
Abstract
Background
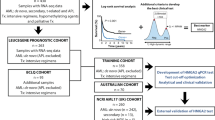
Identifying reliable predictive markers is important to make therapeutic decisions, and determine the prognosis for acute myeloid leukemia (AML) patients. However, approximately 50% patients could not be accurately predicted by existing risk factors. It is necessary to identify novel prognostic factors to subdivide the intermediate-risk group or patients without any cytogenetic and molecular abnormalities.
Methods
Kaplan–Meier and Cox regression were used for survival analyses in three independent AML datasets. Analyses integrating both bioinformatics and ChIP-qPCR experiments were performed to explore the role of CEBPE in regulating the expression of known prognostic factors.
Results
CEBPE expression was an independent predictor for both overall survival (OS) and event-free survival (EFS) of AML patients. Moreover, low-expression of CEBPE was found to be associated with high relapse rate. We also proved that differential expression of CEBPE stratified the wild-type patients of multiple genes into good and poor outcomes. In addition, the results showed that no obvious improvement was achieved by allogeneic transplantation in CEBPE high-expressed group, while the survival rate (both OS and EFS) was significantly increased in transplanted patients that with low expression of CEBPE. Finally, we found that CEBPE might regulate the expression of known prognostic factors by localizing on their promoters.
Conclusion
Our findings indicated that CEBPE expression was an independent prognostic factor for AML survival, relapse and allogeneic transplantation, which will provide useful information for outcome prediction and therapeutic decisions.
Similar content being viewed by others


Background
Acute myeloid leukemia (AML) is an aggressive malignancy and the most typical leukemia in adults, which is characterized by excessive proliferation, differentiation failure and apoptosis disorder, resulted in the abnormal accumulation of myeloblasts in the bone marrow and peripheral blood [1]. A majority of patients with AML will relapse after achieving complete remission [2]. At present, chemotherapy and/or allogeneic transplantation are the major treatments of AML [3, 4]. According to the acquired cytogenetic and molecular alterations at diagnosis, we could stratify the patients into different prognostic categories, and predict the relapse risk, survival time, drug response and whether a potentially curative allogeneic transplantation is possible [5]. Therefore, identifying reliable predictive markers is important in personalized therapy of AML.
Some risk factors were identified by previous studies, and used to predict treatment outcome for AML patients. For example, patients with genomic translocations such as t(15;17) (lead to PML–RARa fusion protein), t(8;21) (lead to AML1–ETO fusion protein) and inv(16) (lead to CBFb–MYH11 fusion protein) were classified in the favorable-risk group, cytogenetically normal AML (CN-AML) were in the intermediate-risk group, and those with a complex karyotype were classified in the adverse-risk group [6]. Moreover, some molecular abnormalities, including mutations of TP53, CEBPA, FLT3, DNMT3A were also found to provide important prognostic information, especially for CN-AML patients [7, 8]. For example, mutations in CEBPA are associated with a good outcome [9]; internal tandem duplications in FLT3 (FLT3–ITD) adversely affect the clinical outcome [10]. Mutations with prognostic implications in a number of other genes (e.g., TET2 [11], ASXL1 [12, 13], DNMT3A [14], p53 [15] and KIT [12]) have also been identified. To facilitate the prediction of treatment outcome of AML, a standardized system was proposed by an international expert panel in 2010 (working on behalf of the European LeukemiaNet (ELN)) [16]. Based on the published data on the prognostic significance of cytogenetic and molecular alterations, ELN stratified the patients into four groups: favorable, intermediate-I, intermediate-II and adverse. This system refined the classification of AML prognosis [6].
