
Abstract
Background
Carcinoma of unknown primary (CUP) is a metastatic epithelial malignancy in the absence of an identifiable primary tumour. Prognosis for patients with CUP is poor because treatment options are generally limited to broad spectrum chemotherapy. A shift towards personalised cancer management based on mutation profiling offers the possibility of new treatment paradigms. This study has explored whether actionable, oncogenic driver mutations are present in CUP that have potential to better inform treatment decisions.
Methods
Carcinoma of unknown primary cases (n = 21) were selected and DNA was isolated from formalin-fixed paraffin embedded sections prior to amplification and sequencing. Two distinct yet complementary targeted gene panels were used to assess variants in up to 76 known cancer-related genes for the identification of biologically relevant and actionable mutations.
Results
Variants were detected in 17/21 cases (81%) of which 11 (52%) were potentially actionable with drugs currently approved for use in known primary cancer types or undergoing clinical trials. The most common variants detected were in TP53 (47%), KRAS (12%), MET (12%) and MYC (12%). Differences at the molecular level were seen between common CUP histological subtypes. CUP adenocarcinomas and poorly differentiated carcinomas harboured the highest frequency of variants in genes involved in signal transduction pathways (e.g. MET, EGFR, HRAS, KRAS, and BRAF). In contrast, squamous cell carcinoma exhibited a higher frequency of variants in cell cycle control and DNA repair genes (e.g. TP53, CDKN2A and MLH1).
Conclusion
Taken together, mutations in biologically relevant genes were detected in the vast majority of CUP tumours, of which half provided a potentially novel treatment option not generally considered in CUP.
Similar content being viewed by others
Background
Carcinoma of unknown primary (CUP) is classified as any type of metastatic epithelial tumour where, following extensive clinical history, physical examination, radiological studies and histopathological (including immunohistochemical) investigations, no primary site can be identified [1]. CUP is the eighth most common cancer diagnosed and is the fourth most common cause of cancer-related death in both sexes worldwide [2,3,4]. The overall age-standardised incidence of CUP ranges between 4 and 9 cases per 100,000 people annually worldwide [3,4,5]. CUP is an aggressive, highly heterogeneous disease with a variable biology. There is no standard treatment and broad-spectrum chemotherapy (e.g. paclitaxel, carboplatin) is generally used [6, 7]. Clinical trials are difficult to perform because of the heterogeneity in tumour types within CUP. As a result, CUP has a poor prognosis with a median survival of less than 12 months and 5-year survival only 14% [8]. There is an urgent need to improve treatment and prolong survival for patients with CUP. The new era of personalised medicine through the use of next-generation sequencing (NGS) technologies offers such an opportunity based on the identification of targeted therapies [9].
Next-generation sequencing technology allows whole-genome sequencing, whole-exome sequencing, or mutation analysis with specific (“targeted”) panels of genes. Many genomic studies are limited by the availability of poor quality formalin-fixed paraffin-embedded (FFPE) tissue which make up the main source of sample preparation and storage in routine diagnostics [10, 11]. As such, for NGS to be useful in the clinical setting, small amounts of FFPE tissue from variable sources needs to be successfully evaluated. With advancements in knowledge about important targetable mutations across various cancers, targeted sequencing approaches allow selective screening of known druggable targets using relatively small amounts of DNA input. Focusing on specific regions of interest through targeted sequencing leads to a greater depth of coverage, increasing the confidence of identifying low-level variants in cancer samples [10, 12].
Recent studies have demonstrated that precision medicine may play a crucial role in optimising treatment for patients with malignant disease [13,14,15,16]. Furthermore studies have shown improved overall survival in patients with advanced cancers who have received genotype-matched targeted therapies [15, 17,18,19]. This approach, if applied to CUP, may lead to personalised approaches to treatment by targeting tumour-specific somatic variants. This could become highly relevant in CUP, as genomic studies have revealed that these metastatic tumours commonly have a complex mutational landscape [3, 20]. In the present study, we assessed the genomic profile of CUP to provide insight into the genetic makeup of these tumours, and determined whether there were potentially actionable targets by performing targeted NGS.
Methods
Patients and samples
Formalin-fixed paraffin-embedded specimens from 21 cases of CUP were included in the study. Cases were selected based on the archival histopathological report and subsequent review by experienced pathologists. Cases were diagnosed as CUP following review of clinicopathological details and a complete histopathological work-up [including immunohistochemistry (IHC)] revealed a metastatic lesion without a specific site of origin. The clinicopathological details of cases are outlined in Table 1. Ethical approval for this study was obtained from the Sir Charles Gardiner Hospital Human Research Ethics Committee (SCGH HREC Number 2014-025) and the Department of Health WA Human Research Ethics Committee (DOH HREC Number 2015-40).
