
Abstract
Csn5 is subunit 5 of the COP9 signalosome (CSN), but the mechanism by which it strictly controls the pathogenicity of pathogenic fungi through autophagy remains unclear. Here, we found that Csn5 deficiency attenuated pathogenicity and enhanced autophagy in Magnaporthe oryzae. MoCSN5 knockout led to overubiquitination and overdegradation of MoTor (the core protein of the TORC1 complex [target of rapamycin]) thereby promoted autophagy. In addition, we identified MoCsn5 as a new interactor of MoAtg6. Atg6 was found to be ubiquitinated through linkage with lysine 48 (K48) in cells, which is necessary for infection-associated autophagy in pathogenic fungi. K48-ubiquitination of Atg6 enhanced its degradation and thereby inhibited autophagic activity. Our experimental results indicated that MoCsn5 promoted K48-ubiquitination of MoAtg6, which reduced the MoAtg6 protein content and thus inhibited autophagy. Aberrant ubiquitination and autophagy in ΔMocsn5 led to pleiotropic defects in the growth, development, stress resistance, and pathogenicity of M. oryzae. In summary, our study revealed a novel mechanism by which Csn5 regulates autophagy and pathogenicity in rice blast fungus through ubiquitination.
Similar content being viewed by others
Introduction
Rice blast, caused by Magnaporthe oryzae, is a worldwide fungal disease [1, 2]. The conidium is the infection weapon of the rice blast fungus and germinates to form a germ tube under suitable environmental conditions; then, the tip of the germ tube expands to form an appressorium [3, 4]. The mature appressorium generates a mechanical force of up to 8.0 MPa and penetrates the plant epidermis through infection nails to form infection hypha [5]. M. oryzae has high genetic operability and exhibits the typical growth, development, and infection mechanism characteristics and is a model organism for studying the interaction between pathogens and host plants. The entire infection cycle of rice blast fungus, from surface recognition, adhesion, and germ tube germination to appressorium formation, infectious growth, and pathogenicity, is closely related to the protein degradation process and signal transduction pathways [6, 7]. Protein degradation systems include the autophagy‒lysosome pathway (ALP) and the ubiquitin‒proteasome system (UPS) [8, 9]. The typical signal transduction pathways include the mitogen-activated protein kinase signaling pathway (MAPK), calcium signaling pathway, G protein signaling pathway, and TOR signaling pathway, which are critical to the pathogenicity of pathogenic fungi [10,11,12,13,14].
The COP9 (constitutive morphogenesis number 9) signalosome (usually called the CSN) is a highly conserved protein complex found in almost all multicellular eukaryotes [15, 16]. The CSN consists of eight subunits, Csn1-Csn8, and the recently discovered ninth subunit, CSNAP; among these subunits, Csn5 and Csn6 contain the feature domain MPN (Mpr1 and Pad1 N-terminal) domain, and Csn1-4 and Csn7-8 contain the feature domain PCI (proteasome lid-CSN-initiation factor 3) domain [17, 18]. The most studied function of the CSN is to coordinate the activity of cullin-RING E3 ubiquitin ligases (CRLs) [19]. CRLs are an E3 ubiquitin-ligating enzyme family that has a significant impact on cellular regulation by binding ubiquitin to target proteins [20]. Specifically, CSN negatively regulates the E3 ubiquitin-ligating enzyme activity of CRLs by uncoupling the ubiquitin-like protein Nedd8 (neural precursor cell expressed, developmentally downregulated 8) from the cullin subunit (deneddylation) or combining with deneddylated CRLs, thus relieving the interaction between CRLs and E2 ubiquitin-conjugating enzymes and ubiquitin substrates [21]. Recent studies have shown that the CSN plays a key role in the development and cell processes of eukaryotes. The mammalian CSN plays a complex regulatory role in a series of different biological processes, such as signal transduction, autophagy, circadian rhythm, and cell development [22,23,24]. In pathogenic fungi, CSNs play indispensable roles in autophagy, ubiquitin protein degradation, aerial hyphal growth, and pathogenicity [25]. The absence of any CSN subunit is fatal for higher eukaryotes but not for fungi; thus, fungi are good models for studying the molecular mechanism of the CSN. Previous studies have shown that the absence of Csn5 in Arabidopsis thaliana or Beauveria bassiana, the absence of Csn6 in M. oryzae, and the absence of any CSN in Fusarium graminearum are correlated with the growth, development, and pathogenicity of pathogenic fungi [26,27,28,29]. To our knowledge, apart from the interaction between Csn6 and Atg6 in rice blast fungus, which regulates autophagy and virulence [26], little is known about the biological function of the CSN in regulation of autophagy and pathogenicity in pathogenic fungi.