However, the existing risk factors still could not effectively predict the outcome of AML patients for the following reasons. Firstly, approximately 50% patients are CN-AML which is not associated with large chromosomal abnormalities [17]. The relapse rate and survival time of these CN-AML patients are difficult to predict because of high heterogeneity [18]. Secondly, although some gene mutations have statistical significance in predicting survival time of AML (especially for CN-AML), the mutation rates of these genes are relatively low. For example, AML patients with TP53 mutation are predicted to have adverse outcome, but only approximately 5% AML patients are with TP53 mutation [5]. The majority of patients are unpredictable based on gene mutation. Moreover, a significant proportion of patients are classified in intermediate-risk group according to the ELN standardized system [6], but the prognosis of these patients varies, some individuals respond well to chemotherapy based consolidation regimens while others may require allogeneic transplantation. Therefore, it is necessary to identify novel prognostic factors to subdivide the intermediate-risk group or patients without any cytogenetic and molecular abnormalities.
In this study, we found that CEBPE, as a master transcription regulator of myeloid differentiation, was an independent predictor for both overall survival (OS) and event-free survival (EFS) of AML patients. Moreover, CEBPE expression was observed to have prognostic power for AML relapse. Also, CEBPE expression was a potential factor for directing allogeneic transplantation.
Materials and methods
Gene expression data of AML patients
We used three independent AML datasets in this study, including The Cancer Genome Atlas (TCGA), GSE1159 and GSE10358. Only samples with both gene expression data and clinical annotations were kept. RNA-Seq data of 184 clinically annotated adult cases of AML were downloaded from TCGA [5]. Microarray data of 260 AML patients were downloaded from GSE1159 [19, 20]. And microarray data of 91 AML patients were downloaded from GSE10358 [21]. Microarray data and cytogenetic risk of each sample in GSE14468 [22, 23] were also used in this study.
Cell culture
The AML cell lines NB4 and Kasumi-1 were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA), and cultured in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% heat-inactivated fetal bovine serum (GIBCO-BRL), 100 U/mL penicillin and 100 mg/mL streptomycin (GIBCO-BRL). All cells were incubated in a humified 5% CO2 at 37 °C.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay of NB4 and Kasumi-1 cells was conducted by the manufacturer’s Active Motif protocol. Chromatin extracts were immunoprecipitated with anti-CEBPE (Santa Cruz Biotechnology, sc-158) and rabbit IgG (Abcam, ab172730) was used as negative control antibodies. ChIP-qPCR was conducted to analyze immunoprecipitated DNA using SYBR Green PCR Master Mix (Toyobo, Osaka, Japan) and the ABI Prism 7900HT detection system (Thermo Fisher Scientific). Fold enrichment of ChIP DNA vs. input DNA was calculated. The primers were designed to cover regions that are shown in Additional file 1: Table S1.
Statistical analyses
Survival was estimated according to the Kaplan–Meier method. The log-rank test was used to assess statistical significance. Cox regression was used to assess the association of a given variable with OS or EFS. Multivariable testing was performed using Cox proportional hazards models. P values < 0.05 were considered statistically significant. All of the statistical analyses were conducted using R package “Survival”.
Results
CEBPE is actively expressed in AML patients with favorable outcome
We collected AML gene expression data from TCGA, GSE14468 and GSE1159. The three independent datasets contained 184, 186 and 260 samples, respectively. The information of prognosis classification based on cytogenetic factors was also obtained. The results showed that CEBPE was highly expressed in patients with good prognosis. And this observation was confirmed in all of the three independent datasets. The t-test P-values of CEBPE differential expression between good and poor patients were 5.021e−05, 2.813e−11, 1.217e−6, respectively (Fig. 1a). Moreover, we also found that patients with high expression of CEBPE tended to have good prognosis in TCGA datasets (Fig. 1b).

CEBPE expression in AML patients with different prognosis. a CEBPE expression of AML patients with good and poor outcomes in three independent datasets TCGA, GSE14468 and GSE1159. b CEBPE expression and prognosis classification based on cytogenetic factors in TCGA database
CEBPE expression is an independent predictor of AML
We then validated the prognostic impact of CEBPE expression in three independent AML datasets, namely TCGA (n = 184), GSE1159 (n = 260) and GSE10358 (n = 91). In each dataset, we ranked the samples according to CEBPE expression, and samples of the top quartile were classified in high-expressed group, while others were classified in low-expressed group. As expected, Kaplan–Meier survival analyses demonstrated that decreased expression value of CEBPE was significantly (P < 0.05) associated with shorter OS and EFS (Fig. 2). In datasets of TCGA, GSE1159 and GSE10358, the 5-year overall survival rates were 38%, 47% and 59% in CEBPE high-expressed group, while 17%, 29% and 35% in CEBPE low-expressed group. Significant difference was also observed in OFS analysis.

