
Abstract
Background
Early life epigenetic programming influences adult health outcomes. Moreover, DNA methylation levels have been found to change more rapidly during the first years of life. Our aim was the identification and characterization of the CpG sites that are modified with time during the first years of life. We hypothesize that these DNA methylation changes would lead to the detection of genes that might be epigenetically modulated by environmental factors during early childhood and which, if disturbed, might contribute to susceptibility to diseases later in life.
Methods
The study of the DNA methylation pattern of 485577 CpG sites was performed on 30 blood samples from 15 subjects, collected both at birth and at 5 years old, using Illumina® Infinium 450 k array. To identify differentially methylated CpG (dmCpG) sites, the methylation status of each probe was examined using linear models and the Empirical Bayes Moderated t test implemented in the limma package of R/Bioconductor. Surogate variable analysis was used to account for batch effects.
Results
DNA methylation levels significantly changed from birth to 5 years of age in 6641 CpG sites. Of these, 36.79 % were hypermethylated and were associated with genes related mainly to developmental ontology terms, while 63.21 % were hypomethylated probes and associated with genes related to immune function.
Conclusions
Our results suggest that DNA methylation alterations with age during the first years of life might play a significant role in development and the regulation of leukocyte-specific functions. This supports the idea that blood leukocytes experience genome remodeling related to their interaction with environmental factors, underlining the importance of environmental exposures during the first years of life and suggesting that new strategies should be take into consideration for disease prevention.
Similar content being viewed by others
Background
DNA methylation is an epigenetic mechanism that regulates different genome functions, including gene expression, which may intervene in physiological events such as cell lineage determination, cell differentiation, cell maturation and tissue-specific gene expression [1, 2]. Much of a person’s epigenomic pattern is established during embryogenesis and early development of the fetus [3]. However, genomic DNA methylation is known to be sensitive to environmental stimuli and changes during lifetime and with aging [4]. Some epigenomic modifications over time are important in development, but others occur stochastically [5, 6]. These alterations in DNA methylation patterns have been suggested to account for many age-related diseases [7–10]. For instance, age-associated alterations in DNA methylation have been found to be involved in the initiation and progression of cancer and certain chronic diseases [11].
The relationship between DNA methylation levels and age has already been demonstrated [12–16]. In fact, the use of DNA methylation data has been proposed as a method of measuring biological aging and it is possible to predict the age of a tissue based on its methylation pattern at specific CpG sites [12, 13, 17–19]. However, most studies exploring age-associated DNA methylation changes have been carried out on adults and have focused on aspects such as cell senescence, longevity, cancer, stem cell functions and chronological age [18, 20–25]. Reports on DNA methylation patterns during early childhood are still scarce [26–30]. The characterization of DNA methylation patterns during the first years of life is an ongoing task, and data from longitudinal studies are more revealing. DNA methylation levels have been shown to change rapidly during early development, with more pronounced changes in the immediate postnatal years, while methylation levels at many sites tend to stabilize beyond age 7 [31].
Additionally, early life conditions can predispose the fetus to a range of adult health outcomes, and DNA methylation seems to play an important role in this process [32, 33]. For instance, the time immediately before and after birth may be a sensitive period related to programming cardiometabolic risk [34, 35]. Adult health outcomes are therefore determined not only by conventional risk factors experienced in adult life, but also by early life programming [36], which has been shown to be mediated by DNA methylation [37].
Due to the influence of early life epigenetic programming on health outcomes and the fact that DNA methylation levels seem to change more rapidly during the first years of life, the identification of CpG sites that are modified by age in infants would lead to the detection of genes that might be epigenetically modulated by environmental factors during early childhood. If disturbed, these might contribute to susceptibility to specific diseases later in life [38, 39].
Thus, the aims of this study were: (1) the identification of CpG sites with changes in DNA methylation levels measured longitudinally between cord blood samples and peripheral blood samples at 5 years after birth in a group of 15 children. Children who were small (SGA), appropriate (AGA), and large for gestational age (LGA), and normal weight or overweight/obese at 5 years old were included; and (2) the characterization of the genomic distribution and functional relationships of age-modified CpG sites during early childhood.
Results
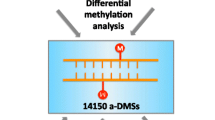
In order to identify DNA methylation changes with time during the first 5 years of life, the methylation patterns of 484103 CpG sites in cord and 5-year-old blood samples from the same 15 subjects were compared. We found that the DNA methylation levels of 6641 CpG sites changed as a function of age. Specifically, 2443 probes (36.79 %) were hypermethylated with time, corresponding to 1407 genes; and 4198 probes (63.21 %) corresponding to 2640 genes, were hypomethylated with time. Hierarchical clustering of all samples using the dmCpGs enabled each sample to be correctly classified into its corresponding age group (Fig. 1).

