
Abstract
Background
Neurogenin3 (Ngn3) and neurogenic differentiation 1 (NeuroD1), two crucial transcriptional factors involved in human diabetes (OMIM: 601724) and islet development, have been previously found to directly target to the E-boxes of the insulinoma-associated 2 (Insm2) gene promoter, thereby activating the expression of Insm2 in insulin-secretion cells. However, little is known about the function of Insm2 in pancreatic islets and glucose metabolisms.
Methods
Homozygous Insm2−/− mice were generated by using the CRISPR-Cas9 method. Glucose-stimulated insulin secretion and islet morphology were analyzed by ELISA and immunostainings. Expression levels of Insm2-associated molecules were measured using quantitative RT-PCR and Western blots.
Results
Fasting blood glucose levels of Insm2−/− mice were higher than wild-type counterparts. Insm2−/− mice also showed reduction in glucose tolerance and insulin/C-peptide levels when compared to the wild-type mice. RT-PCR and Western blot analysis revealed that expression of Insm1 was significantly increased in Insm2−/− mice, suggesting a compensatory response of the homolog gene Insm1. Similarly, transcriptional levels of Ngn3 and NeuroD1 were also increased in Insm2−/− mice. Moreover, Insm2−/− female mice showed a significantly decreased reproductive capacity.
Conclusions
Our findings suggest that Insm2 is important in glucose-stimulated insulin secretion and is involved in the development pathway of neuroendocrine tissues which are regulated by the transcription factors Ngn3, NeuroD1 and Insm1.
Similar content being viewed by others

Background
Pancreatic endocrine cells play a key role in glucose metabolism by regulating the synthesis and secretions of islet hormones, such as insulin and glucagon. Mature islets contain five different types of endocrine cells. Ngn3, a proendocrine marker, is essential for islet cell development [1]. Biallelic mutations in Ngn3 have been identified to cause permanent neonatal diabetes [2, 3]. NeuroD1, which is directly activated by Ngn3, participates in the maintenance and differentiation of mature islet cells [4]. Heterozygous mutations in NeuroD1 have been identified to be associated with maturity-onset diabetes (OMIM: 601724) and type 2 diabetes mellitus [5, 6]. Although much efforts have been made to decipher the mechanism of differentiation and maintenance of pancreatic endocrine cells, the molecular basis of how Ngn3 and NeuroD1 function in the islets remains under investigation.
Previously we reported the isolation of two homologous genes, INSM1 (a.k.a. IA-1) [7] and INSM2 (INSM transcriptional repressor 2; a.k.a. IA-6) [8], from human pancreatic islet cells. Two additional genes, named PTPRN and PTPRN2 (a.k.a., IA-2 and IA-2beta), were also isolated from human islet cells, which have been turned out to be major autoantigens in patients with type 1 diabetes and are involved in the secretion of hormones and neurotransmitters [9, 10]. The encoded C2H2 zinc-finger proteins by INSM1 and INSM2 belong to the SNAIL/GFI1/INSM transcriptional repressor superfamily (i.e., SNAG-domain mediated transcription factors) that plays an important role in the developmental processes and molecular pathogenesis of various human conditions [11, 12]. For instance, mutations in GFI1 and GFI1B have been identified in patients with neutropenia (OMIM: 600871) [13] and affected members of a family with platelet-type bleeding disorder-17 (OMIM: 604383) [14, 15], respectively.
Functional studies demonstrated both INSM1 and INSM2 are direct targets of Ngn3 and NeuroD1 [8, 16]. During the embryonic development, the Insm1−/− knockout mice showed a significantly reduced insulin production, suggesting a key role for Insm1 in islet β-cell differentiation and maturation [17, 18]. Recent studies demonstrated that Insm1 cooperates with Neurod1 and Foxa2 to maintain mature pancreatic beta-cell function [19]. Insm2 also was found to be expressed in developing endocrine cells peaking from E11.5 to E13.5 and activated in Ngn3/NeuroD1-transduced pancreatic epithelial duct cells [8]. Numerous clinical studies showed that Insm1 is a sensitive and highly specific marker for various tumors, such as neuroendocrine differentiation in primary lung neoplasms [20], Merkel cell carcinoma [21], small cell carcinoma of the prostate [22], head and neck tumors [23] as well as insulinoma.