Autophagy is a catabolic membrane transport process that is highly conserved in eukaryotes. Damaged or excess cell components are encapsulated in double-membrane autophagy vesicles and sent to vacuoles (fungi and plants) or lysosomes (mammals) for degradation and recycling, which helps maintain cell homeostasis and organism survival under various stresses [7, 30, 31]. Moderate autophagy is necessary for the complete virulence of pathogenic fungi [32]. Five main systems are involved in the autophagy process [33]. Inhibition of TOR activity by rapamycin (TORC1 inhibitor) leads to dephosphorylation of Atg13 (autophagy-related protein 13), which then binds to Atg17 to form the Atg1-Atg13-Atg17 complex localized at the phagophore assembly site (PAS), marking the initiation of autophagy [34]. After the induction of autophagy, the autophagy membrane extend to the PAS to form vesicles, which eventually form mature autophagosomes that fuse with vacuoles or lysosomes [31]. The ubiquitin-like Atg8/Atg12 conjugation system [35], the phosphatidylinositol 3-phosphate kinase complex 3 (PI3KC3) [36], and the Atg9 recycling complex are involved in this process [37]. Atg6, a homolog of the vacuole protein sorting (Vps) 30 in yeast and Beclin1 in mammals, participates in the assembly of the PI3KC3 complex by interacting with Atg14 and Vps34, which is crucial for the activity of the PI3KC3 complex and the localization of autophagy-related proteins to the PAS [38]. Although the biological functions of autophagy have gradually been revealed, the molecular and regulatory mechanisms of autophagy in phytopathogenic fungi remain poorly understood.
Although the importance of Csn5 has been confirmed in different species, to our knowledge, its relationship with autophagy in filamentous fungi has not been reported. In this study, we found that MoCsn5 is a novel autophagy inhibitor in M. oryzae that inhibits autophagy by regulating ubiquitination, thereby mediating pathogenicity. Further analyses revealed the following mechanism: MoCsn5 promoted the K48-ubiquitination of MoAtg6 by interacting with MoAtg6, which was found to be more susceptible to degradation by the ubiquitin‒proteasome pathway and thereby hindered the formation of autophagosomes. Moreover, MoCsn5 inhibited the K48-ubiquitination and UPS-mediated degradation of MoTor (the core protein of the TORC1 complex [target of rapamycin]), thereby maintaining the inhibitory effect of the TORC1 complex on autophagy. In addition, abnormal ubiquitination and autophagy induced by MoCsn5 deficiency led to pleiotropic defects in growth, development, stress resistance, and pathogenicity in M. oryzae. In summary, we characterized the novel function of Csn5 in autophagy and provided a new perspective on the mechanism by which ubiquitination regulates autophagy.
Results
Targeted gene deletion of the MoCSN subunit reduces the growth, sporulation, and pathogenicity of M. oryzae
Previously, we identified the CSN complex in M. oryzae and found that its component Csn6 is involved in fungal development, autophagy, ubiquitination, and pathogenicity [26]. To further investigate the biological function of the CSN complex in M. oryzae, high-throughput target gene deletion strategies were used to knock out MoCSN1, MoCSN2, MoCSN3, MoCSN4, MoCSN5 and MoCSN7a in M. oryzae 70-15 (wild type) (Figure S1A). At least four positive knockout transformants with similar phenotypes were obtained for each CSN gene, and single-copy validation was performed (Figure S1B and C). To determine the roles of various CSN subunits in the growth of rice blast fungus, the obtained CSN mutants and 70-15 were inoculated on complete medium (CM) or minimum medium (MM) for 9 days. As shown in Figure S2A and B, compared with 70-15, the destruction of any CSN subunit led to a sharp decrease in mycelial growth. After the corresponding CSN gene was replenished, the mycelial growth rates of all replenished strains (Mocsn1-C, Mocsn2-C, Mocsn3-C, Mocsn4-C, Mocsn5-C and Mocsn7a-C) were restored to those of the wild type (WT) strain (Figure S2A and B). Further phenotypic analysis indicated that all subunits of the CSN are necessary for spore production and complete pathogenicity (Figure S2C and D), indicating that the CSN complex is a key regulator of nutritional growth, spore production, and pathogenicity in rice blast fungus.