Clustered heatmap showing the methylation levels across all samples for the hyper- and hypo-methylated CpG sites. Methylation levels range from dark blue (no methylation) to light yellow (100 % methylated). Dendrograms were computed using Euclidean distance and a complete cluster agglomeration method. Both age groups are clustered correctly and methylation values have a homogeneous intra-group profile. YEAR5 stands for samples of DNA isolated from blood that was collected from individuals when they were 5 years-old. CORD stands for samples of DNA isolated from cord blood that was collected at birth from the same individuals
To characterize these dmCpG sites from a functional genomics point of view, we first determined their distribution within the different regions of the CpG islands [40]. Hypermethylated probes were enriched in CpG island shores, while hypomethylated CpG sites were enriched in non CpG islands (CGIs) (Pearson’s Chi squared test; p < 0.001, OR = 1.60 and p < 0.001, OR = 1.91, respectively) (Fig. 2a). In terms of genomic location, hypermethylated CpG sites were enriched mainly in exons (Pearson’s Chi squared test; p < 0.001, OR = 1.37), and hypomethylated probes in introns (Pearson’s Chi squared test; p < 0.001, OR = 1.41) (Fig. 2b). There was no statistically significant relationship between both hyper- and hypomethylated CpG sites and their respective distances to centromeres. On the other hand, only hypomethylated probes have a statistically significant change in their distance to telomeres (Fig. 3), but with a minimal effect size measured by Cliff’s Delta (D) (Wilcoxon test; p < 0.001, D = −0.0017), which seems to be non-biologically relevant.

Genomic characterization of the dmCpGs with time. a Stacked bar chart describing the proportion of CpG sites in the selected subsets of interest according to their CpG Island status and relative to the background Illumina® 450 k (All) proportions. Hypermethylated probes are enriched in CpG island shores while hypomethylated CpG sites are enriched in non CpG islands (CGIs) (Pearson’s Chi squared test; p < 0.001, OR = 1.60 and p < 0.001, OR = 1.91, respectively). b Stacked bar chart showing the proportion of selected CpG sites with respect to their genomic location and relative to the background (All). Hypermethylated CpG sites are enriched mainly in exons (Pearson’s Chi squared test; p < 0.001, OR = 1.37), and hypomethylated probes in introns (Pearson’s Chi squared test; p < 0.001, OR = 1.41)

Distance to centromere (a, b) and telomere (c, d) of differentially methylated probes. a, b Violin plots showing the distribution of the distance to centromeres for the hypermethylated (a) and hypomethylated (b) CpG sites (In) with respect to those sites belonging to the Illumina® 450 k microarray but not included in the corresponding subset of interest (Out). There is no statistically significant relationship between both hyper- and hypomethylated CpG sites and their distance to centromeres. c, d Violin plots showing the distribution of the distance to telomeres for the hypermethylated (c) and hypomethylated (d) CpG sites (In) with respect to those sites belonging to the Illumina® 450 k microarray but not included in the corresponding subset of interest (Out). Only hypomethylated probes have a statistically significant change in their distance to telomeres, but with a minimal effect size (Wilcoxon test; p < 0.001, D = −0.0017)
To distinguish the chromatin marks associated with the dmCpG sites showing changes over time, the DNA sequences identified in our study were analyzed against previously reported data on a collection of histone modifications and chromatin modifiers in 10 different cell types obtained from healthy individuals, (see “Methods” section), where hematologic cells are also represented. In the present study, we found statistically significant associations of hypermethylated CpGs with the repressive histone mark H3K27me3 and the polycomb group protein EZH2 in most differentiated ENCODE cell lines (Fisher's exact test; p < 0.05) (Fig. 4). This is in line with previously published data [41, 42]. Similarly, hypomethylated probes were here associated with regions enriched in H3K4me1 (Fisher's exact test; p < 0.05) (Fig. 4). This has been shown previously, where age-associated changes of DNA methylation were studied in differentiated and adult stem cells [41].