To explore further the biological and pathophysiological role of Insm2 in pancreatic islets, we developed an Insm2−/− mouse using the CRSPR-Cas9 technique, and measured blood glucose, insulin and C-peptide levels of the Insm2−/− mice [9]. Furthermore, we measured transcriptional levels of the transcription factors Ngn3, NeuroD1 as well as the homolog gene Insm1 in Insm2−/− mice.
Methods
Generation of Insm2-deficient mice
The pST1374-NLS-flag-linker-Cas9 (#44758) and pUC57-sgR (#51132) plasmids were commercially obtained (http://www.addgene.org/). Single guide RNA (sgRNA) was designed to target the coding region of the mouse Insm2 gene (GenBank accession number: NM_020287.2). Mouse embryos were injected with Cas9 and sgRNA and then transferred into pseudo-pregnant mice to give birth to chimeric mice. Homozygous Insm2−/− global knockout mice were obtained by breeding of Insm2+/− mice and wild-type C57BL/6J. All experimental procedures were carried out in accordance with protocols approved by the NIDCR Animal Care and Use Committee (#12-641) and the Animal Usage Committee of Wenzhou Medical University (#31571301). Statistical analysis was performed using the Student’s t test for unpaired comparisons.
Genotyping and quantitative real-time PCR
Primers for genotyping (forward: 5′-gtctcagctataaagcgggc-3′, reverse: 5′-aattggaacggatacaggga-3′) were located on flanking sides of the deleted region of Insm2. PCR products were genotyped by Sanger sequencing. Total RNAs were isolated from mouse brain and pancreas tissues using TRIZOL reagents (Life Technology, Rockville, MD, USA). The concentration of RNAs was determined by NANO 2000 (Thermal Scientific, Amarillo, TX, USA). Real-time PCR was performed using Mx3000p System with a two-step cycling program (Stratagene): 95 for 10 min, 40 cycles of 95 °C for 1 min and 60 °C for 30 s. Primers included for following genes: Insm2 (forward: 5′-gctccggcagctcctacc-3′; reverse: 5′-ggctcctccggtgaggatt-3′), Insm1 (5′-ggagtacgctgacccgttcg-3′; 5′-aagaccttggcgcactctgg-3′), Ngn3 (5′-aagagcgagttggcatgagcaag-3′; 5′-gcgttggtccgctatgcgcag-3′), NeuroD1 (5′-cttggccaagaactacatctgg-3′; 5′-ggagtagggatgcaccgggaa-3′), mouse Insulin 1 gene (5′-ccttagtgaccagctataatcagag-3′; 5′-cacttgtgggtcctccactt-3′) and Insulin 2 gene (5′-tcagcaagcaggaagcctatcttcc-3′; 5′- cacttgtgggtcctccactt-3′) [24]. Gapdh PCR product was used as a loading control.
Western blots and antibodies
Proteins were isolated from mouse brain tissues for Western blots with following antibodies: anti-INSM2 rabbit antibody (1:800) [8], INSM1 rabbit antibody (1:1000, ab170876, Abcam, Shanghai, China), and anti-GAPDH antibody (1:1000, Santa Cruz (6C5), USA). The bands were visualized using enhanced chemiluminescence detection reagents (Applygen Technologies, Beijing, China) and detected by the FluorChem E imaging system (Cell Biosciences, Santa Clara, USA).
Histological staining and immunostaining
Genotyped mice aged 10–23 weeks were sacrificed for isolation of pancreatic tissues, which were then treated in 10% formalin buffer for paraffin embedding. 5 μm thick sections on slides were stained with hematoxylin and eosin (H&E) for further analysis. Monoclonal anti-insulin antibody (1:250, Clone K36AC10, Sigma, USA) and anti-INSM2 rabbit antibody (1: 100) were also tested for immunostainings of mouse pancreas tissues [8].
Intraperitoneal glucose tolerance test
Targeted mice aged 10–23 weeks were fasted for 14 h, followed by glucose injection (2 g/kg body weight via intraperitoneal injection). Venous blood was drawn from the tail vein at 0 (just before the injection), 15, 30, 60, 90 and 120 min after the injection of glucose. Blood glucose level was measured using a portable glucometer (Bayer, Elkhart, IN) as described previously [25].
