5.
Lodde BM, Mineshiba F, Kok MR, Wang J, Zheng C, Schmidt M, Cotrim AP, Kriete M, Tak PP, Baum BJ. NOD mouse model for Sjogren’s syndrome: lack of longitudinal stability. Oral Dis. 2006;12:566–72.
Gervais EM, Desantis KA, Pagendarm N, Nelson DA, Enger T, Skarstein K, Liaaen Jensen J, Larsen M. Changes in the submandibular salivary gland epithelial cell subpopulations during progression of Sjogren’s syndrome-like disease in the NOD/ShiLtJ mouse model. Anat Rec. 2015;298:1622–34.
Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmstrom P, Wahren-Herlenius M, Jonsson R. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren’s syndrome. Arthritis Rheum. 2003;48:3187–201.
Kroese FG, Abdulahad WH, Haacke E, Bos NA, Vissink A, Bootsma H. B-cell hyperactivity in primary Sjogren’s syndrome. Expert Rev Clin Immunol. 2014;10:483–99.
Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol. 2016;34:335–68.
Andersen CJ, Murphy KE, Fernandez ML. Impact of obesity and metabolic syndrome on immunity. Adv Nutr. 2016;7:66–75.
Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162.
Bremer AA, Jialal I. Adipose tissue dysfunction in nascent metabolic syndrome. J Obes. 2013;2013:393192.
Freitag J, Berod L, Kamradt T, Sparwasser T. Immunometabolism and autoimmunity. Immunol Cell Biol. 2016;94:925–34.
Ramos-Casals M, Brito-Zeron P, Siso A, Vargas A, Ros E, Bove A, Belenguer R, Plaza J, Benavent J, Font J. High prevalence of serum metabolic alterations in primary Sjogren’s syndrome: influence on clinical and immunological expression. J Rheumatol. 2007;34:754–61.
Perez-De-Lis M, Akasbi M, Siso A, Diez-Cascon P, Brito-Zeron P, Diaz-Lagares C, Ortiz J, Perez-Alvarez R, Ramos-Casals M, Coca A. Cardiovascular risk factors in primary Sjogren’s syndrome: a case-control study in 624 patients. Lupus. 2010;19:941–8.
Augusto KL, Bonfa E, Pereira RM, Bueno C, Leon EP, Viana VS, Pasoto SG. Metabolic syndrome in Sjogren’s syndrome patients: a relevant concern for clinical monitoring. Clin Rheumatol. 2016;35:639–47.
Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. 2008;51:216–26.
Shim GJ, Warner M, Kim HJ, Andersson S, Liu L, Ekman J, Imamov O, Jones ME, Simpson ER, Gustafsson JA. Aromatase-deficient mice spontaneously develop a lymphoproliferative autoimmune disease resembling Sjogren’s syndrome. Proc Natl Acad Sci USA. 2004;101:12628–33.
Han X, Tao YL, Deng YP, Yu JW, Cai J, Ren GF, Sun YN, Jiang GJ. Metformin ameliorates insulitis in STZ-induced diabetic mice. PeerJ. 2017;5:e3155.
Szasz T, Wenceslau CF, Burgess B, Nunes KP, Webb RC. Toll-like receptor 4 activation contributes to diabetic bladder dysfunction in a murine model of Type 1 diabetes. Diabetes. 2016;65:3754–64.
Patterson E, Marques TM, O’Sullivan O, Fitzgerald P, Fitzgerald GF, Cotter PD, Dinan TG, Cryan JF, Stanton C, Ross RP. Streptozotocin-induced type-1-diabetes disease onset in Sprague-Dawley rats is associated with an altered intestinal microbiota composition and decreased diversity. Microbiology. 2015;161:182–93.
Gonzalez-Gay MA, Gonzalez-Juanatey C, Pineiro A, Garcia-Porrua C, Testa A, Llorca J. High-grade C-reactive protein elevation correlates with accelerated atherogenesis in patients with rheumatoid arthritis. J Rheumatol. 2005;32:1219–23.
Sinicato NA, da Silva Cardoso PA, Appenzeller S. Risk factors in cardiovascular disease in systemic lupus erythematosus. Curr Cardiol Rev. 2013;9:15–9.
Sherer Y, Zinger H, Shoenfeld Y. Atherosclerosis in systemic lupus erythematosus. Autoimmunity. 2010;43:98–102.