Heatmaps showing the association between the location of hypermethylated (a) and hypomethylated (b) CpG sites and enriched regions for several chromatin marks and cell lines. Chromatin marks peak location information for each cell line was extracted from the ENCODE BROAD Histone project information available at the UCSC Genome Browser. Associations between CpG site and chromatin mark peak locations were tested using a Fisher's exact test. P values were adjusted for multiple comparisons and only those falling under a 0.05 FDR threshold are shown as colored spots in the heatmap. The base-2 logarithm of the odds ratio (OR) was used as a measure of effect size. Associations with higher effect sizes are drawn in darker shades of red. There are statistically significant associations of hypermethylated (a) CpGs with the repressive histone mark H3K27me3 and the polycomb group protein EZH2 in most differentiated ENCODE cell lines. On the other hand, hypomethylated probes are associated with regions enriched in H3K4me1 (b)
The analysis of the gene ontology (GO) of the genes associated with the differentially methylated probes showed that both hyper- and hypomethylated genes were significantly enriched (FDR <0.05) in specific GO terms of biological processes, molecular functions and cellular components (Tables 1, 2). Hypermethylated genes were associated with biological processes related to development and cell adhesion, with molecular functions related to sequence-specific DNA binding, and cellular components such as dendrite or axon (Table 1). On the other hand, hypomethylated genes were associated with biological processes related to immune system regulation, with molecular functions related to antigen binding and intracellular signalling, and with cellular components related to the MHC protein complex and cytoskeleton (Table 2).
To analyze whether methylation changes with time were associated with birth weight or being overweight at 5 years of age, the comparative analysis was performed considering these two variables. No significant DNA methylation changes were found in relation to SGA group, or to being overweight at 5 years old (normal weight/overweight). It was not possible to determine whether there was an association between the SGA and being overweight at 5 years old, due to the fact that none of the individuals that were overweight at 5 years old belonged to the group of subjects who were SGA.
Discussion
The present longitudinal study focuses on the dynamics and the context of DNA methylation changes during early childhood in peripheral blood leukocytes. Data were compiled from 30 blood samples corresponding to 15 individuals at two time points (umbilical cord at birth, and 5 years after birth). It was shown that DNA methylation levels are modified as a function of age in 6641 CpG sites, most of them being hypomethylated. In hyper- and hypomethylated CpG sites, DNA methylation changes were significantly associated with intragenic regions, with exons and introns respectively. This implies that these DNA methylation changes are non-randomly distributed and specifically occur in discrete regions of the genome.
To further examine the features of the identified dmCpGs, the GO terms related to the genes associated with the differentially methylated probes were characterized. Both hyper- and hypomethylated sites were significantly enriched in specific GO terms of biological processes, molecular functions and cellular components. Specifically, it was found that genes with age-hypermethylated CpG sites were enriched in biological processes related to different tissue morphogenesis and development. This is in line with previous studies where increased DNA methylation was involved in silencing developmental genes [43].
Regarding hypomethylation with age, this study supports findings from previous reports where CpG sites, which are age-hypomethylated in the first 2 or 5 years following birth, are enriched in immune-related genes [26, 29]. Taking into account that a decrease in DNA methylation levels at promoter regions is known to enable gene expression [44], DNA methylation changes with age during the first years of life in human leukocytes may be closely associated with cell differentiation, and commitment to lymphoid and myeloid lineages [45]. These results thus denote that differences in DNA methylation associated with age may not only be triggered by stochastic DNA methylation changes [24, 46], but may also be related to the immune system function [26]. Additionally, age-hypomethylated CpGs sites were enriched in genes related to the MHC protein complex. This finding is in line with a recent study in samples from birth to 5 years old, where DNA methylation levels in class I and class II MHC molecules were found to decrease with age [26].
To explain the mechanisms that mediate the DNA methylation changes observed during aging, an increasing number of studies have focused on the identification of the factors determining the dynamics of DNA methylation. For instance, genes that are hypermethylated in blood during aging have been recently associated with the presence of bivalent chromatin domains in embryonic stem cells [21, 42, 47, 48], as well as with repressive histone marks (H3K27me3/H3K9me3) in differentiated cells [41, 42]. The results of the present study indicate the presence of the same repressive histone marks found in differentiated cells in the sequences that are hypermethylated with time during the first 5 years of life in leukocytes. This finding supports the notion that these repressive histone marks are related to DNA methylation gain during aging, independent of the type of cell or its potential, as previously described [41].