Although previous studies have shown that the CSN complex is closely related to the pathogenicity of phytopathogenic fungi, the specific regulatory mechanisms involved are still unclear. MoCsn6 is involved in the regulation of autophagy in M. oryzae [26]. Therefore, we used yeast two-hybrid methods to preliminarily identify interactions between CSN subunits and autophagy-related proteins. Yeast two-hybrid experiments showed that MoCsn1, MoCsn3, and MoCsn4 all interacted with MoAtg5, MoAtg6, MoAtg14, MoAtg16, and MoAtg17, that MoCsn4 interacted with MoAtg5, MoAtg12, and MoAtg18, that MoCsn5 interacted with MoAtg6 and MoAtg14, and that MoCsn7a interacted with MoAtg5 and MoAtg6 (Figure S3A). These results suggest that the CSN complex may mediate the pathogenic ability of M. oryzae by regulating autophagy, but the exact mechanism still needs to be explored.
Identification of the Csn5 protein in rice blast fungus
In this study, we focused on MoCsn5, the key subunit of the CSN complex, to explore its function in M. oryzae. The amino acid sequence (344 aa) encoded by MGG_05274 was compared in the NCBI database. The results showed that this protein had 53.31% homology with Homo sapiens Csn5 and 66.08% homology with F. graminearum Csn5 (Figure S4A); therefore, we named this protein MoCsn5. A phylogenetic tree was constructed based on the Csn5 amino acid sequences of M. oryzae (XP_003712833.1), F. graminearum (XP_011318525.1), Mus musculus (NP_038743.1), Caenorhabditis elegans (NP_500841.1), Aspergillus fumigatus (XP_755961.2), H. sapiens (NP_006828.2), and Neurospora crassa (XP_956786.1) (Figure S4B). The phylogenetic tree showed that the Csn5 of M. oryzae had greater homology with the Csn5 of F. graminearum than with those of C. elegans, M. musculus, and H. sapiens (Figure S4B). Csn5 is the fifth subunit of the CSN, which is highly conserved in eukaryotes and contains a conserved MPN domain (Figure S4C).
ΔMocsn5 is deficient in vegetative growth and sporulation
To further explore the basic biological functions of MoCsn5, growth and spore production assays were performed. The ΔMocsn5 strain showed a significantly slower growth rate on both CM and MM (Fig. 1A). As shown in Fig. 1B, the aerial hyphae of ΔMocsn5 were thinner than those of 70-15 and Mocsn5-C. Compared with those of 70-15 and Mocsn5-C, the colony diameter of ΔMocsn5 on CM was decreased by 22.10%, and that on MM was decreased by 51.38% (Fig. 1C). The conidium pedicels of 70-15 and Mocsn5-C show typical branching structures and bear more conidium, whereas ΔMocsn5 has few fascicular conidial pedicels, and most of them are single-branching structures with few spores (Fig. 1D). Statistical analysis revealed that the spore production of ΔMocsn5 was significantly decreased to only 0.73% of that of 70-15 (Fig. 1E). In summary, MoCsn5 was essential for the vegetative growth and sporulation of rice blast fungus.

MoCsn5 is required for vegetative growth and conidiation. A Colony morphology of 70-15, ΔMocsn5 and Mocsn5-C. The strains were grown on CM and MM plates for 9 days. B Hyphal growth of 70-15, ΔMocsn5 and Mocsn5-C on solid CM. C Statistical analysis of the colony growth diameter. The data were analyzed using GraphPad Prism 8.0 software. The error bars represent the standard deviations. ***P < 0.001. D Conidiophores of 70-15, ΔMocsn5 and Mocsn5-C. The strains were cultivated in an incubator at 25 ℃ for 9 days and observed under an optical microscope. E Statistical analysis of conidia production. The data were analyzed with GraphPad Prism 8.0 software. ***P < 0.001. F Localization of MoCsn5 in the conidium, appressorium and invasive hypha. The MoH2B-mCherry vector was transformed into the complementation strain with the MoCsn5-GFP label. Conidia were harvested from colonies that were cultivated in solid CM for 9 days. The conidial suspension (5 × 104 ml−1) was dripped onto a hydrophobic film and incubated in a humid chamber at 22 °C for 24 h. The red fluorescence and green fluorescence in the spores and appressoria were observed under a fluorescence microscope. Bar: 10 μm. G The fluorescence densities of MoH2B-mCherry and MoCsn5-GFP were analyzed using ImageJ software
Then, we investigated the subcellular localization of MoCsn5. The MoCsn5-GFP (green fluorescent protein) vector was constructed and transformed into the ΔMocsn5 mutant. As shown in Fig. 1F, the green fluorescence of MoCsn5-GFP was observed in the conidium, appressorium and invasive hypha. To confirm whether MoCsn5-GFP is localized in the nucleus and cytoplasm, the nuclear location marker protein MoH2B-mCherry was transformed into the complemented strain with the MoCsn5-GFP label through an Agrobacterium tumefaciens-mediated transformation (ATMT) strategy. The green fluorescence of MoCsn5-GFP overlapped with that of the nucleus labeled with MoH2B-mCherry (Fig. 1G). These data indicate that MoCsn5 is localized in the cytoplasm and nucleus of M. oryzae.