Tumour cell isolation from FFPE specimens
The tumour content of the FFPE samples was assessed on haematoxylin and eosin (H&E) stained slides. Each case was processed in three different ways depending on the percentage of tumour content. For cases with > 70% tumour content (n = 11), 2 × 10 µM sections were collected in microcentrifuge tubes (Axygen, Australia) for DNA extraction. For cases with < 70% tumour content (n = 21), matched H&E slides were used to guide either macro or micro-dissection of tumour rich regions. Specifically, for cases with 50–70% tumour content (n = 6), macro-dissection was performed to obtain > 90% tumour rich material for DNA extraction. In brief, sections were mounted onto double positive charged slides (Hurst Scientific, Australia) and air-dried for 1 h. A sterile scalpel blade was used to scrape the tumour-rich tissue off the unstained slides into LoBind Eppendorf tubes (Eppendorf, Australia) for DNA extraction. Micro-dissection was performed on cases with < 50% tumour content (n = 4) using 4 × 8 µM sections. Briefly, each slide was de-paraffinised, stained and dehydrated through a series of xylene/ethanol washes. Tumour rich areas were micro-dissected using an ArcturusXT laser capture micro-dissection (LCM) instrument (ThermoFisher Scientific, USA) with CapSure Macro LCM caps (ThermoFisher Scientific, USA). Each section was selectively captured by focal melting of the caps polymer membrane with a small to medium sized infrared (IR) pulse (range between 45 and 82 mW) and an ultra-violet (UV) laser beam (range between 5 and 35 mW/ms). The power and duration of the laser were adjusted each time a new section of tissue was selected.
DNA extraction
For all three tissue processing methods, genomic DNA was isolated using a GeneRead DNA FFPE kit (Qiagen, Germany) according to manufacturer’s instructions, with minor modifications. In brief, once the tissue from each specimen was de-paraffinised, samples were incubated at 56 °C for 1 h with proteinase K, then again at 90 °C for 1 h to partially reverse formaldehyde modification of nucleic acids. Following tissue digestion, the samples were treated with Uracil-DNA-Glycosylase (UNG; Qiagen, Germany) for the specific removal of artificially induced uracils introduced by fixation and embedding. Silica-gel membrane spin columns were then used to bind the DNA, facilitating the removal of any contaminants. Purified DNA was then eluted from the spin column using nuclease-free water to minimise salt carry-over. The DNA template was quantitatively assessed using a Qubit Fluorometer (ThermoFisher Scientific, USA) and Qubit dsDNA HS Assay Kit (Life Technologies) according to manufacturer’s instructions.
Library preparation
Two different targeted panels were used to generate sequencing libraries from 10 ng of DNA. The Oncomine Focus Assay (OFA) and Cancer Hotspot v2 (CHPv2) panel (Thermofisher Scientific, USA) are specifically optimised for detection of up to 50 genes commonly implicated in human cancers and relevant to targeted treatment of solid tumours (Additional file 1: Table S1). Library preparation for each sample differed slightly between the two panels. Several OFA libraries and one CHPv2 assay library were prepared using Ion PGM Select and Ion AmpliSeq reagents (ThermoFisher Scientific, USA) respectively, according to the manufacturer’s instructions. For both panels, unique barcode adapters 1–32 (Ion PGM Select Adaptors; ThermoFisher Scientific, USA) were ligated to the amplicons and subsequently purified to ensure each individual sample had a unique ID. The final amplicon libraries were then amplified, purified and equalised to ~ 100 pM using AMPure beads (Ion PGM Select Library Equaliser; ThermoFisher Scientific, USA).
Emulsion PCR and semiconductor sequencing
Uniquely barcoded library samples were pooled for sequencing on either an Ion 318 chip (for the OFA) or an Ion PI v3 chip (for the CHPv2 panel). Each pool was clonally amplified onto Ion Sphere Particles (TMPL ISP; Ion OneTouch Select Template Reagents, ThermoFisher Scientific, USA) by emulsion PCR. For the OFA this was carried out using a One Touch 2 System (ThermoFisher Scientific, USA), and each pool was manually loaded onto an Ion 318 Select chip. In contrast, for the CHPv2 assay, the Ion Chef System (ThermoFisher Scientific, USA) was used for fully automated template preparation and Ion PI v3 chip loading. Single-end sequence analysis was carried out either on the Ion PGM (OFA) or Ion Proton Sequencer (Cancer Hotpsot v2) (ThermoFisher Scientific) for 200-base-read-sequencing.