The present data also show a strong enrichment in the active chromatin mark H3K4me1 in age-hypomethylated sequences, which is in line with the data provided for hypomethylated sequences in MSCs and differentiated cells during aging [41]. This finding points towards this histone modification being of use as a cell-type-independent chromatin signature of DNA hypomethylation during aging. Additionally, a recent study indicated correspondence of H3K4me1 with enhancers [49], and an association between DNA hypomethylation within specific transposable elements and tissue-specific enhancer marks [50]. This suggests that H3K4me1-associated DNA hypomethylation could play a role in tissue-specific epigenetic gene regulation and the deregulation of gene expression during aging [51]. More studies, however, are required to clarify the mechanisms managing the methylation machinery to the age-modified loci during this time window.
A major issue in age-related DNA methylation studies is hematologic cell heterogeneity [52, 53], due to the fact that DNA methylation is usually measured in unfractionated blood. In order to adjust the model of analysis, a Surogate Variable Analysis (SVA)-based approach was applied, as described in Leek et al. [54] (see “Methods” section for details). This ensured that cell heterogeneity had a minimal impact on the blood DNA methylation data.
One limitation of the present study was that between-group differences with respect to variables such as SGA or being overweight at 5 years of age could not be adequately evaluated. This was due to the number of individuals belonging to each category analyzed being insufficient or too unequally distributed between groups to allow for comparisons and analysis. A larger number of individuals should be incorporated to a future study to address this issue.
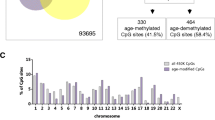
Several studies have explored DNA methylation patterns during early childhood [26–31]. For instance, blood samples from 3 months and 5 years of age were analyzed using the HumanMethylation450 BeadChip in the longitudinal study performed by Acevedo et al. [26]. This provided a total of 794 CpG sites where 330 CpG sites (41.5 %) were age-methylated and 464 CpG sites (58.4 %) were age-demethylated. When comparing with their results, it was found that 144 (43.64 %) of their hypermethylated and 208 (44.83 %) of their hypomethylated probes were identified by the present study (Fig. 5). Common GO terms (related to MHC protein complex) were also found when analyzing probes that were hypomethylated in both studies (Table 2). Furthermore, the tendency of a loss of methylation with age was corroborated in our set of samples. Additionally, other studies have identified different regions with changes in DNA methylation with age [27–31]. The possible differences in the CpG sites found in the literature could be explained due to disparities in the cell type (buccal epithelium, mononuclear cells, blood…), methodologies (HumanMethylation450 and/or 27 BeadChip), ages included in the study, methods of analysis, or purpose of the studies, for instance.

Venn diagram showing the intersections of the identified hyper- and hypo-methylated CpG sites and those described in Acevedo et al. [26]. There is a general consensus over the methylation direction of change between our results and the selected literature. Similarities and differences among the corresponding subsets are also shown
Methods
Selection of participants
Parents of newborns born at term (gestational age ≥37 weeks) in the General Hospital, University of Valencia, Spain, after uncomplicated pregnancies and in the absence of perinatal illness were randomly invited to participate in the study. Exclusion criteria were multiple gestations, cesarean section, and that parents were planning to move out of the area after delivery. Gestational age at birth was ascertained according to the method of Ballard et al. [55], and the general characteristics of gestation and delivery for each participant were obtained from routine obstetrical records. Subjects were divided according to birth weight (BW) and gestational age—SGA, <10th percentile for their sex; AGA, between 10th–90th percentile; and LGA, >90th percentile [56]. The subjects were followed-up at 5 years of age and all measurements were taken at birth and at 5 years. At birth all parents gave informed consent for their children to participate in the study, and the Committee for the Protection of Human Subjects of the Hospital General approved the study according to the Declaration of Helsinki.
Anthropometric parameters
At 5 years, body weight was recorded to the nearest 0.1 kg using a standard beam balance scale with the subjects wearing light indoor clothing and no shoes. Height was recorded to the nearest 0.5 cm using a standardized wall-mounted height board. Body mass index (BMI) and the corresponding standard deviation were calculated, with BMI being the weight in kilograms divided by the square of the height in meters. Subjects with a BMI ranging from the 85th to 95th percentile were defined as being overweight [57] while they were defined as obese when having a BMI above the 95th percentile [58].
In total, 15 subjects were enrolled in this study. Eight of the 15 subjects (53.3 %) (2 boys/6 girls) were SGA and the other seven (46.7 %) (4 boys/3 girls) were AGA or LGA. Four individuals were overweight/obese at 5 years old (BMI between 17 and 21.4), none of whom were SGA (Table 3).