MoCsn5 is involved in conidial germination, appressorium formation, and pathogenicity in M. oryzae
We then performed mycelial plug pathogenicity experiments on two susceptible hosts (barley and rice). As expected, the disease spots were smaller in the ΔMocsn5 mutant than in the 70-15 and complemented strains (Fig. 2A and B). In addition, we inoculated wounded rice and barley leaves with mycelial plugs of 70-15, ΔMocsn5 and Mocsn5-C for 3 days. Moreover, 70-15 and Mocsn5-C caused severe lesions, whereas ΔMocsn5 caused smaller disease lesions than 70-15 and Mocsn5-C (Fig. 2C and D). As the infectious weapon of rice blast fungus, conidia germinate to form appressoria with sufficient penetration turgor to infect the host and thus play a vital role in virulence. To investigate whether MoCsn5 regulates the development of appressoria in M. oryzae, we induced appressoria on an artificially hydrophobic surface and observed their morphology under a microscope. As shown in Fig. 2F, the conidial morphology was not affected by MoCSN5 deletion, and typical three-celled spores remained. Compared with those of 70-15 and Mocsn5-C, the germ tube germination rate and appressorium formation rate of ΔMocsn5 were significantly decreased at 4 h postinoculation (hpi) and 24 hpi (Fig. 2G-J). At 4 hpi, the germination rate of ΔMocsn5 was only 25.3%, and the appressorium formation rate was only 6.3%, whereas the germination rate of 70-15 was 90.0%, and its appressorium formation rate was 84.3% (F ig. 2I and J). Although the observation time was extended to 24 h, the tube germination rate (37.3%) and appressorium formation rate (16.7%) of ΔMocsn5 were still significantly lower than those of 70-15 (97.3%/93%) and Mocsn5-C (96.3%/94.3%) (Fig. 2I and J). Pmk1-MAPK pathways have been reported to regulate appressorium formation in M. oryzae. The decreased phosphorylation levels of MoPmk1 in ΔMocsn5 indicate that MoCsn5 is involved in appressorium formation through the MAPK pathway (Fig. 2K).

MoCsn5 is involved in conidial germination, appressorium formation, and pathogenicity in M. oryzae. A Disease spots of detached barley leaves inoculated with mycelial plugs from the 70-15, ΔMocsn5 and Mocsn5-C strains. Leaves were cultured at 25 ℃ for 4 days after inoculation. B Mycelial plugs from the 70-15, ΔMocsn5 and Mocsn5-C strains were inoculated on detached rice leaves. C Disease symptoms on wounded leaves of rice inoculated with mycelial plugs of the 70-15, ΔMocsn5 mutant and Mocsn5-C strains. D Disease symptoms on wounded leaves of barley inoculated with mycelial plugs of 70-15, ΔMocsn5 mutant and Mocsn5-C strains. E Disease spots of detached barley leaves inoculated with conidial suspensions of the 70-15, ΔMocsn5 and Mocsn5-C strains. Leaves were cultured at 25 ℃ for 4 days after inoculation. F Conidial morphology of 70-15, ΔMocsn5 and Mocsn5-C. Bar: 10 μm. G Germ tube germination assays and appressorium formation assays on hydrophobic surfaces at 4 hpi. Bar: 10 μm. H Germ tube germination assays and appressorium formation assays on hydrophobic surfaces at 24 hpi. Bar: 10 μm. I Statistical analysis of germ tube germination rates of 70-15, ΔMocsn5 and Mocsn5-C when conidia were dropped onto hydrophobic surfaces at 4 hpi and 24 hpi. The data were analyzed with GraphPad Prism 8.0. ***P < 0.001. J Statistical analysis of the appressorium formation rates of 70-15, ΔMocsn5 and Mocsn5-C when conidia were dropped onto hydrophobic surfaces at 4 hpi and 24 hpi. The data were analyzed using GraphPad Prism 8.0. ***P < 0.001. K Phosphorylation analysis of Pmk1 in 70-15 and ΔMocsn5. Pmk1 phosphorylation and Pmk1 levels were detected with phospho-Pmk1 and Pmk1 antibodies, respectively. The protein actin was used as a loading control. L The conidial suspension (5 × 104 ml−1) was dripped onto isolated leaves and incubated in a humid chamber at 25 °C. After 3 days, the leaves were decolorized with methanol and observed under an optical microscope. Bar: 10 μm. M The infection rate was quantified and statistically analyzed using GraphPad Prism 8.0 software