Coverage and data analysis
Raw data from both panels was collected, processed and trimmed using the Ion Torrent platform-specific software. Removal of polyclonal and low-quality reads, as well as 3′ quality trimming of reads was performed using TorrentSuite v4.6 (ThermoFisher Scientific, USA). Reads were aligned to the reference genome (human genome hg19) and Ion Reporter v5.0 software package (ThermoFisher Scientific, USA) was used to detect and annotate variants for both panels. Specifically, the ‘Oncomine Focus Panel v1—DNA—Single Sample’ automatic workflow in Ion Reporter was used to identify and annotate the copy number variants from the OFA. This workflow has preconfigured parameter settings for copy number calling, including a 5% confidence interval and CNV ploidy ≥ gain of 2 over normal. Alternatively, the ‘CHPv2—Annotate variants single sample’ automatic workflow was used to identify and annotate variants from the CHPv2. Ion Reporter was also used to identify a subset of variants previously reported in publicly available databases (namely, the 1000 Genomes Project). The resulting annotated variant data detected with the OFA were further analysed using the Ion Torrent Oncomine Knowledgebase Reporter v2.0.3 (ThermoFisher Scientific, USA), providing details of the clinically relevant targeted therapies currently on the market or undergoing clinical trials for the associated Oncomine variants detected.
Results
The cohort comprised 67% (14/21) female and 33% (7/21) male patients with a median age at diagnosis of 71 (range 36–91). Tumours were classified into four histological subgroups: squamous cell carcinoma (SCC; 38%), poorly differentiated (PD) carcinoma (29%), adenocarcinoma (24%), and neuroendocrine carcinoma (9%). The most common anatomical site where CUP presented was in lymph nodes (38%) (Table 1).
DNA was extracted from tissue sections of 11 cases, macro-dissected tissue from 6 cases and LCM tissue from 4 cases (Table 2). The concentration of DNA collected ranged between 0.5 and 176 ng/µL which varied depending on tumour cell isolation type. The average concentration of DNA extracted from tissue sections was 45 ng/µL (range 3–176 ng/µL); from macro-dissected tumour tissue it was 84 ng/µL (range 36–157 ng/µL); and from LCM cases it was 3 ng/µL (range 0.5–10 ng/µL). Although DNA concentration varied by sampling method, there was a sufficient amount of DNA extracted for sequencing with both panels, with the exception of LCM cases which were re-captured and re-extracted for the second sequencing panel.
With the OFA, all samples were sequenced to an average mean depth of 1396 reads per nucleotide position within the ~ 27 kb target region, whereas with the CHPv2 panel, the samples were sequenced to an average mean depth of 9105. The distribution of reads across the 269 amplicons was consistent among samples from both panels, with an average uniformity coverage of 98.25% and 98.81% for the OFA and CHPv2 panels respectively. Approximately 94% of the sequence reads from both panels were mapped to the targeted gene regions (aligned to human genome reference 19), demonstrating the high specificity of the amplicon-based amplification method. Successful sequencing of the samples was measured by using a minimum of 250,000 reads with a quality score of AQ20, a sequencing coverage of 1000×, as well as a variant frequency of at least 5% in a background of wildtype alleles.
From the combined results of both panels, a total of 608 variants were detected in 41 genes (10 genes for which no variants were identified in), with an average of 14 variants detected per case (range 0–26 variants). Stringent variant detection criteria were used to identify likely somatic variants. First, we filtered out variants with a minor allele frequency (MAF) > 5% (according to the 1000 Genomes Project) and synonymous exonic mutations. After frequency filtering, a total of 26 variants remained, of which 14 were known gain-of-function variants; 10 actionable single nucleotide variants, and 4 actionable copy number variants (Fig. 1); the remaining 12 variants were known cancer related hotspot variants. The allele ratios at which the hotspot variants occurred at ranged from 0.08 to 0.79. Six samples had reportable variants within regions covered by both panels, and these variants were detected at similar allele frequencies, emphasising the validity of the variants detected (Table 2).