Sample collection, DNA extraction, and quantification
Blood samples were collected from 15 subjects at the two testing times. First, cord blood samples were taken at birth, and second, peripheral venous blood samples were taken from each child during their fifth year of life. Genomic DNA was extracted with the RealPure kit (RealPure, REAL, Durviz, Ref: RBMEG01) and quantified with the Nanodrop-2000C Spectrophotometer. A DNA quality check was performed with Quant-iT PicoGreen dsDNA reagent.
Illumina® Infinium 450 k data preprocessing
The study of the DNA methylation pattern of 485577 CpG sites was performed using Illumina® Infinium 450 k array and the IDAT files from the microarray were processed further using the R/Bioconductor package minfi [59]. In order to adjust for the different probe design types present in the 450 k architecture, red and green signals from the IDAT files were corrected using the SWAN algorithm [60]. No background correction or control probe normalization was applied. Probes where at least two samples had detection p values over 0.01 were filtered out. In accordance with Du et al. [61], both Beta values and M values were computed and employed across the analysis pipeline. M values were used for all the statistical analyses, assuming homoscedasticity, while Beta values were mostly used for the intuitive interpretation and visualization of the results.
Batch effect correction
Surrogate Variable Analysis (SVA) [54] was employed to capture the heterogeneity of the underlying methylation data and to account for possible batch effects or confounding variables that might be of interest. Coefficients for the detected surrogate variables (SVs) were later added to the phenotypical data and included in the definition of a model in order to detect differentially methylated probes (DMPs). The R/Bioconductor package sva [62] implementation was used to estimate the number of SVs and their coefficients, using both age group (newborns/five year olds) and gender as covariates of interest, and only one intercept term as a null background model. Multidimensional scaling (MDS) was employed as a visualization tool whenever there was a need to illustrate the influence of possible confounders on the data.
White blood cell heterogeneity adjustment
Cellular heterogeneity is a main source of variation in Epigenomic studies [63]. Each cell type has a different Epigenomic profile, and variations of the different subpopulations can often be confounded with the phenotype of interest, resulting in a higher rate of both false positives and negatives. This is especially true when using whole blood as our main tissue, due to its highly variable subpopulation composition. This is especially relevant if we take into account the number of Epigenomic studies that have been published and that use whole blood as their main tissue.
One of the most common approaches to dealing with blood cellular heterogeneity is the Houseman method [53], which uses a methylation database of several, pure-lineage samples in order to compute an approximation to the real subpopulation percentages. Using this information, the method is able to adjust the original methylation dataset and generate a new one where the confounder influence has been removed. Several other methods have been proposed that expand on this concept, and some of them do not even require having a purified samples methylation database in advance [64].
However, there are also other approaches to the detection of confounding factors that do not need information about the Epigenomic profiles of the different cell subtypes. SVA [54], for example, is a general framework for the detection of structured variability patterns over the residuals of a previously fitted model using the main phenotype of interest. In general, SVA is not only able to capture the variation due to cellular heterogeneity, but also due to other factors, some of them possibly unknown to the researcher.
After an exploratory analysis of the data, it was decided to use SVA to capture the main confounding factors in our dataset, and to include them in our model. This resulted in a better fitted model than those based on the Houseman method alone, which suggested that in our case SVA is able to capture the cellular subpopulations proportion variations occurring in our data.
Detection of differentially methylated probes
Significant methylation of a probe was determined by the moderated t test implemented in the R/Bioconductor package limma [65]. A linear model, with methylation level as response and all the combinations of age group, birth weight and overweight at 5 years as the main covariate of interest, was fitted to the methylation data. Surrogate Variables generated using SVA and information regarding the gender and pair ID of the samples was also included in the model definition. Contrasts were then defined as the linear combinations of the different values the main covariate of interest could take, in order to represent the different questions arising from the model design. Each contrast generated a coefficient and P value for each probe. P values were corrected for multiple testing using the Benjamini-Hochberg method for controlling the false discovery rate (FDR). A FDR threshold of 0.05 was employed to determine DMPs.
Histone enrichment analysis
In order to analyze the enrichment of histone marks for a subset of probes, the information contained in the UCSC Genome Browser Broad Histone track from the ENCODE Project was used. Histone mark peaks were downloaded for every combination of cell line and antibody. For each track, a 2 × 2 contingency table was built to represent the partition of the whole set of possible probes in the microarray with respect to their membership of the subset of interest and the overlap between the probes and the histone peaks. A Fisher's exact test was used to determine whether there was a significant enrichment of the selected histone mark for the subset of interest. P values were adjusted for multiple comparisons using the Benjamini-Hochberg method for controlling the FDR. A significance level of 0.05 was used to determine whether the given combination of histone mark and cell line presented a significant change in proportion. Additionally, the base-2 logarithm of the odds ratio (OR) was used as a measure of effect size.