Survival analyses of AML patients with differential expression of CEBPE. a Overall survival (OS) analyses of three independent datasets TCGA, GSE1159, GSE10358. b Event-free survival (EFS) analyses of three independent datasets TCGA, GSE1159, GSE10358
In addition, univariable Cox regression analysis demonstrated that patients with higher CEBPE expression showed lower risk. The following variables were evaluated in univariable Cox regression models for outcome: CEBPE expression, age, sex, white blood cell (WBC), peripheral blood (PB) or bone marrow (BM) blasts, the presence or absence of various chromosomal translocations [i.e., inv(16), t(8;21), t(15;17), t(9;11), t(11q23) and t(9;22)] and other abnormalities [+8, −3/inv(3)/t(3;3), −7/del(7q), −5/del(5)], and the presence or absence of gene mutations (FLT3-ITD or FLT3-TKD, DNMT3A, IDH1, IDH2, RUNX1, TET2, TP53, NRAS, CEBPA, KRAS, NPM1, KIT, PHF6 and ASXL1). Variables for which P < 0.1 in univariable analysis were shown in the Table 1 (OS) and Table 2 (EFS). Hazard ratios (HR) > 1 or < 1 indicate, respectively, a higher or lower risk of an event for higher values of continuous variables or for the first category listed for categorical variables in OS or EFS models. Accordingly, we found that age, TP53 mutation, DNMT3A mutation, WBC, t(9;11), RUNX1 mutation were risk factors, while CEBPE expression, t(15;17) and inv(16) were protective factors for AML OS and EFS. Through multivariable testing, we showed that the CEBPE low-expression remained significantly associated with worse OS and EFS in TCGA datasets, after adjusting for all other variables that had P < 0.1 in univariable analyses. Variables for which P < 0.05 in multivariable models were also shown in the Table 3 (OS) and Table 4 (EFS). It turned out that age, TP53 mutation, WBC and CEBPE expression were independent predictors for AML OS and EFS.
Low-expression of CEBPE predicts high relapse rate
We evaluated the association between CEBPE expression and relapse rates after complete remission using datasets of TCGA and GSE1159, which contained the information of relapse. All of the samples were classified into CEBPE high-expressed and low-expressed groups based on k-Nearest Neighbor (KNN) approach. The results showed that CEBPE expression had significant predictive power for AML relapse (P < 0.05). Low expression of CEBPE resulted in an increased incidence of relapse (Fig. 3).

Kaplan-Meier analyses of AML relapse rates after complete remission in TCGA and GSE1159 datasets
CEBPE expression has prognostic significance for wild-type AML patients of multiple genes
Some gene mutations were reported to be associated with poor outcome of AML, such as mutations of TP53 [24], FLT3 [25], DNMT3A [26], RUNX1 [27]. However, the frequency of patients with these mutations was relatively low. Novel prognostic factors were required to predict the outcome of wild-type patients. We evaluated the prognostic power of CEBPE expression for AML wild-type patients in TCGA datasets. For each gene mutation, samples were divided into four classes, namely mutated/CEBPE high, mutated/CEBPE low, wild-type/CEBPE high, wild-type/CEBPE low. The results showed that CEBPE expression differences in wild-type patients of TP53, FLT3, DNMT3A, KRAS, RUNX1 and NRAS were strongly associated with survival time (Fig. 4). Wild-type patients with high-expression of CEBPE showed longer survival than low-expressed wild-type patients. Thus, CEBPE expression could provide useful prognosis information by subdividing the wild-type patients.