Insulin secretion
Glucose (3 g/kg body weight) was injected intraperitoneally into male and female mice aged 10–23 weeks. Insulin levels were measured using rat Insulin Ultrasensitive ELISA Kit and mouse C-peptide ELISA Kit (ALPCO, Salem, NH, USA). Blood samples were collected from the mouse orbital vein and centrifuged at the speed of 3000 rpm for 10 min at 4 °C and stored at − 80 °C until use.
Results
Characterization of Insm2 −/− mice
The offsprings of the CRISP-Cas9 mice were genotyped by PCR and Sanger sequencing, which showed a 25-bp deletion in coding region of the sole exon of the Insm2 gene (Fig. 1a). This deletion resulted in a frameshift mutation p.S36fsX67 (FWGPPASHLRRTTPSGARVAATAPAPAPRGRRAPSCAARSWSAACA LRSLRSPSPAPPP SAPRRPRP)*, thereby disrupting the reading frame of Insm2. Wild-type (Insm2+/+), heterozygous (Insm2+/−), and homozygous (Insm2−/−) mice were identified by tail DNA PCR and Sanger sequencing with the allele-specific primers (Fig. 1b).

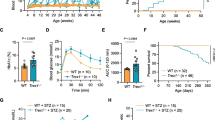
Genotyping and characterization of Insm2−/− mice. a Targeted deletion of 25-bp (c.108_132delAGTCCCCGACCGGGCTCCTCCGGTG) in the coding sequence region (CDS) of Insm2 (GenBank acc. no., NM_020287.2). b Sanger chromatograms show the target sequence regions in wild-type (WT), heterozygous (HET), and homozygous (HO) Insm2−/− mice. c Quantitative RT-PCR analysis indicates that Insm2 mRNA is markedly decreased in the Insm2−/− mouse brain as compared to normal controls. d Western blotting shows no detectable INSM2 protein in Insm2−/− mouse brain tissues. e, f H&E staining reveals no significant structure alterations in Insm2−/− mouse pancreatic islets
Quantitative RT-PCR analysis of mRNAs extracted from the brain tissues of Insm2−/− mice showed that the mRNA levels of Insm2 were dramatically decreased, probably due to unstable Insm2 mRNAs with the frameshifted deletion, compared to the wild type mice (Fig. 1c). The mRNA level of Insm2 in pancreas tissue was also significantly decreased as compared to the wild type mice (not shown). Western blot analysis, using rabbit anti-INSM2 sera to measure protein expression level in mouse brain extracts, clearly showed no visible expression of INSM2 protein in the homozygous Insm2−/− as compared to Insm2+/+ mice (Fig. 1d).
Phenotype analysis
Physical examination of the Insm2−/− mice revealed no gross development abnormalities. Body weight of Insm2−/− male or female mice at 8, 16, and 24 weeks of age did not differ from the Insm2+/+ mice. However, fertility of Insm2−/− female mice was significantly reduced. Specifically, after mating with Insm2−/− male mice, of the 16 mating pairs of Insm2−/− female mice, only five (31.25%) gave birth but with a smaller litter size (4 vs. 9 in normal mice, P < 0.0001). In a sharp contrast, all Insm2−/− male mice had normal reproductive capacity when mating with Insm2+/– female mice. However, the underlying pathogenesis of the significantly reduced fertility observed here needs further investigation.
Pancreatic tissue analysis using H&E staining in more than 30 slides did not show any significant alterations in terms of the number and size of islets in Insm2−/− mice (Fig. 1e, f). However, fasting blood glucose levels of the Insm2−/− male mice at 16 weeks of age (Fig. 2a) and female mice at 16- and 24 weeks of ages were elevated (Fig. 2b) as compared to the Insm2+/+ mice.