Then, we tested the pathogenicity of ΔMocsn5 on barley leaves using conidia. As expected, only some nonexpanding small necrotic lesions were observed on the leaves inoculated with ΔMocsn5, while the leaves inoculated with 70-15 and Mocsn5-C produced a large number of connected brown necrotic lesions (Fig. 2E). Finally, we carried out infection experiments with isolated barley leaves to further explore the progression of infection in the ΔMocsn5 mutant. Seventy-two hours after spore infection, the leaves were observed under a microscope. The main growth morphology of infected hyphae in leaves infected with 70-15, ΔMocsn5 and Mocsn5-C is shown in Fig. 2L. The infected hyphae of 70-15 and Mocsn5-C expand into multiple adjacent plant cells, whereas the appressoria of ΔMocsn5 are not infected, or the infected hyphae are limited to a single plant cell (Fig. 2L). At 72 hpi, nearly 86% of the infection structures were detected in the 70-15 strains, and 76% had expanded to neighboring cells. However, with ΔMocsn5, only approximately 12% of the infected structures formed, and 9% were still limited to the initially infected cells (Fig. 2M). These results indicate that MoCsn5 is essential for germ tube germination, appressorium development, and pathogenicity in M. oryzae.
Tolerance to external stresses is critical for the survival, appressorium formation, and invasion of rice blast fungus [39,40,41]. We found that ΔMocsn5 was more sensitive to hyperosmotic stress (KCl, NaCl, and sorbitol), oxidative stress (menadione and H2O2), and amphotericin B (an antifungal agent that disrupts membrane permeability by binding to sterols on the fungal cell membrane) than 70-15 and was not sensitive to myriocin (a sphingolipid synthesis inhibitor) (Fig. 3A-F). Fungi have been reported to respond to hyperosmotic stress through two mechanisms: the Osm1-MAPK signaling pathway and the TORC2-Ypk1 signaling pathway [42,43,44]. We further determined the phosphorylation levels of Osm1 and Ypk1 under 0.6 M NaCl treatment. The ΔMocsn5 mutant and 70-15 strains showed a consistent trend in Ypk1 phosphorylation (Fig. 3G). However, the level of Osm1 phosphorylation was lower in the ΔMocsn5 mutant than in the 70-15 strain (Fig. 3H). The phosphorylation level of Osm1 in the ΔMocsn5 mutant was also significantly lower than 70-15 after treatment with 2 μM amphotericin B (Fig. 3I), which disrupts cell membrane permeability. The above-described experimental results indicate that MoCsn5 participates in the response to hyperosmotic stress by participating in the Osm1-MAPK signaling pathway.

ΔMocsn5 is defective in the maintenance of external stresses. A and B Colony morphology and relative growth rate of 70-15, ΔMocsn5 and Mocsn5-C on CM supplemented with 2 mM hydrogen peroxide (H2O2) and 100 μM menadione (VK3). ***P < 0.001. C and D Colony morphology and relative growth rate of 70-15, ΔMocsn5 and Mocsn5-C on CM supplemented with 2 μM amphotericin B (AmB) and 1.5 μM myriocin. ***P < 0.001. E and F Colony morphology and relative growth rate of 70-15, ΔMocsn5 and Mocsn5-C on CM supplemented with 0.4 M KCl, 0.6 M NaCl, and 0.8 M sorbitol. Pictures were taken at 9 days. The data were analyzed with GraphPad Prism 8.0 software. ***P < 0.001, **P < 0.01. (G) Phosphorylation level of MoYpk1 in 70-15 and ΔMocsn5. The strains were cultured in CM supplemented with 0.6 M NaCl before analysis. H Phosphorylation level of MoOsm1 in 70-15 and ΔMocsn5 strains. The strains were cultured in CM supplemented with 0.6 M NaCl before analysis. I Phosphorylation level of MoOsm1 in 70-15 and ΔMocsn5 strains. The strains were cultured in CM or treated with 2 μM amphotericin B before analysis
MoCsn5 regulates the ubiquitin‒proteasome pathway in rice blast fungus
Our previous experimental results showed that MoCsn5 is a subunit of the CSN complex and participates in the assembly of the CSN complex through interaction with other CSN subunits, mediating the ubiquitin‒proteasome pathway. To further investigate the role of MoCsn5 in ubiquitination, we detected the ubiquitination levels of 70-15 and ∆Mocsn5 using an anti-ubiquitin antibody. The lower level of ubiquitination in ∆Mocsn5 suggests that MoCsn5 promotes ubiquitination (Fig. 4A). This result was further confirmed by the recovery of the ubiquitination levels in the complemented strain Mocsn5-C (Fig. 4A). MG132 is a proteasome inhibitor that inhibits the degradation of ubiquitinated proteins. After induction with MG132 for 4 h, the levels of ubiquitination of 70-15 and ∆Mocsn5 significantly increased due to the accumulation of ubiquitinated proteins (Fig. 4A). However, regardless of MG132 induction, the ubiquitination protein level of ∆Mocsn5 was significantly lower than that of 70-15 and Mocsn5-C. This finding indicates that the ability of MoCsn5 to promote ubiquitination in rice blast fungus was sustained.