CEBPE expression has prognostic significance for wild-type patients of multiple genes. “+” indicates mutation and “−” indicates wild-type. Differential expression of CEBPE stratified the wild-type patients into good and poor outcomes
CEBPE expression was a potential prognostic factor for allogeneic transplantation
We analyzed the association between CEBPE expression and allogeneic transplantation to explore whether CEBPE expression could provide useful information for directing allogeneic transplantation. All samples were classified into CEBPE high-expressed and low-expressed groups based on KNN approach. Then, in each group, Kaplan–Meier survival analyses were applied to compare the survival difference between individuals received and not received transplants. The results showed that no obvious improvement was achieved by allogeneic transplantation in CEBPE high-expressed group, while the survival rate (both OS and EFS) was significantly increased in transplanted patients that with low expression of CEBPE (Fig. 5). These results suggested that CEBPE expression would be a potential predictor for outcome of allogeneic transplantation in AML patients.

CEBPE expression was a potential prognostic factor for allogeneic transplantation. a Overall survival analyses for CEBPE low-expressed patients received or not received allogeneic transplantation. b Overall survival analyses for CEBPE high-expressed patients received or not received allogeneic transplantation. c Event-free survival analyses for CEBPE low-expressed patients received or not received allogeneic transplantation. d Event-free survival analyses for CEBPE high-expressed patients received or not received allogeneic transplantation
CEBPE regulates known predictors of AML
According to the above results, we showed that CEBPE expression was an independent prognostic factor for AML survival, relapse and allogeneic transplantation. Then, we attempted to explain the molecular mechanism of favorable outcome induced by increase of CEBPE expression. An international collaborative study reported by Li et al. [28] identified a 24-gene prognostic signature based on the data analyses of 1324 AML patients, and improved the established risk classification of AML prognosis. The identified 24 genes were ALS2CR8, ANGEL1, ARL6IP5, BSPRY, BTBD3, C1RL, CPT1A, DAPK1, ETFB, FGFR1, HEATR6, LAPTM4B, MAP7, NDFIP1, PBX3, PLA2G4A, PLOD3, PTP4A3, SLC25A12, SLC2A5, TMEM159, TRIM44, TRPS1, and VAV3, the increased expression levels of which were significantly associated with worse (22 genes) or favorable (two genes: FGFR1 and PLOD3) OS of AML. We found that CEBPE expression was significantly correlated with these known predictors of AML. As many as 13 genes were co-expressed with CEBPE in TCGA dataset (P-value < 0.05, Fig. 6a left panel), and 15 genes were co-expressed with CEBPE in GSE1159 dataset (P-value < 0.05, Fig. 6a right panel). Interestingly, CEBPE expression level was positively correlated with FGFR1 and PLOD3 in both datasets, which were reported as favorable factors, while negatively correlated with other genes which reported as predictors for poor outcome. This observation was consistent with our results that high expression of CEBPE predicted longer survival and lower relapse rate. Given the fact that CEBPE was an important transcription factor in regulating myeloid differentiation [29, 30], we hypothesized that CEBPE might regulate the expression of these known prognostic factors by localizing on their promoters, and verified using ChIP-qPCR assay in NB4 and Kasumi-1 cells. The results showed that CEBPE actually occupied on the promoters of known predictors, suggesting the regulation role of CEBPE on genes associated with AML prognosis.