Dessein PH, Joffe BI, Veller MG, Stevens BA, Tobias M, Reddi K, Stanwix AE. Traditional and nontraditional cardiovascular risk factors are associated with atherosclerosis in rheumatoid arthritis. J Rheumatol. 2005;32:435–42.
Bartlett HS, Million RP. Targeting the IL-17-T(H)17 pathway. Nat Rev Drug Discov. 2015;14:11–2.
Katz Y, Nadiv O, Beer Y. Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: a possible role as a “fine-tuning cytokine” in inflammation processes. Arthritis Rheum. 2001;44:2176–84.
Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–52.
Alunno A, Carubbi F, Bartoloni E, Bistoni O, Caterbi S, Cipriani P, Giacomelli R, Gerli R. Unmasking the pathogenic role of IL-17 axis in primary Sjogren’s syndrome: a new era for therapeutic targeting? Autoimmun Rev. 2014;13:1167–73.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8.
Wu HY, Quintana FJ, Weiner HL. Nasal anti-CD3 antibody ameliorates lupus by inducing an IL-10-secreting CD4 + CD25- LAP + regulatory T cell and is associated with down-regulation of IL-17 + CD4 + ICOS + CXCR5 + follicular helper T cells. J Immunol. 2008;181:6038–50.
Bermejo DA, Jackson SW, Gorosito-Serran M, Acosta-Rodriguez EV, Amezcua-Vesely MC, Sather BD, Singh AK, Khim S, Mucci J, Liggitt D, et al. Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORgammat and Ahr that leads to IL-17 production by activated B cells. Nat Immunol. 2013;14:514–22.
Nguyen CQ, Hu MH, Li Y, Stewart C, Peck AB. Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjogren’s syndrome: findings in humans and mice. Arthritis Rheum. 2008;58:734–43.
Sakai A, Sugawara Y, Kuroishi T, Sasano T, Sugawara S. Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren’s syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J Immunol. 2008;181:2898–906.
Lin X, Rui K, Deng J, Tian J, Wang X, Wang S, Ko KH, Jiao Z, Chan VS, Lau CS, et al. Th17 cells play a critical role in the development of experimental Sjogren’s syndrome. Ann Rheum Dis. 2015;74:1302–10.
del Lopez Valle LM, Ocasio-Lopez C, Steffen M. Comparison of levels of salivary cytokines in diabetic and nondiabetic puerto rican children: a case-control pilot study. Pediatr Dent. 2015;37:30–4.
Azuma MM, Gomes-Filho JE, Prieto AKC, Samuel RO, de Lima VMF, Sumida DH, Ervolino E, Cintra LTA. Diabetes increases interleukin-17 levels in periapical, hepatic, and renal tissues in rats. Arch Oral Biol. 2017;83:230–5.
Xiao E, Mattos M, Vieira GHA, Chen S, Correa JD, Wu Y, Albiero ML, Bittinger K, Graves DT. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe. 2017;22(120–128):e124.
Tong Z, Liu W, Yan H, Dong C. Interleukin-17A deficiency ameliorates streptozotocin-induced diabetes. Immunology. 2015;146:339–46.
Sun X, Feng X, Tan W, Lin N, Hua M, Wei Y, Wang F, Li N, Zhang M. Adiponectin exacerbates collagen-induced arthritis via enhancing Th17 response and prompting RANKL expression. Sci Rep. 2015;5:11296.
Endo Y, Asou HK, Matsugae N, Hirahara K, Shinoda K, Tumes DJ, Tokuyama H, Yokote K, Nakayama T. Obesity drives Th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep. 2015;12:1042–55.
Acknowledgements
None.
Funding
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI13C0016) and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI15C1062).
Author information
Authors and Affiliations
Contributions
SHH designed the experiments and analyzed the data. JSP designed the study and wrote the manuscript. SCY, KAJ and JWC performed the experiments. SKK analyzed the data and reviewed the manuscript. SHP and MLC conceived and designed the study, analyzed the data, and prepared the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Animals: The procedures were approved by the Animal Research Ethics Committee of the Catholic University of Korea and conformed to the guidelines of the National Institutes of Health of the United States (Permit Number: CUMS- 2019-0001-01).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hwang, SH., Park, JS., Yang, S. et al. Metabolic abnormalities exacerbate Sjögren’s syndrome by and is associated with increased the population of interleukin–17–producing cells in NOD/ShiLtJ mice. J Transl Med 18, 186 (2020). https://doi.org/10.1186/s12967-020-02343-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02343-7