Analysis of glucose tolerance, insulin secretion, and Insm2 homologous or regulatory genes. a, b Fasting glucose levels are elevated at 24 weeks Insm2−/− male and at 16 and 24 weeks old Insm2−/− female mice. Values are presented in mean ± SEM from eight animals per group in three separate experiments (**P < 0.01; ***P < 0.001). c, d Glucose tolerance tests in male and female Insm2+/+ and Insm2−/− mice. After overnight fasting, d-glucose (2 g/kg body weight) was injected intraperitoneally, and blood glucose levels were measured at different times as indicated. Values are presented in mean ± SEM from 14 mice per group in three separate experiments (**P < 0.01; ***P < 0.001). e, f Blood insulin and C-peptide levels in response to intraperitoneal glucose in male and female Insm2−/− mice aged 16–24 weeks were measured and compared to Insm2+/+ mice at 15 min. Blood samples were drawn from the tail vein using heparinized capillary tubes before and after glucose injection
Glucose tolerance tests also showed significantly elevated glucose levels in both male and female Insm2−/− mice (Fig. 2c, d). ELISA analysis showed that serum insulin levels in these mice were significantly lower than that in the Insm2+/+ mice (i.e., 0.56 ng/ml vs 1.37 ng/ml in male mice and 0.57 ng/ml vs 1.31 ng/ml in female mice, respectively, Fig. 2e). Similarly, basal blood C-peptide levels in the Insm2−/− mice also were significantly decreased compared to the wild type mice (0.14 ng/ml vs 0.31 ng/ml in males and 0.11 ng/ml vs 0.23 ng/ml in female, respectively, Fig. 2f).
In fact, quantitative RT-PCR using primers of either the mouse insulin 1 gene (Ins1) or insulin 2 gene (Ins2) showed that their transcriptional levels in pancreatic tissues of Insm2−/− mice were only 60–65% of the wild-type mice (Fig. 3a, b). Immunostainings using anti-mouse insulin antibody also showed reduced insulin expression in Insm2−/− mouse islet beta cells compared to the wild-type islet cells (Fig. 3c).

Insulin expression levels in pancreatic islet cells. a, b Expression of the Ins1 and Ins2 gene is decreased in the pancreas tissues of the Insm2−/− mice compared to the wildtype controls. c Immunostainings show lower expression of insulin in the islet cells of Insm2−/− mice compared to the wildtype controls
Expression levels of Insm2-associated regulators
Since Insm2-encoded protein shares significant homology as well as similar protein structure with Insm1 [8], this suggested that the expression of Insm1 might be increased as a compensatory response to the deficiency of Insm2. RT-PCR analysis of the Insm2−/− brain tissue mRNAs showed a 2.5-fold increase of Insm1 expression as compared to that of wild type mice (Fig. 4a). Western blotting with the anti-INSM1 antibody showed that the INSM1 protein levels in Insm2−/− mice were 2 to 3-folds higher than that of wild type mice (Fig. 4b). Islet tissue was not examined because there is no detectable expression of Insm1/Ngn3/NeuroD1 in adult mouse islet cells.

Expression levels of Insm1, Ngn3, and NeuroD1. a, b Insm1 mRNA and its encoded protein levels extracted from brain tissues and measured by quantitative RT-PCR and Western blots. c, d Transcriptional levels of Ngn3 and NeuroD1 extracted from brains and analyzed by qRT-PCR. The results represent the average of three independent experiments in triplicates. Data are means ± SEM. *P < 0.01; **P < 0.001; ***P < 0.0001
Given that the transcription factors Ngn3 and NeuroD1 were previously found to promote Insm2 expression via binding to the proximal E-boxes of the Insm2 promoter [8], we examined their transcriptional response to the deletion of Insm2. RT-PCR analysis showed that expression levels of Ngn3 and NeuroD1 mRNAs were significantly increased in the Insm2−/− mice compared to that of Insm2+/+ mice (Fig. 4c, d). Taken together, our findings suggest that the Ngn3 and NeuroD1 will elicit the expression of Insm2 in a number of neuroendocrine tissues.