Genomic region analysis
The probes in the microarray were assigned to a genomic region according to their position relative to the transcript information extracted from the R/Bioconductor package TxDb.Hsapiens.UCSC. hg19.knownGene (package version 3.0.0). A probe was said to be in a promoter region if it was located in a region up to 2 kb upstream of the transcription start site (TSS) of any given transcript. Similarly, a set of mutually exclusive regions were defined inside the transcripts, namely 5UTR, 3UTR, first exon, exon and intron. A probe could only belong to one of these categories, and when anyone overlapped with two or more of these regions in different transcripts, it was assigned to the region with the higher level of precedence (i.e. in the same order as stated above). If a probe was not assigned to any of these special regions, it was labeled by default as intergenic. A contingency table was built for each of the subsets, partitioning the complete set of probes according to membership of a given category and the subset of interest. A Pearson’s χ2 test was used to determine if there was a significant change in proportion between the number of probes marked as belonging to a given region inside and outside the subset of interest. A significance level of 0.05 and the effect size as measured by the odds ratio (OR) were employed for this test.
CpG Island status analysis
The CpG island locations used in the analyses were obtained from the R/Bioconductor package FDb.InfiniumMethylation.hg19 [66]. The generation procedure of these CpG Islands is described by Wu et al. [40] CpG shores were defined as the 2kbp regions flanking a CpG island. CpG shelves were defined as the 2kbp region either upstream or downstream of each CpG shore. Probes not belonging to any of the regions previously mentioned were assigned to the special category non-CpG island. Each probe was assigned to only one of the categories. A 4 × 2 contingency table was constructed for every subset of probes in order to study the association between the given subset and the different CpG island categories. A Chi squared test was used to determine whether any of the categories had a significant association with the given subset. For each of the CpG island status levels, a 2 × 2 contingency table was defined and another Chi squared test was used to independently evaluate the association of the given subset with each status level. A significance level of 0.05 was employed for all tests. Effect size was reported as the Odds Ratio for each of the individual tests.
Gap distance analysis
Distance from both the centromere and telomere was measured for each of the probes in the Human Methylation450 microarray. In order to find significant differences between the probes inside the subset of interest and those in the background, a Wilcoxon non-parametric test was used. Again, a significance level of 0.05 was employed for all tests, and Cliff’s Delta (D) was used as a measure of effect size.
Microarray background correction
Although it is sometimes referred to as a genome wide solution, the Infinium450 k microarray only covers a fraction of the entire genome. In its 27 k predecessor, the probes were mainly located at gene promoter regions, while in addition to the promoter probes, the Infinium450 k includes probes located inside genes and in intergenic regions [67]. The irregular distribution of probes can lead to unwanted biases when studying whether a selected subset of probes is enriched with respect to any functional or clinical mark. In this study, a reference to the background distribution of features was included in every type of statistical test performed in order to prevent the conclusions from being driven by the irregular distribution of probes. In qualitative tests (CpG island status, genomic region and histone mark enrichment), the contingency matrix was built to represent the background distribution of the microarray. Thus any significant result would indicate a departure from the fixed background distribution, and so avoid any manufacturer bias.
Gene ontology analysis and annotation
Probe sets were converted to gene sets by using the annotation information present in the R/Bioconductor package TxDb.Hsapiens.UCSC.hg19.knownGene (Carlson M. TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation package for TxDb object(s).). A probe was assigned to a gene if the probe was contained within the union of all the genomic regions represented by the different transcripts belonging to that gene, or in a 2kbp region upstream of the corresponding TSS. Probes converted in this way can be assigned to zero (intergenic probes) or more genes. After gene conversion, each subset of interest was analyzed using the HOMER software tool [68]. The software was configured to use the whole set of genes represented in the HumanMethylation450 architecture as a background. HOMER tested the genes in each subset of interest against 21 different databases, including the Gene Ontology (GO) Biological Process, Molecular Function and Cellular Component ontologies, as well as KEGG and Reactome pathway databases, among others.
Conclusions
The present study provides a group of 6641 CpG sites that change their methylation levels from birth to 5 years of age in human blood leukocytes. Age-hypermethylated CpG sites are associated with genes related mainly to development, suggesting that DNA methylation-changes with age during the first years of life might play a significant role in the regulation of differentiation and leukocyte-specific functions. Conversely, genes with age-hypomethylated sites both reveal an immunological window of opportunity in childhood and indicate that blood leukocytes experience a genome remodeling, which is related to interaction with environmental factors. This underlines the importance of environmental exposures during the first years of life and highlights the need to take this into consideration in new strategies for disease prevention.