CEBPE regulates known predictors of AML. a Heatmaps for gene expression of CEBPE and known prognostic factors of AML in TCGA and GSE1159 datasets. The Pearson correlation coefficient between expression values of CEBPE and each known predictor was listed in the brackets. b ChIP-qPCR was performed using the anti-CEBPE in Kasumi-1 and NB4 cell lines. Data are shown as fold enrichment of ChIPed DNA vs. input DNA. Error bars represent SD of triplicate measurements. NC: negative control
Discussion
In the clinical setting, it is important to identify prognostic factors to direct the appropriate treatments and predict outcomes. Patients with a molecular profile that is associated with a favorable risk have relatively good outcomes with chemotherapy, whereas patients with an unfavorable-risk profile require allogeneic transplantation during the first remission to improve their prognosis [5, 31]. Based on the analyses of several independent datasets, we identified CEBPE expression as an independent prognostic factors for AML patients. Low-expression of CEBPE was found to be associated with shorter OS, EFS and higher relapse rate, indicating adverse outcome of AML. Importantly, both RNA-Seq and microarray data supported this results, suggesting that CEBPE expression was a reliable prognostic factor. In addition, CEBPE expression was proved to have prognostic significance for wild type patients of various genes, providing useful information for prognosis of patients without molecular alterations. Moreover, CEBPE expression was also a potential prognostic factor for allogeneic transplantation. This observation could be easily used in routine clinical practice, as CEBPE expression could be tested before deciding if allogeneic transplantation should be implemented, and allogeneic transplantation surgery would be recommended only for CEBPE low-expressed patients, which will provide accurate information for therapeutic decisions.
The generation and development of AML are associated with the disregulation of various transcription factors (TFs) [32]. Especially, the abnormal expression of TFs which are important in hematopoietic or myeloid differentiations would lead to the accumulation of myeloblasts in the bone marrow and peripheral blood [33]. Previous studies suggested that CEBPE was indispensable for myeloid normal differentiation progress [30, 34]. For example, CEBPE knockout mice die within a few months of birth due to the loss of mature neutrophils or eosinophils [35]. Similarly, patients with a frame-shift mutation in CEBPE are suffered from specific granulocyte deficiency disease [36]. These observations imply that CEBPE may play a pivotal role in granulocytic maturation and exert an important function in myeloid differentiation. Our observations suggested that CEBPE localized on the promoters of a series of known predictors of AML survival, and had positive or negative co-expression relationship with these target genes. This result highlighted the reasons of why CEBPE expression showed significant prognostic power. Importantly, it is much more practical and economical to test the expression of one driver gene (CEBPE) than to test several passenger genes. Therefore, CEBPE expression holds great potential for clinical application in risk stratification and outcome prediction in AML.
Conclusion
Our findings indicated that CEBPE expression was an independent prognostic factor for AML survival, relapse and allogeneic transplantation, which will provide useful information for outcome prediction and therapeutic decisions.
Availability of data and materials
The datasets analyzed in the current study are available in The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) and Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/).
Abbreviations
- AML:
-
acute myeloid leukemia
- OS:
-
overall survival
- EFS:
-
event-free survival
- ChIP:
-
chromatin immunoprecipitation
- WBC:
-
white blood cell
- PB:
-
peripheral blood
- BM:
-
bone marrow
- HR:
-
hazard ratios
- KNN:
-
k-Nearest Neighbor
- TFs:
-
transcription factors
- ELN:
-
European LeukemiaNet
- ITD:
-
internal tandem duplication
- CI:
-
confidence interval
References
Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35:934–46.
Szer J. The prevalent predicament of relapsed acute myeloid leukemia. Hematol Am Soc Hematol Educ Progr. 2012;2012:43–8.
Burnett AK, Hills RK, Milligan DW, Goldstone AH, Prentice AG, McMullin MF, Duncombe A, Gibson B, Wheatley K. Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: results of the MRC AML12 trial. J Clin Oncol. 2010;28:586–95.
O’Donnell MR, Tallman MS, Abboud CN, Altman JK, Appelbaum FR, Arber DA, Bhatt V, Bixby D, Blum W, Coutre SE, De Lima M, Fathi AT, Fiorella M, Foran JM, Gore SD, Hall AC, Kropf P, Lancet J, Maness LJ, Marcucci G, Martin MG, Moore JO, Olin R, Peker D, Pollyea DA, Pratz K, Ravandi F, Shami PJ, Stone RM, Strickland SA, Wang ES, Wieduwilt M, Gregory K, Ogba N. Acute myeloid leukemia, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw. 2017;15:926–57.