Discussion
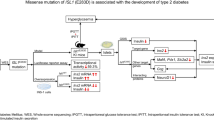
The human INSM2 gene was mapped to chromosome 14q13.2 and showed a broader spatial-temporal expression pattern in neuroendocrine, heart, and liver tissues [8], whereas the INSM1 expression was restricted in neuroendocrine tissues and tumors [26, 27]. INSM2 belongs to the SNAI1 (OMIM: 604238)/GFI1 (OMIM: 600871)/INSM1 (OMIM: 600010) family of transcriptional repressors (OMIM: 614027). Intriguingly, the mouse Insm2 gene was found to be methylated and silenced in liver tumors of SV40 T antigen transgenic mice [28]. In contrast to the earlier death of Insm1−/− mice with disrupted development of insulin-producing beta cells [17], the phenotypes we observed in Insm2−/− mice were relatively subtle. Although no obvious diabetic symptoms were observed in Insm2−/− mice, elevated fasting glucose, elevated glucose levels in glucose tolerance test, lower serum insulin secretion, and lower C-peptide levels are indicators that deletion of Insm2 affects pancreatic islet cell functions (Fig. 5). Understanding of the regulatory mechanisms of critical transcriptional factors, such as Ngn3, NeuroD1, and Insm1, in islet development is important for making insulin-producing cells for prospective stem cell therapy to treat diabetes [29,30,31].

Interactions of several crucial transcriptional factors in pancreatic islet insulin-producing cells. Arrows in black indicate direct or regulatory interactions; arrows in red point to the phenotypic alterations due to deleterious mutations previously identified in affected individuals or specific knockout of the corresponding gene in mice
In depth analyses of Insm1, Ngn3, and NeuroD1 in Insm2−/− mice revealed that their increased expressions could be a compensatory response to the deficiency of Insm2 because both Insm1 and Insm2 share a common activation pathway through Ngn3 and NeuroD1 in islet cells [8]. Previous study also demonstrated that endogenous Insm2 expression was activated in Ngn3/NeuroD1-transduced pancreatic epithelial duct cells [8]. Therefore, the phenotype severity in pancreatic islets of Insm2−/− mice could be lessened through the increased Insm1 expression. In fact, similar to the expression pattern of Insm2 in brain [8], Ngn3 and NeuroD1 are also abundantly expressed in brain tissues, especially in developing and adult hippocampus [32, 33]. Therefore, the increased expression levels of Ngn3 and NeuroD1 in brain tissues should be a compensatory response to the deletion of Insm2 in mice. As previously reported, Ngn3 and NeuroD1 positively regulate Insm2 expression through the binding of the E-box of the Insm2 promoter [8]. It would be interesting to search additional insulin-pathway related phenotypes in the double knockout mice harboring homozygous Insm2−/− and heterozygous Insm1+/− alleles.
Furthermore, genetic analysis by GWAS in a cohort of Africa Americans with type 2 diabetes revealed a significant association of the disease to a SNP (rs1952392, MAF = 0.0188; P < 0.001) at the proximal promoter region of the human INSM2 gene (personal communication) [34]. A previous genetic study also showed an association of microsatellite polymorphisms at 14q13.2 with type 2 diabetes mellitus in Latvian and Finnish populations [35]. Interestingly, INSM2 was recently found to upregulate the expression of the ultraconserved (uc) RNA uc.372, which in turn suppresses the maturation of miR-195/miR-4668 to regulate expression of genes related to lipid synthesis and uptake in liver [19]. Given the risk of non-alcoholic fatty liver disease in type 2 diabetes mellitus (see recent meta-analysis) [36, 37], it is worth noting whether the deletion of Insm2 in mice affects the hepatic lipid accumulation and thus attributes to the risk of type 2 diabetes.
Based on the important role of the INSM2 gene in neuroendocrine tissues as well as the preliminary clinical findings, potential mutations and altered expressions of the INSM2 gene in neuroendocrine-associated disorders, such as diabetes and infertility, can be screened utilizing next-generation sequencing technologies. In a cancer-related study, Insm2 was found to be methylated and silenced in liver tumors of SV40 T antigen transgenic mice [28], suggesting its role in tumor inhibition. Among 64 different mammalian cell lines, intriguingly, INSM2 appears to be exclusively expressed in SH-SY5Y cells (a neuroblastoma cell line, Human Protein Atlas, https://www.proteinatlas.org). Further studies are needed to learn whether or not INSM2 plays a role in tumorigenesis and functions as a prognostic marker in neuroblastoma.
Conclusion
In conclusion, we demonstrated that deletion of Insm2 affects glucose-stimulated insulin secretion and glucose tolerance. We also showed that Insm2 is involved in the Ngn3/NeuroD1 pathway in neuroendocrine tissues. Whether Insm2−/− mice induce islet cell differentiation and proliferation defects and/or develop type 2 diabetes, further experiments under more challenging conditions, such as high-fat and high-sugar diet [38, 39], are required to test this hypothesis.