Data access
The Illumina® Infinium 450 k DNA methylation data sets from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSEXXXXX (SubSeries GSEXXXXX and GSEXXXXX).
References
Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42.
Nagae G, Isagawa T, Shiraki N, Fujita T, Yamamoto S, Tsutsumi S, Nonaka A, Yoshiba S, Matsusaka K, Midorikawa Y, et al. Tissue-specific demethylation in CpG-poor promoters during cellular differentiation. Hum Mol Genet. 2011;20:2710–21.
Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109.
Fraga MF. Genetic and epigenetic regulation of aging. Curr Opin Immunol. 2009;21:446–53.
Bjornsson HT, Cui H, Gius D, Fallin MD, Feinberg AP. The new field of epigenomics: implications for cancer and other common disease research. Cold Spring Harb Symp Quant Biol. 2004;69:447–56.
Heyn H, Moran S, Esteller M. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics. 2013;8:28–33.
Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13:497–510.
Feinberg AP. Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nat Biotechnol. 2010;28:1049–52.
Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138–62.
Florath I, Butterbach K, Muller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. 2014;23:1186–201.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Weidner CI, Wagner W. The epigenetic tracks of aging. Biol Chem. 2014;395:1307–14.
West J, Widschwendter M, Teschendorff AE. Distinctive topology of age-associated epigenetic drift in the human interactome. Proc Natl Acad Sci USA. 2013;110:14138–43.
Xu Z, Taylor JA. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis. 2014;35:356–64.
Bocklandt S, Lin W, Sehl ME, Sanchez FJ, Sinsheimer JS, Horvath S, Vilain E. Epigenetic predictor of age. PLoS ONE. 2011;6:e14821.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67.
Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jockel KH, Erbel R, Muhleisen TW, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15:R24.
Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629.
Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci USA. 2012;109:10522–7.
Johnson KC, Koestler DC, Cheng C, Christensen BC. Age-related DNA methylation in normal breast tissue and its relationship with invasive breast tumor methylation. Epigenetics. 2014;9:268–75.
Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, Suchiman HE, Slagboom PE, Boomsma DI, Heijmans BT. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11:694–703.
Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet. 2013;22:R7–15.
West J, Beck S, Wang X, Teschendorff AE. An integrative network algorithm identifies age-associated differential methylation interactome hotspots targeting stem-cell differentiation pathways. Sci Rep. 2013;3:1630.
Acevedo N, Reinius LE, Vitezic M, Fortino V, Soderhall C, Honkanen H, Veijola R, Simell O, Toppari J, Ilonen J, et al. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clin Epigenetics. 2015;7:34.
Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22:623–32.
Martino D, Loke YJ, Gordon L, Ollikainen M, Cruickshank MN, Saffery R, Craig JM. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013;14:R42.
Martino DJ, Tulic MK, Gordon L, Hodder M, Richman TR, Metcalfe J, Prescott SL, Saffery R. Evidence for age-related and individual-specific changes in DNA methylation profile of mononuclear cells during early immune development in humans. Epigenetics. 2011;6:1085–94.
Wang D, Liu X, Zhou Y, Xie H, Hong X, Tsai HJ, Wang G, Liu R, Wang X. Individual variation and longitudinal pattern of genome-wide DNA methylation from birth to the first two years of life. Epigenetics. 2012;7:594–605.
Simpkin AJ, Suderman M, Gaunt TR, Lyttleton O, McArdle WL, Ring SM, Tilling K, Davey Smith G, Relton CL. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum Mol Genet. 2015;24:3752–63.
Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–41.
Burdge GC, Lillycrop KA. Nutrition, epigenetics, and developmental plasticity: implications for understanding human disease. Annu Rev Nutr. 2010;30:315–39.
Lurbe E, Carvajal E, Torro I, Aguilar F, Alvarez J, Redon J. Influence of concurrent obesity and low birth weight on blood pressure phenotype in youth. Hypertension. 2009;53:912–7.
Lurbe E, Garcia-Vicent C, Torro MI, Aguilar F, Redon J. Associations of birth weight and postnatal weight gain with cardiometabolic risk parameters at 5 years of age. Hypertension. 2014;63:1326–32.
Singhal A, Lucas A. Early origins of cardiovascular disease: is there a unifying hypothesis? Lancet. 2004;363:1642–5.