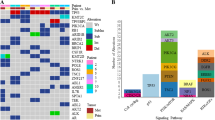
Frequency of clinically relevant gene targets. Total number of druggable single nucleotide variants (SNVs) (dark grey), druggable amplifications (light grey) and hotspot mutations (black) identified across the CUP cases (total number of variants n = 26). BRAF, MET, KRAS and HRAS were the most common druggable SNVs; MYC was the most common druggable copy number variant (CNV) detected; and TP53 was the most common hotspot gene detected
Variants were identified in cases with DNA isolated by all three methods. Specifically, variants were identified in 9 cases with DNA extracted from tissue section, in 5 cases from macro-dissected tissue and in 3 cases with DNA extracted from LCM tissue (Table 2). There was no correlation between the number of variants and allele frequencies detected and the different percentages of tumour cellularity (50–100%) or the sampling methods used (tissue sections versus macro-dissection versus LCM) (Table 2).
Following the filtering process, biologically relevant and therapeutically actionable variants were identified in 81% (17/21) of the cohort. The mean number of variants was 1 per case (range 0–4) (Table 2 and Fig. 2). Hotspot mutations were identified in 59% (10/17) of cases. These were in genes associated with various cell signalling, cell cycle control and DNA repair pathways. The gene with the most common hotspot mutations was TP53 identified in 47% (8/17) of cases, and the most common copy number variation (CNV) was MYC amplification identified in 12% (2/17) of cases. Correlation with the tumour pathology showed that the CUP cases with morphology of adenocarcinoma and PD carcinoma had the largest number of cell signalling pathway variants (EGFR, MET, JAK3, KRAS, HRAS, BRAF, PIK3CA, PTPN11 and APC). In contrast, SCC tumours showed a higher number of variants in cell cycle regulation genes (TP53 and CDKN2A). There were no associations with other clinicopathological parameters (i.e. age, gender or anatomical site of presentation).

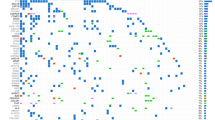
Integrated gene map of the variant data from the Oncomine Focus Assay panels (OFA) and Cancer Hotspot v2 (CHPv2) panel (ThermoFisher Scientific, USA) identified in the CUP cohort. This gene map shows the pattern of case-specific, concurrent and mutually exclusive mutations identified in the CUP cases. Each column represents an individual case and each row denotes a specific gene assigned to one of two functional categories (black—hotspot variant; grey—druggable target). The list of genes includes only those found to be aberrantly expressed within the cohort, and represents a subset of the total number of genes interrogated by both panels. No variants were identified in four cases and were not included in the figure
Potentially actionable targets were detected in 52% (11/21) of cases. The most common variants were in the KRAS, HRAS and BRAF genes. Although the mean number of actionable variants was 1 per case (range 1–3), one sample presented with 3 possible druggable targets (CCND1, FGFR1 and MYC) (Table 2). When the number of variants were normalised by site (i.e. \(\frac{{ {\text{number of cases with variants presenting in a site}}}}{\text{total number of CUP cases }} \times {\text{number of variants detected in that site}}\)), the highest number of variants were detected in lymph nodes (all lymph nodes) (0.7 variants), followed by brain (0.2 variants). When the number of variants were normalised by histological morphology, the subtype associated with the greatest number of variants was SCC (1.8 variants), followed by PD carcinoma (0.8 variants). When the number of variants were normalised by gender, the highest number of variants were detected in female CUP patients (3.8 variants) compared with male CUP patients (0.4 variants).
Discussion
A recent shift in treatment focus towards personalising cancer management has encouraged mutational profiling [21,22,23,24]. This is the first study to utilise two complementary gene panels to identify variants in CUP that are aligned to known oncogenic driver mutations and approved therapies with published evidence of targeted on-the-market drugs or therapeutics currently in clinical trial. We have identified relevant variants in 81% (n = 17) of CUP cases, of which over half (65%) were to potentially actionable targets. This is a highly significant result as the identification of variants for which there is a known therapeutic agent available may offer a potential new and “personalised” treatment approach for patients with CUP. This is important considering the limited therapeutic benefit current CUP patients receive with generic chemotherapy. Support for this concept is offered by several small studies of patients with CUP that have reported durable treatment responses with the use of mutation matched (e.g. EGFR, KIT, MET and BRAF) targeted therapies [18, 19, 25,26,27,28]. Currently approved existing therapeutic agents are available for 2 of the mutations detected (BRAF V600E, ERBB2 S310F), whilst therapeutic agents for the other gain-of-function variants detected (i.e. CCND1, FGFR1, MYC, PIK3CA, MET, KRAS and HRAS) are currently being investigated in active, ongoing clinical trials. A large proportion of the variants detected in this study are known to be associated with various signal transduction pathways, apoptotic regulation and cell cycle progression. These results are promising as the majority of available targeted drugs target act through one of these pathways, which are commonly altered in many cancers [29,30,31].