MoCsn5 regulates the ubiquitin‒proteasome pathway in rice blast fungus. A Ubiquitination levels of 70-15, ΔMocsn5 and Mocsn5-C at 0 h and 3 h after MG132 induction. Hyphae were grown in liquid CM for 36 h and then shifted to CM supplemented with 20 μM MG132 (a proteasome inhibitor, a tool for studying cellular degradation of the ubiquitin‒proteasome pathway) for 0 h and 3 h. B Transcript levels of MoSKP1 (MGG_04978), MoCULLIN1 (MGG_07145), MoCULLIN3 (MGG_07731) and MoCULLIN4b (MGG_14763) in 70-15 and ΔMocsn5. The error bars represent the standard deviations. Tukey’s test was used to determine significance. **P < 0.01. C The interaction between MoCsn5 and MoCullin3 was detected by yeast two-hybrid assays. pGADT7-T and pGBKT7-53 were used as positive controls. pGADT7-T and MoCsn5-BD and pGBKT7 and MoCullin3-AD served as two pairs of negative controls. D The relationship between MoCsn5 and MoCullin3 in vivo was examined by coimmunoprecipitation assays. The MoCullin3-Flag bands were detected following MoCsn5-GFP immunoprecipitation
To explore the mechanism by which MoCsn5 promotes ubiquitination, we examined the mRNA levels of ubiquitin-associated proteins in M. oryzae. The qPCR results showed that the expression levels of MoCULLIN1, MoCULLIN3, MoCULLIN4b, and MoSKP1 were significantly decreased by the elimination of MoCsn5 (Fig. 4B). This finding explains the reduced level of ubiquitination in ∆Mocsn5. In addition, the results of the interaction experiment indicated that MoCsn5 directly interacts with MoCullin3 in vivo and in vitro to regulate CRLs (Fig. 4C and D). In summary, we conclude that MoCsn5 promotes the ubiquitination of M. oryzae by regulating the components of CRLs.
MoCsn5 negatively regulates autophagy
The ubiquitin‒proteasome system and autophagy‒lysosome pathway, which are responsible for the degradation of cellular proteins, are crucial for various cellular processes, such as the growth and development of organisms [8]. Due to the important function of CSN in the ubiquitin‒proteasome system and the interaction between MoCsn5 and autophagy-related proteins, we examined the autophagy levels in 70-15 and ΔMocsn5. The GFP-MoAtg8 fusion protein was used to determine whether there was a connection between MoCsn5 and autophagy. During the fusion of the autophagosome with the lysosome, MoAtg8 on the inner membrane of the autophagosome is delivered to the vacuole for degradation, where it produces free GFP (in the vacuole); therefore, the [(GFP)/(GFP + GFP-MoAtg8)] ratio is widely used to indicate the level of autophagy. First, the subcellular localization of GFP-MoAtg8 in 70-15 and ΔMocsn5 was observed. As shown in Fig. 5A, under nutritional conditions, GFP-MoAtg8 in 70-15 was positioned around the vacuole in a spot-like manner, whereas the vacuoles in ΔMocsn5 appeared bright and uniform green without obvious dot-like localization. After 3 h of starvation induction, the vacuoles in the 70-15 strain were uniformly green overall, and the same trend was observed in the ΔMocsn5 strain (Fig. 5A). For further confirmation, a western blot analysis was conducted to detect full-length GFP-MoAtg8 and free GFP in 70–15 and ΔMocsn5. As shown in Fig. 5B, the autophagic flux in the ΔMocsn5 mutant was greater than that in the wild type.