References
Rukstalis JM, Habener JF. Neurogenin3: a master regulator of pancreatic islet differentiation and regeneration. Islets. 2009;1(3):177–84.
Rubio-Cabezas O, et al. Permanent neonatal diabetes and enteric anendocrinosis associated with biallelic mutations in NEUROG3. Diabetes. 2011;60(4):1349–53.
Hancili S, et al. A novel NEUROG3 mutation in neonatal diabetes associated with a neuro-intestinal syndrome. Pediatr Diabetes. 2018;19(3):381–7.
Mastracci TL, et al. Regulation of Neurod1 contributes to the lineage potential of Neurogenin3+ endocrine precursor cells in the pancreas. PLoS Genet. 2013;9(2):e1003278.
Malecki MT, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23(3):323–8.
Horikawa Y, et al. NEUROD1-deficient diabetes (MODY6): identification of the first cases in Japanese and the clinical features. Pediatr Diabetes. 2018;19(2):236–42.
Xie J, et al. The zinc-finger transcription factor INSM1 is expressed during embryo development and interacts with the Cbl-associated protein. Genomics. 2002;80(1):54–61.
Cai T, et al. Expression of insulinoma-associated 2 (INSM2) in pancreatic islet cells is regulated by the transcription factors Ngn3 and NeuroD1. Endocrinology. 2011;152(5):1961–9.
Cai T, Notkins AL. Pathophysiologic changes in IA-2/IA-2beta null mice are secondary to alterations in the secretion of hormones and neurotransmitters. Acta Diabetol. 2016;53(1):7–12.
Cai T, et al. Deletion of Ia-2 and/or Ia-2beta in mice decreases insulin secretion by reducing the number of dense core vesicles. Diabetologia. 2011;54(9):2347–57.
Lan MS, Breslin MB. Structure, expression, and biological function of INSM1 transcription factor in neuroendocrine differentiation. Faseb J. 2009;23(7):2024–33.
Chiang C, Ayyanathan K. Snail/Gfi-1 (SNAG) family zinc finger proteins in transcription regulation, chromatin dynamics, cell signaling, development, and disease. Cytokine Growth Factor Rev. 2013;24(2):123–31.
Person RE, et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet. 2003;34(3):308–12.
Monteferrario D, et al. A dominant-negative GFI1B mutation in the gray platelet syndrome. N Engl J Med. 2014;370(3):245–53.
Anguita E, et al. Transcription factor GFI1B in health and disease. Front Oncol. 2017;7:54.
Mellitzer G, et al. IA1 is NGN3-dependent and essential for differentiation of the endocrine pancreas. EMBO J. 2006;25(6):1344–52.
Gierl MS, et al. The zinc-finger factor Insm1 (IA-1) is essential for the development of pancreatic beta cells and intestinal endocrine cells. Genes Dev. 2006;20(17):2465–78.
Zhang T, et al. Insulinoma-associated antigen-1 zinc-finger transcription factor promotes pancreatic duct cell trans-differentiation. Endocrinology. 2010;151(5):2030–9.
Guo J, et al. Ultraconserved element uc.372 drives hepatic lipid accumulation by suppressing miR-195/miR4668 maturation. Nat Commun. 2018;9(1):612.
Mukhopadhyay S, et al. Insulinoma-associated protein 1 (INSM1) is a sensitive and highly specific marker of neuroendocrine differentiation in primary lung neoplasms: an immunohistochemical study of 345 cases, including 292 whole-tissue sections. Mod Pathol. 2018. https://doi.org/10.1038/s41379-018-0122-7.
Lilo MT, et al. INSM1 is more sensitive and interpretable than conventional immunohistochemical stains used to diagnose merkel cell carcinoma. Am J Surg Pathol. 2018;42(11):1541–8.
Xue W, et al. Insulinoma-associated protein 1 is a novel sensitive and specific marker for small cell carcinoma of the prostate. Hum Pathol. 2018;79:151–9.
Rooper LM, et al. INSM1 is a sensitive and specific marker of neuroendocrine differentiation in head and neck tumors. Am J Surg Pathol. 2018;42(5):665–71.