The most commonly mutated gene identified in this study was TP53 (47%, 8/17) with 9 different non-synonymous coding region variants. This is unsurprising, because TP53 mutations have been described to contribute to metastatic progression in multiple cancer types, supporting the high percentage of TP53 variants reported in CUP [32]. Other common variants detected in this cohort were observed in genes involved in the activation and regulation of key signal transduction pathways (i.e. BRAF, HRAS and KRAS). This is the first study to report HRAS variants and codon 12 KRAS variants (G12A) in CUP [22, 23]. Although activating mutations in codons 12 and 13 of KRAS are the most commonly occurring isoforms in human cancers, variants were limited to codon 12 in the present cohort [33, 34]. KRAS codon 12 mutations confer a more aggressive tumour phenotype with stronger transforming abilities compared with codon 13 mutations [33, 35,36,37]. The detection of codon 12 mutations in this cohort is consistent with the highly aggressive nature of CUP tumours. Furthermore, characterising the mutational status of KRAS has become clinically relevant in some malignancies, because the presence of a KRAS mutation is known to confer poor response to some tyrosine kinase inhibitors (e.g. EGFR inhibitors) [38, 39]. Although there is currently no therapeutic agent to target and inhibit mutant KRAS activity, a recent case study reported a partial response in a CUP patient treated with a MEK inhibitor (trametinib) following the detection of a KRAS G12D mutation [28, 40]. This encourages the detection of KRAS as a possible druggable target in CUP.
Activating BRAF V600E mutations were identified, in keeping with other reports [6, 21, 22, 24]. This offers the prospect of treatment with BRAF inhibitors (e.g. vemurafenib and dabrafenib) for CUP with BRAF V600E mutations. This has been exemplified in a case of CUP with a BRAF V600E inguinal nodal mass mutation that showed successful treatment (complete clinical response) with BRAF targeted therapy (vemurafenib) coupled with immunotherapy (ipilimumab) [25]. Mutations in MET and ERBB2 were detected in 3 cases, providing the possibility of targeting these receptor tyrosine kinases (RTK). Targeted MET therapy (crizotinib) has been used with success in CUP patients in combination with HER2 targeted therapy (trastuzumab). The current success of HER2 and MET targeted therapies in advanced and/or metastatic malignancies, and the recent success of trastuzumab and crizotinib demonstrating a positive response in a HER2 and MET-mutant CUP tumour, provides evidence for future evaluation of these genes as druggable targets in patients with CUP.
Our results support those of other CUP studies which have demonstrated the value of sequencing techniques for the identification of actionable targets [6, 14, 21, 23]. These studies similarly identified actionable variants in 75% (n = 16), 85% (n = 200), 55% (n = 87) and 30% (n = 150) of CUP cases, including activating variants in core mitogenic and cell growth pathways. Comparable to our study, the most common clinically relevant alterations detected by these studies included ERBB2, EGFR, KRAS, PIK3CA and BRAF. Previous studies detected additional actionable variants in other genes involved in cell proliferation, cell cycle progression and apoptotic regulation (AKT1, FGFR3, JAK2, BRCA1, PTEN, RICTOR, NF1, CDKN2A, CTNNB1 and MCL1). Variants in these genes may have not been detected in the present study due to the use of specific gene panels, one of which enables a more translatable definition of actionable ability, which is not taken into consideration by prior CUP sequencing studies [6, 21, 23, 24].
This is the first study to have successfully compared different tissue processing techniques (i.e. tissue sections versus macro-dissection versus LCM) for the enrichment of tumour cells from CUP tissue with the subsequent detection of actionable targets by targeted NGS. Variants were successfully identified in cases with DNA isolated by all three methods, highlighting the use of low input DNA for accurate targeted sequencing. Previous studies have used hybridisation capture techniques that are optimal for samples possessing a high tumour burden, requiring larger concentrations of input DNA (minimum of 50 ng). The use of an amplicon-based approach (as used in this study) tolerates even lower concentrations of input DNA and is advantageous because it is amenable to sequencing low levels of enriched tumour cell populations (isolated by LCM). This is highly relevant in the context of CUP, because a large proportion of CUP cases have limited tissue available as they are generally core biopsies and fine needle aspirate FFPE cell blocks. Furthermore, amplicon targeted sequencing has a quicker turnaround processing time, leading to less expensive sequencing costs compared with hybridization capture methods, proving diagnostically feasible for routine molecular pathology laboratories [10, 12].