MoCsn5 negatively regulates autophagy. A Subcellular localization of GFP-MoAtg8 in 70-15 and ΔMocsn5 under nutrient and starvation conditions. Bar: 10 μm. Mycelia were stained with CMAC to label the vacuoles. B Autophagic flux analysis of GFP-MoAtg8 in 70-15 and ΔMocsn5. Total GFP-MoAtg8 and free GFP were detected by western blot. The gray values of the protein bands were analyzed with ImageJ software. The degradation rate was expressed as [GFP/(GFP + GFP-MoAtg8)]. The protein content of actin was used as a control. C Analysis of MoAtg8/MoAtg8-PE turnover in 70-15 and ΔMocsn5. The ratio of MoAtg8-PE to actin was calculated to assess the rate of MoAtg8 lipidation to yield MoAtg8-PE
Next, we evaluated the turnover rate of endogenous MoAtg8/MoAtg8-phosphatidylethanolamine (MoAtg8-PE). The conversion of microtubule-associated protein 1 light chain 3 (LC3-I, Atg8 homology) to a lapidated form (LC3-II, Atg8-PE) is a committed step in autophagosome formation. Accordingly, the LC3-II protein level is commonly used as a marker of the autophagosome number [45]. As shown in Fig. 5C, under nutritional conditions, the MoAtg8 band was stronger, whereas the MoAtg8-PE band obtained with both 70-15 and ΔMocsn5 was weaker. After 3 and 6 h of starvation induction, the MoAtg8-PE and MoAtg8 band strengths were enhanced in both the 70-15 and ΔMocsn5 strains, indicating that starvation stimulated the formation of autophagosomes (Fig. 5C). However, it is worth noting that compared to 70-15, ΔMocsn5 has stronger MoAtg8-PE and MoAtg8 bands. These results indicated that the total conversion of MoAtg8 to MoAtg8-PE was increased in the ΔMocsn5 strain, and the number of autophagosomes and autophagic flux were greater in the ΔMocsn5 strain than in the 70-15 strain.
Because MoCsn5 and MoCsn6 are involved in the regulation of autophagy and other subunits also interact with autophagy-related proteins, we are very interested in whether other subunits of the CSN complex generally have regulatory functions in autophagy. Therefore, we transfected GFP-MoAtg8 into the corresponding mutants to detect the autophagy levels. Several transfection experiments were performed, and GFP-MoAtg8 was not transfected into the ΔMocsn2 and ΔMocsn3 mutants. Compared with 70-15, the ΔMocsn1, ΔMocsn4, and ΔMocsn7a strains all showed increased levels of autophagy (Figure S3B and C). Thus, other subunits of the CSN complex also regulate autophagy in M. oryzae.
MoCsn5 inhibits autophagy by promoting K48-ubiquitination of MoAtg6
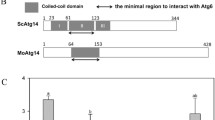
Figure 5 shows increased autophagosome formation and accelerated autophagosome transport in ΔMocsn5. Yeast two-hybrid and pull-down assays revealed that MoCsn5 interacts with the autophagy-related proteins MoAtg6 and MoAtg14 in vitro (Fig. 6A-C). Co-IP experiments also confirmed that MoCsn5 interacts with MoAtg6 and MoAtg14 in vivo (Fig. 6D and E). These results indicate that Csn5 may participate in the autophagy regulation pathway in M. oryzae by regulating autophagy-related proteins.

MoCsn5 inhibits autophagy by promoting the K48-ubiquitination of MoAtg6. A The interaction between MoCsn5 and MoAtg6 was detected by yeast two-hybrid assays. pGADT7-T and pGBKT7-53 were used as positive controls. pGADT7-T and MoCsn5-BD and pGBKT7 and MoAtg6-AD served as two pairs of negative controls. B The interaction between MoCsn5 and MoAtg14 was detected by yeast two-hybrid assays. pGADT7-T and pGBKT7-53 were used as positive controls, and pGADT7-T and MoCsn5-BD and pGBKT7 and MoAtg14-AD were used as two pairs of negative controls. C Pulldown assays to detect the interaction between MoCsn5 and MoAtg14 in vitro. GST-MoAtg14 and His-MoCn5, empty GST, and His-MoCsn5 were incubated sequentially with glutathione agarose gel beads with GST labels for 2 h. The final eluents were detected by western blot assays. D The relationship between MoCsn5 and MoAtg14 was examined by in vivo coimmunoprecipitation assays. The MoCsn5-Flag bands were detected following MoAtg14-GFP immunoprecipitation. E The relationship between MoCsn5 and MoAtg6 was examined by in vivo coimmunoprecipitation assays. The MoCsn5-Flag bands were detected following