Rangel M, dos Santos JC, Ortiz PH, Hirata M, Jasiulionis MG, Araujo RC, Ierardi DF. Franco Mdo C: modification of epigenetic patterns in low birth weight children: importance of hypomethylation of the ACE gene promoter. PLoS ONE. 2014;9:e106138.
Morales E, Bustamante M, Vilahur N, Escaramis G, Montfort M, de Cid R, Garcia-Esteban R, Torrent M, Estivill X, Grimalt JO, Sunyer J. DNA hypomethylation at ALOX12 is associated with persistent wheezing in childhood. Am J Respir Crit Care Med. 2012;185:937–43.
Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, Ho SM. Relation of DNA methylation of 5′-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS ONE. 2009;4:e4488.
Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. Redefining CpG islands using hidden Markov models. Biostatistics. 2010;11:499–514.
Fernandez AF, Bayon GF, Urdinguio RG, Torano EG, Garcia MG, Carella A, Petrus-Reurer S, Ferrero C, Martinez-Camblor P, Cubillo I, et al. H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res. 2014;25:27–40.
Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–9.
Oda M, Yamagiwa A, Yamamoto S, Nakayama T, Tsumura A, Sasaki H, Nakao K, Li E, Okano M. DNA methylation regulates long-range gene silencing of an X-linked homeobox gene cluster in a lineage-specific manner. Genes Dev. 2006;20:3382–94.
Thomas RM, Sai H, Wells AD. Conserved intergenic elements and DNA methylation cooperate to regulate transcription at the il17 locus. J Biol Chem. 2012;287:25049–59.
Bocker MT, Hellwig I, Breiling A, Eckstein V, Ho AD, Lyko F. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood. 2011;117:e182–9.
Issa JP. Aging and epigenetic drift: a vicious cycle. J Clin Invest. 2014;124:24–9.
Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, Yamamoto H, Hidalgo M, Tan AC, Galm O, et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2012;22:407–19.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–6.
Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2010;470:279–83.
Xie M, Hong C, Zhang B, Lowdon RF, Xing X, Li D, Zhou X, Lee HJ, Maire CL, Ligon KL, et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat Genet. 2013;45:836–41.
Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441:1011–4.
Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8:290–302.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–35.
Ballard JL, Novak KK, Driver M. A simplified score for assessment of fetal maturation of newly born infants. J Pediatr. 1979;95:769–74.
Battaglia FC, Lubchenco LO. A practical classification of newborn infants by weight and gestational age. J Pediatr. 1967;71:159–63.
Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, Wei R, Curtin LR, Roche AF, Johnson CL. CDC Growth Charts for the United States: methods and development. Vital Health Stat. 2000;11(2002):1–190.
Barlow SE. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics. 2007;120(Suppl 4):S164–92.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Makismovic J, Gordon L, Oshlack A. SWAN: subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13(6):R44.
Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–3.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31.
Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30:1431–9.
Smyth GK. Limma: linear models for microarray data. In: bioinformatics and computational biology solutions using R and bioconductor. New York: Springer. 2005; 397–420.
Triche TJ Jr, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41:e90.
Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450 K technology. Epigenomics. 2011;3:771–84.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89.
Authors’ contributions
RGU, MFF, GFB, and AFF carried out the methylation studies and participated in drafting the manuscript. RGU, MFF, GFB and AFF performed the statistical analysis. EL conceived of the study and participated in its design and coordination of the manuscript. MIT and JAP participated in collecting and qualifying mothers and children. PR performed critical revision of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Ronnie Lendrum for editorial assistance. This work has been financially supported by Plan Nacional de I + D+I 2008–2011/2013–2016/FEDER (PI14/01781 and PI11/00144 to M.I.T., J.A., P.R., and E.L.; PI12/01080 to M.F.F.; and PI11/01728 to AF.F.); the Fundación Cientifica de la AECC (to R.G.U.); Centros de Investigación Biomédica en Red de Fisiopatología Obesidad y Nutrición (CB06/03), Instituto de Salud Carlos III (to M.I.T., J.A., P.R., and E.L.); IUOPA (to G.F.B.); and Fundación Ramón Areces (to M.F.F). A.F.F. is sponsored by ISCIII-Subdirección General de Evaluación y Fomento de la Investigación (CP11/00131). The IUOPA is supported by the Obra Social Cajastur, Spain.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Urdinguio, R.G., Torró, M.I., Bayón, G.F. et al. Longitudinal study of DNA methylation during the first 5 years of life. J Transl Med 14, 160 (2016). https://doi.org/10.1186/s12967-016-0913-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-016-0913-x