The use of two different but complementary NGS panels provided an internal, orthogonal method for validating the sequencing results, whereby several variants covered by both panels were similarly identified (Fig. 3). We demonstrated that variants detected in our study that were included in both the CHPv2 and OFA panels were 100% concordant. This confirms that the workflow and platform utilised in this study with either panel generated results that were reproductible, accurate and precise to each sample. Seven different variants covered by both sequencing panels were detected in six different CUP specimens. Specifically, this included common hotspot variants in PIK3CA, MET, HRAS, KRAS, and BRAF that were detected at similar allele frequencies (Table 2). Although 8 hotspot targets were identified by both panels (Fig. 3), other important hotspot variants commonly reported in cancer were found exclusively by either panel. For example, the detection of ERBB2 p.310F gain-of-function variant was only covered by the OFA, whilst TP53 and CDKN2A hotspot variants were only covered by the CHPv2 panel. It is important to note that additional clinically relevant copy number alterations were detected by the OFA panel only. This included the detection of amplifications (8 or more copies) in CCND1, FGFR1 and MYC across two different CUP cases.

Venn diagram representing the variants exclusively detected by each panel, and the overlap of variants detected by both panels. Five variants were only detected by the Oncomine Focus Assay (OFA); 13 variants were only detected by the Cancer Hotspot v2 panel (CHPv2); 8 variants were commonly detected by both panels
Although mutations in driver genes were identified in the vast majority of cases (81%, 17/21), there was no single common CUP-specific molecular profile. In fact, over 80% of cases harboured exclusive variants. This highlights the genetic heterogeneity of CUP and supports the theory that these are not a discrete group of malignancies. They have different clinico-pathological characteristics which may be modulated by distinct biological mechanisms differing at a molecular level [21, 41, 42]. In support of this, and despite the small number of cases analysed, we did identify some difference in variants between pathological subtypes. Adenocarcinoma and PD carcinoma more commonly showed variants in genes involved in signal transduction pathways (e.g. MET, JAK3, EGFR, HRAS, KRAS and BRAF), whereas alterations in cell cycle control and DNA repair pathway genes (e.g. TP53, CDKN2A and MLH1) were more commonly seen in SCC. This is in keeping with other reports assessing histological subtypes in a range of different cancers including CUP [21, 43,44,45]. This not only indicates biological differences but could suggest differences in therapeutic responses. However, further studies are needed to expand and elucidate the relationship between specific mutations in CUP presenting at differing anatomical sites and the efficacy of targeted drug activity, as it is well established that, for example, not all BRAF mutant malignancies respond to BRAF targeted therapies [46,47,48,49].
Conclusion
The poorly differentiated nature of CUP tumours and lack of specific antigen detection, prevents primary tissue of origin diagnoses in these patients. Without the identification of a primary origin site, treatment is restricted to generic chemotherapy with limited benefit. The detection of mutations across the majority of CUP cases included in this study highlights not only the genomic instability present in these tumours, but also offers the possibility of targeted therapies for a significant percentage of patients with CUP. The opportunity for alternative therapeutic options has the potential to improve the prognosis for CUP. Identification of actionable targets could prove useful in complementing routine diagnostic work-up and guiding therapeutic decisions for patients with CUP.
Abbreviations
- CNV:
-
copy number variation
- CHPv2:
-
cancer hotspot v2 panel
- CUP:
-
carcinoma of unknown primary
- DOH:
-
Department of Health
- FFPE:
-
formalin-fixed paraffin embedded
- H&E:
-
haematoxylin and eosin
- HREC:
-
Human Research Ethics Committee
- IHC:
-
immunohistochemistry
- IR:
-
infrared
- ISP:
-
ion sphere particles
- LCM:
-
laser capture micro-dissection
- MAF:
-
minor allele frequency
- NGS:
-
next-generation sequencing
- OFA:
-
oncomine focus assay
- PD:
-
poorly differentiated
- SCC:
-
squamous cell carcinoma
- SCGH:
-
Sir Charles Gardiner Hospital
- SNVs:
-
single nucleotide variants
- UNG:
-
uracil-DNA-glycosylase
- UV:
-
ultra-violet
- WA:
-
Western Australian
References
Fizazi K, Greco F, Pavlidis N, et al. Cancers of unknown primary site: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(suppl 5):v133–8.