Cancer Genome Atlas Research, N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ, Jr. Laird PW, Baty JD, Fulton LL, Fulton R, Heath SE, Kalicki-Veizer J, Kandoth C, Klco JM, Koboldt DC, Kanchi KL, Kulkarni S, Lamprecht TL, Larson DE, Lin L, Lu C, McLellan MD, McMichael JF, Payton J, Schmidt H, Spencer DH, Tomasson MH, Wallis JW, Wartman LD, Watson MA, Welch J, Wendl MC, Ally A, Balasundaram M, Birol I, Butterfield Y, Chiu R, Chu A, Chuah E, Chun HJ, Corbett R, Dhalla N, Guin R, He A, Hirst C, Hirst M, Holt RA, Jones S, Karsan A, Lee D, Li HI, Marra MA, Mayo M, Moore RA, Mungall K, Parker J, Pleasance E, Plettner P, Schein J, Stoll D, Swanson L, Tam A, Thiessen N, Varhol R, Wye N, Zhao Y, Gabriel S, Getz G, Sougnez C, Zou L, Leiserson MD, Vandin F, Wu HT, Applebaum F, Baylin SB, Akbani R, Broom BM, Chen K, Motter TC, Nguyen K, Weinstein JN, Zhang N, Ferguson ML, Adams C, Black A, Bowen J, Gastier-Foster J, Grossman T, Lichtenberg T, Wise L, Davidsen T, Demchok JA, Shaw KR, Sheth M, Sofia HJ, Yang L, Downing JR, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, Metzeler KH, Schwind S, Wu YZ, Kohlschmidt J, Pettenati MJ, Heerema NA, Block AW, Patil SR, Baer MR, Kolitz JE, Moore JO, Carroll AJ, Stone RM, Larson RA, Bloomfield CD. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012;30:4515–23.
Komanduri KV, Levine RL. Diagnosis and therapy of acute myeloid leukemia in the era of molecular risk stratification. Annu Rev Med. 2016;67:59–72.
Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Gorlich D, Schneider S, Konstandin NP, Dufour A, Braundl K, Ksienzyk B, Zellmeier E, Hartmann L, Greif PA, Fiegl M, Subklewe M, Bohlander SK, Krug U, Faldum A, Berdel WE, Wormann B, Buchner T, Hiddemann W, Braess J, Spiekermann K, Group AS. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128:686–98.
Theis F, Corbacioglu A, Gaidzik VI, Paschka P, Weber D, Bullinger L, Heuser M, Ganser A, Thol F, Schlegelberger B, Gohring G, Kohne CH, Germing U, Brossart P, Horst HA, Haase D, Gotze K, Ringhoffer M, Fiedler W, Nachbaur D, Kindler T, Held G, Lubbert M, Wattad M, Salih HR, Krauter J, Dohner H, Schlenk RF, Dohner K. Clinical impact of GATA2 mutations in acute myeloid leukemia patients harboring CEBPA mutations: a study of the AML study group. Leukemia. 2016;30:2248–50.
Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312.
Dohner H, Dolnik A, Tang L, Seymour JF, Minden MD, Stone RM, Del Castillo TB, Al-Ali HK, Santini V, Vyas P, Beach CL, MacBeth KJ, Skikne BS, Songer S, Tu N, Bullinger L, Dombret H. Cytogenetics and gene mutations influence survival in older patients with acute myeloid leukemia treated with azacitidine or conventional care. Leukemia. 2018;32:2546–57.
Jawhar M, Dohner K, Kreil S, Schwaab J, Shoumariyeh K, Meggendorfer M, Span LLF, Fuhrmann S, Naumann N, Horny HP, Sotlar K, Kubuschok B, von Bubnoff N, Spiekermann K, Heuser M, Metzgeroth G, Fabarius A, Klein S, Hofmann WK, Kluin-Nelemans HC, Haferlach T, Dohner H, Cross NCP, Sperr WR, Valent P, Reiter A. KIT D816 mutated/CBF-negative acute myeloid leukemia: a poor-risk subtype associated with systemic mastocytosis. Leukemia. 2019;33:1124–34.
Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrozek K, Nicolet D, Whitman SP, Wu YZ, Schwind S, Powell BL, Carter TH, Wetzler M, Moore JO, Kolitz JE, Baer MR, Carroll AJ, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN favorable genetic category. Blood. 2011;118:6920–9.
Morita K, Kantarjian HM, Wang F, Yan Y, Bueso-Ramos C, Sasaki K, Issa GC, Wang S, Jorgensen J, Song X, Zhang J, Tippen S, Thornton R, Coyle M, Little L, Gumbs C, Pemmaraju N, Daver N, DiNardo CD, Konopleva M, Andreeff M, Ravandi F, Cortes JE, Kadia T, Jabbour E, Garcia-Manero G, Patel KP, Futreal PA, Takahashi K. Clearance of somatic mutations at remission and the risk of relapse in acute myeloid leukemia. J Clin Oncol. 2018;36:1788–97.
Prochazka KT, Pregartner G, Rucker FG, Heitzer E, Pabst G, Wolfler A, Zebisch A, Berghold A, Dohner K, Sill H. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica. 2019;104:516–23.
Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Lowenberg B, Bloomfield CD, European L. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74.
Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–48.
Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, Usui N, Kanamori H, Kiguchi T, Imai K, Uike N, Kimura F, Kitamura K, Nakaseko C, Onizuka M, Takeshita A, Ishida F, Suzushima H, Kato Y, Miwa H, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Ogawa S, Naoe T. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28:1586–95.
Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, van Waalwijk Barjesteh, van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–28.
Stirewalt DL, Meshinchi S, Kopecky KJ, Fan W, Pogosova-Agadjanyan EL, Engel JH, Cronk MR, Dorcy KS, McQuary AR, Hockenbery D, Wood B, Heimfeld S, Radich JP. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer. 2008;47:8–20.
Tomasson MH, Xiang Z, Walgren R, Zhao Y, Kasai Y, Miner T, Ries RE, Lubman O, Fremont DH, McLellan MD, Payton JE, Westervelt P, DiPersio JF, Link DC, Walter MJ, Graubert TA, Watson M, Baty J, Heath S, Shannon WD, Nagarajan R, Bloomfield CD, Mardis ER, Wilson RK, Ley TJ. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood. 2008;111:4797–808.
Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–91.
Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CA, Wouters BJ, van der Poel-van de Luytgaarde SC, Damm F, Krauter J, Ganser A, Schlenk RF, Lowenberg B, Delwel R, Dohner H, Valk PJ, Dohner K. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 2011;117:2469–75.
Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI, Paschka P, Held G, von Lilienfeld-Toal M, Lubbert M, Frohling S, Zenz T, Krauter J, Schlegelberger B, Ganser A, Lichter P, Dohner K, Dohner H. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–21.
Cucchi DGJ, Denys B, Kaspers GJL, Janssen J, Ossenkoppele GJ, de Haas V, Zwaan CM, van den Heuvel-Eibrink MM, Philippe J, Csikos T, Kwidama Z, de Moerloose B, de Bont E, Lissenberg-Witte BI, Zweegman S, Verwer F, Vandepoele K, Schuurhuis GJ, Sonneveld E, Cloos J. RNA-based FLT3-ITD allelic ratio is associated with outcome and ex vivo response to FLT3 inhibitors in pediatric AML. Blood. 2018;131:2485–9.
Saygin C, Hirsch C, Przychodzen B, Sekeres MA, Hamilton BK, Kalaycio M, Carraway HE, Gerds AT, Mukherjee S, Nazha A, Sobecks R, Goebel C, Abounader D, Maciejewski JP, Advani AS. Mutations in DNMT3A, U2AF1, and EZH2 identify intermediate-risk acute myeloid leukemia patients with poor outcome after CR26. Blood Cancer J. 2018;8:4.