Panda AC, et al. Novel splice variant of mouse insulin2 mRNA: implications for insulin expression. FEBS Lett. 2010;584(6):1169–73.
Saeki K, et al. Targeted disruption of the protein tyrosine phosphatase-like molecule IA-2 results in alterations in glucose tolerance tests and insulin secretion. Diabetes. 2002;51(6):1842–50.
Chen C, et al. Sonic hedgehog signaling pathway promotes INSM1 transcription factor in neuroendocrine lung cancer. Cell Signal. 2018;46:83–91.
Goto Y, et al. A novel human insulinoma-associated cDNA, IA-1, encodes a protein with “zinc-finger” DNA-binding motifs. J Biol Chem. 1992;267(21):15252–7.
Tateno M, et al. Identification of a novel member of the snail/Gfi-1 repressor family, mlt 1, which is methylated and silenced in liver tumors of SV40 T antigen transgenic mice. Cancer Res. 2001;61(3):1144–53.
Bhonde RR, et al. Making surrogate beta-cells from mesenchymal stromal cells: perspectives and future endeavors. Int J Biochem Cell Biol. 2014;46:90–102.
Cheng CW, et al. Fasting-mimicking diet promotes Ngn3-driven beta-cell regeneration to reverse diabetes. Cell. 2017;168(5):775–788 e12.
Masoud MS, et al. Translating the potential of stem cells for diabetes mellitus: challenges and opportunities. Curr Stem Cell Res Ther. 2017;12(8):611–23.
Simon-Areces J, et al. Neurogenin 3 cellular and subcellular localization in the developing and adult hippocampus. J Comp Neurol. 2010;518(10):1814–24.
Brulet R, et al. Mice with conditional NeuroD1 knockout display reduced aberrant hippocampal neurogenesis but no change in epileptic seizures. Exp Neurol. 2017;293:190–8.
Liang J, et al. Single-trait and multi-trait genome-wide association analyses identify novel loci for blood pressure in African-ancestry populations. PLoS Genet. 2017;13(5):e1006728.
Sjakste T, et al. Association of microsatellite polymorphisms of the human 14q13.2 region with type 2 diabetes mellitus in Latvian and Finnish populations. Ann Hum Genet. 2007;71(Pt 6):772–6.
Atan NA, et al. Type 2 diabetes mellitus and non-alcoholic fatty liver disease: a systematic review and meta-analysis. Gastroenterol Hepatol Bed Bench. 2017;10(Suppl1):S1–7.
Mantovani A, et al. Nonalcoholic fatty liver disease and risk of incident type 2 diabetes: a meta-analysis. Diabetes Care. 2018;41(2):372–82.
Gonzalez-Mariscal I, et al. Absence of cannabinoid 1 receptor in beta cells protects against high-fat/high-sugar diet-induced beta cell dysfunction and inflammation in murine islets. Diabetologia. 2018;61(6):1470–83.
Bergeron V, et al. Deletion of protein kinase D1 in pancreatic beta-cells impairs insulin secretion in high-fat diet-fed mice. Diabetes. 2018;67(1):71–7.
Authors’ contributions
TC, ZSS, and ALN conceived the project. LW, YW and KS performed the experiments. LW, BX, CW, GC, GNC, ZSS and TC contributed to the acquisition, analysis and interpretation of the data. TC wrote the paper. All authors were involved in drafting the article or revising it and gave their approval for the final manuscript to be published. TC is the guarantor of this work and takes full responsibility for the content of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank the NIDCR Gene Targeting Facility for technique assistance.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This investigation was carried out according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All animal protocols were approved by the NIDCR Animal Care and Use Committee (#12-641) and the Animal Usage Committee of Wenzhou Medical University (#31571301).
Funding
This study was supported by Zhejiang Province Nature Science Research funding (LY13C070004) and Science and Technology Supporting Program, Ministry of Science and Technology of China (M2014BAI02B01 to ZSS). This research was supported in part by the Intramural Research Program of the NIDCR, NIH.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, L., Sun, Z.S., Xiang, B. et al. Targeted deletion of Insm2 in mice result in reduced insulin secretion and glucose intolerance. J Transl Med 16, 297 (2018). https://doi.org/10.1186/s12967-018-1665-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-018-1665-6