Greco FA. Cancer of unknown primary site: still an entity, a biological mystery and a metastatic model. Nat Rev Cancer. 2013;14(1):3–4.
Pentheroudakis G, Briasoulis E, Pavlidis N. Cancer of unknown primary site: missing primary or missing biology? Oncologist. 2007;12(4):418–25.
Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379(9824):1428–35.
Pavlidis N, Khaled H, Gaafar R. A mini review on cancer of unknown primary site: a clinical puzzle for the oncologists. J Adv Res. 2015;6(3):375–82.
Pentheroudakis G, Kotteas E, Kotoula V, et al. Mutational profiling of the RAS, PI3K, MET and b-catenin pathways in cancer of unknown primary: a retrospective study of the Hellenic Cooperative Oncology Group. Clin Exp Metas. 2014;31(7):761–9.
Pentheroudakis G. CUP: looking for a missing primary site and its biology. Ann Oncol. 2012;23(Suppl 10):x278–81.
Australian Institute of Health and Welfare 2014. Cancer in Australia: an overview. Cancer series no 90. 2014; Cat. No. CAN 88: Canberra: AIHW.
Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31.
Chung M, Lin W, Dong L, et al. Tissue requirements and DNA quality control for clinical targeted next-generation sequencing of formalin-fixed, paraffin-embedded samples: a mini-review of practical issues. J Mol Genet Med. 2017;11(2):1000262.
Munchel S, Hoang Y, Zhao Y, et al. Targeted or whole genome sequencing of formalin fixed tissue samples: potential applications in cancer genomics. Oncotarget. 2015;6(28):25943–61.
Samorodnitsky E, Jewell BM, Hagopian R, et al. Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Hum Mutat. 2015;36(9):903–14.
Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–9.
Varghese AM, Arora A, Capanu M, et al. Clinical and molecular characterization of patients with cancer of unknown primary in the modern era. Ann Oncol. 2017;28(12):3015–21.
Tsimberidou A-M, Wen S, Hong DS, et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin Cancer Res. 2014;20(18):4827–36.
Stockley TL, Oza AM, Berman HK, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016;8:109.
Tsimberidou A-M, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18(22):6373–83.
Tan DSW, Montoya J, Ng QS, et al. Molecular profiling for druggable genetic abnormalities in carcinoma of unknown primary. J Clin Oncol. 2013;31(14):e237-e9.
Palma NA, Ali SM, O’Connor J, et al. Durable response to crizotinib in a MET-amplified, KRAS-mutated carcinoma of unknown primary. Case Rep Oncol. 2014;7(2):503–8.
Kamposioras K, Pentheroudakis G, Pavlidis N. Exploring the biology of cancer of unknown primary: breakthroughs and drawbacks. Eur J Clin Invest. 2013;43(5):491–500.
Ross JS, Wang K, Gay L, et al. Comprehensive genomic profiling of carcinoma of unknown primary site: new routes to targeted therapies. JAMA Oncol. 2015;1(1):40–9.
Gatalica Z, Millis SZ, Vranic S, et al. Comprehensive tumor profiling identifies numerous biomarkers of drug response in cancers of unknown primary site: analysis of 1806 cases. Oncotarget. 2014;5(23):12440–7.
Tothill RW, Li J, Mileshkin L, et al. Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J Pathol. 2013;231(4):413–23.
Löffler H, Pfarr N, Kriegsmann M, et al. Molecular driver alterations and their clinical relevance in cancer of unknown primary site. Oncotarget. 2016;7(28):44322–9.
Røe OD, Wahl SGF. The undifferentiated carcinoma that became a melanoma: re-biopsy of a cancer of an unknown primary site: a case report. J Med Case Rep. 2017;11(1):82.
Yamada T, Ohtsubo K, Ishikawa D, et al. Cancer of unknown primary site with epidermal growth factor receptor mutation for which gefitinib proved effective. Cancer Chemother. 2012;39(8):1291–4.
Herter-Sprie GS, Greulich H, Wong K-K. Activating mutations in ERBB2 and their impact on diagnostics and treatment. Front Oncol. 2013;3(86):1–10.
Kato S, Krishnamurthy N, Banks KC, et al. Utility of genomic analysis in circulating tumor DNA from patients with carcinoma of unknown primary. Can Res. 2017;77(16):4238–46.
Roberts P, Der C. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–310.
Yuan T, Cantley L. PI3 K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–510.
Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905.