Gaidzik VI, Teleanu V, Papaemmanuil E, Weber D, Paschka P, Hahn J, Wallrabenstein T, Kolbinger B, Kohne CH, Horst HA, Brossart P, Held G, Kundgen A, Ringhoffer M, Gotze K, Rummel M, Gerstung M, Campbell P, Kraus JM, Kestler HA, Thol F, Heuser M, Schlegelberger B, Ganser A, Bullinger L, Schlenk RF, Dohner K, Dohner H. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016;30:2160–8.
Li Z, Herold T, He C, Valk PJ, Chen P, Jurinovic V, Mansmann U, Radmacher MD, Maharry KS, Sun M, Yang X, Huang H, Jiang X, Sauerland MC, Buchner T, Hiddemann W, Elkahloun A, Neilly MB, Zhang Y, Larson RA, Le Beau MM, Caligiuri MA, Dohner K, Bullinger L, Liu PP, Delwel R, Marcucci G, Lowenberg B, Bloomfield CD, Rowley JD, Bohlander SK, Chen J. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol. 2013;31:1172–81.
Negrotto S, Ng KP, Jankowska AM, Bodo J, Gopalan B, Guinta K, Mulloy JC, Hsi E, Maciejewski J, Saunthararajah Y. CpG methylation patterns and decitabine treatment response in acute myeloid leukemia cells and normal hematopoietic precursors. Leukemia. 2012;26:244–54.
Marchwicka A, Marcinkowska E. Regulation of expression of CEBP genes by variably expressed vitamin D receptor and retinoic acid receptor alpha in human acute myeloid leukemia cell lines. Int J Mol Sci. 2018;19:E1918.
Maruffi M, Sposto R, Oberley MJ, Kysh L, Orgel E. Therapy for children and adults with mixed phenotype acute leukemia: a systematic review and meta-analysis. Leukemia. 2018;32:1515–28.
Churpek JE, Bresnick EH. Transcription factor mutations as a cause of familial myeloid neoplasms. J Clin Invest. 2019;129:476–88.
Takei H, Kobayashi SS. Targeting transcription factors in acute myeloid leukemia. Int J Hematol. 2019;109:28–34.
Wada T, Akagi T. Role of the Leucine Zipper Domain of CCAAT/enhancer binding protein-Epsilon (C/EBPepsilon) in neutrophil-specific granule deficiency. Crit Rev Immunol. 2016;36:349–58.
Priam P, Krasteva V, Rousseau P, D’Angelo G, Gaboury L, Sauvageau G, Lessard JA. SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPvarepsilon dependent mechanism. Nat Genet. 2017;49:753–64.
Gombart AF, Shiohara M, Kwok SH, Agematsu K, Komiyama A, Koeffler HP. Neutrophil-specific granule deficiency: homozygous recessive inheritance of a frameshift mutation in the gene encoding transcription factor CCAAT/enhancer binding protein–epsilon. Blood. 2001;97:2561–7.
Acknowledgements
Not applicable.
Funding
This work was supported by Grants from the National Natural Science Foundation Grants of China (31801147), the Shanghai Sailing Program (18YF1410800), the startup fund for young teachers of Shanghai Jiao Tong University (17X100040083), the grants from the Key Research Area Grant 2016YFA0501703 of the Ministry of Science and Technology of China, the National Natural Science Foundation of China (Contract no. 61832019, 61503244), the State Key Lab of Microbial Metabolism and Joint Research Funds for Medical and Engineering and Scientific Research at Shanghai Jiao Tong University (YG2017ZD14). The computations were partially performed at the Center for High Performance Computing, Shanghai Jiao Tong University.
Author information
Authors and Affiliations
Contributions
KNL analyzed data and drafted the article. FZ and YXD performed experiments. FZ and DQW designed the project, analyzed data, revised the drafted article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Table S1.
qPCR primer sequences.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, K., Du, Y., Wei, DQ. et al. CEBPE expression is an independent prognostic factor for acute myeloid leukemia. J Transl Med 17, 188 (2019). https://doi.org/10.1186/s12967-019-1944-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-019-1944-x