Powell E, Piwnica-Worms D, Piwnica-Worms H. Contribution of p53 to metastasis. Cancer Discov. 2014;4(4):405–14.
Li W, Qiu T, Zhi W, et al. Colorectal carcinomas with KRAS codon 12 mutation are associated with more advanced tumor stages. BMC Cancer. 2015;15(1):340.
Jančík S, Drábek J, Radzioch D, et al. Clinical relevance of KRAS in human cancers. BioMed Res Int. 2010;2010:1–13 (Article ID 150960).
Imamura Y, Morikawa T, Liao X, et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF-wild-type colorectal cancers. Clin Cancer Res. 2012;18(17):4753–63.
Guerrero S, Casanova I, Farre L, et al. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000;60(23):6750–6.
Stolze B, Reinhart S, Bulllinger L, et al. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci Rep. 2015;5(8535):1–9.
Leonard DG, Bagg A, Caliendo AM, et al. Targeting EGRF signalling pathways in CRC. Molecular pathology in clinical practice, vol. 223. Berlin: Springer; 2007. p. 405–7.
Ludovini V, Bianconi F, Pistola L, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6(4):707–15.
Ross SJ, Revenko AS, Hanson LL, et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. 2017;9(394):eaal5253.
Vikeså J, Møller AKH, Kaczkowski B, et al. Cancers of unknown primary origin (CUP) are characterized by chromosomal instability (CIN) compared to metastasis of know origin. BMC Cancer. 2015;15(1):151.
Krämer A, Löffler H. Cancer of unknown primary. New York: Springer International Publishing; 2016.
Salem ME, Xiu J, Khan SA, et al. Comparative molecular analyses of esophageal cancer: adenocarcinoma vs. squamous cell carcinomas and impact on outcome. J Clin Oncol. 2016;34(15_suppl):4035.
Huang T, Li J, Zhang C, et al. Distinguishing lung adenocarcinoma from lung squamous cell carcinoma by two hypomethylated and three Hypermethylated genes: a meta-analysis. PLoS ONE. 2016;11(2):e0149088.
Kadota K, Yeh Y-C, D’Angelo SP, et al. Associations between mutations and histologic patterns of mucin in lung adenocarcinoma: invasive mucinous pattern and extracellular mucin are associated with KRAS mutation. Am J Surg Pathol. 2014;38(8):1118–27.
Cabanillas M, Patel A, Danysh B, et al. BRAF inhibitors: experience in thyroid cancer and general review of toxicity. Horm Cancer. 2015;6(1):21–36.
Planchard D, Kim TM, Mazieres J, et al. Dabrafenib in BRAF V600E–mutant advanced non-small cell lung cancer: an open-label, single arm, multicenter, phase 2 trial. Lancet Oncol. 2016;17(5):642–50.
Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–8.
Strickler JH, Wu C, Bekaii-Saab T. Targeting BRAF in metastatic colorectal cancer: maximizing molecular approaches. Cancer Treat Rev. 2017;2017(60):109–19.
Authors’ contributions
KM and WE conceived and developed the presented idea. CS helped supervise the project with KM and WE. BC carried out the experiments. BC analysed the results with support from RJNA. BC wrote the manuscript with support from KM, RJNA and WE. BD, NTH, and GS performed pathological evaluation and interpretation of all samples included in the study. All authors provided critical feedback and helped shape the research, analysis and manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Mr. Bob Mirzai, Dr. Belinda Guo and Ms. Lynne Wilson for their time and technical expertise.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Ethical approval for this study was obtained from the Sir Charles Gardiner Hospital Human Research Ethics Committee (HREC Number 2014-025) and the Department of Health Western Australian Human Research Ethics Committee (DOH WA HREC Number 2015-40).
Funding
Britt Clynick was supported by the Richard Walter Gibbon Medical Research Scholarship from the University of Western Australia, and a Ph.D. Top-Up Scholarship from the Cancer Council of Western Australia. This study was funded with department funds from PathWest Laboratory Medicine, Sir Charles Gairdner Hospital and the Translational Cancer Pathology Laboratory, University of Western Australia.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
Combined gene list of the Oncomine Focus Assay (OFA) and Cancer Hotspot v2 (CHPv2) panel (ThermoFisher Scientific).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Clynick, B., Dessauvagie, B., Sterrett, G. et al. Genetic characterisation of molecular targets in carcinoma of unknown primary. J Transl Med 16, 185 (2018). https://doi.org/10.1186/s12967-018-1564-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-018-1564-x