MoAtg6-GFP immunoprecipitation. F Interaction intensity of MoAtg6 and MoAtg14 in 70-15 and ΔMocsn5. MoCsn5 knockout was performed in 70-15 strain expressing MoAtg6-GFP and MoAtg14-Flag to ensure consistent MoAtg6 and MoAtg14 contents. The strains were lysed and immunoprecipitated with anti-GFP beads and then subjected to immunoblotting with anti-GFP and anti-Flag antibodies. The formula MoAtg14-Flag/MoAtg6-GFP was used to calculate the interaction intensity of MoAtg6 and MoAtg14 in the different strains. G Interaction intensity of MoAtg6 and MoAtg14 in 70-15 and ΔMocsn5. MoCsn5 knockout was performed in 70-15 strain expressing MoAtg6-GFP and MoAtg14-Flag to ensure consistent MoAtg6 and MoAtg14 contents. The strains were lysed and immunoprecipitated with anti-Flag beads and then subjected to immunoblotting with anti-GFP and anti-Flag antibodies. The formula MoAtg6-GFP/MoAtg14-Flag was used to calculate the interaction intensity of MoAtg6 and MoAtg14 in the different strains. H Ubiquitination levels of MoAtg14 in 70-15 and ΔMocsn5. Gene knockout of MoCSN5 was performed in 70-15 expressing MoAtg14-Flag to ensure consistent transcript levels of MoATG14. The strain was immunoprecipitated with anti-Flag beads after lysis, and a western blot analysis was then performed with an anti-Flag antibody and an anti-ubiquitin antibody. The formula ubiquitin/MoAtg14-Flag was used to calculate the ubiquitination levels of MoAtg14-Flag. I Deletion of MoCSN5 inhibited the ubiquitination and K48-ubiquitination of MoAtg6. Gene knockout of MoCSN5 was performed in 70-15 expressing MoAtg6-GFP to ensure consistent transcript levels of MoATG6. The strain was immunoprecipitated with anti-GFP beads after lysis, and a western blot analysis was then performed with anti-GFP antibody, anti-ubiquitin antibody, and anti-K48 antibody. The ubiquitination and K48-ubiquitination levels of MoAtg6-GFP were calculated as ubiquitin/MoAtg6-GFP and K48-ubiquitin/MoAtg6-GFP, respectively. J Ubiquitination levels and K48-ubiquitination levels of MoAtg6 in 70-15, ΔMocsn5 and Mocsn5-C. K The protein levels of MoAtg6 in the 70-15, ΔMocsn5 and Mocsn5-C strains were measured with an anti-Beclin1 antibody. The strains were cultured in liquid CM at 25 ℃ for 36 h. The protein content of actin was used as a control. L Transcript levels of MoATG6 in 70-15 and ΔMocsn5. The error bars represent the standard deviations
The PI3KC3-I complex is formed by Atg6/Vps30/Beclin1, Vps34, Vps15, and Atg14 at the phagophore assembly site (PAS) and is essential for proper PAS targeting of other autophagy-related proteins, such as the Atg8, Atg18, and Atg12–Atg5-Atg16 complex [46, 47]. The localization of PI3KC3-I to the PAS is largely dependent on the binding of Atg6 to Atg14 [48]. To investigate the effect of ubiquitination on the PI3KC3-I complex, we examined the ability of Atg14 to target Atg6. Interaction strength tests showed that the amount of MoAtg14 bound to MoAtg6 in ΔMocsn5 was comparable to that bound in 70-15. MoCsn5 did not affect the ability of MoAtg14 to target MoAtg6 to the PAS (Fig. 6F and G). Subsequent analyses revealed that the ubiquitination level of MoAtg14 in ΔMocsn5 was unaffected but that the ubiquitination level of MoAtg6 was lower in ΔMocsn5 than in 70-15, suggesting that deficient ubiquitination modulates the level of MoAtg6 (Fig. 6H and I).
Atg6 is a core protein in the autophagy pathway, and previous studies have shown a positive correlation between its protein expression and autophagic flux [49]. Subsequent experiments showed that the increased MoAtg6 content in ΔMocsn5 resulted in increased autophagy activity (Fig. 6K). However, the mRNA levels of MoATG6 were not significantly different between ΔMocsn5 and 70-15 (Fig. 6L). These results suggested that the increased MoAtg6 content in ΔMocsn5 was caused by decreased degradation rather than increased expression. Different types of anti-ubiquitin antibodies were used in western blotting assays, and we ultimately detected a reduced K48-ubiquitination level of MoAtg6 in ΔMocsn5 (Fig. 6I). Measurements of the ubiquitination and K48-ubiquitination levels of MoAtg6 in 70-15, ΔMocsn5, and Mocsn5-C reconfirmed that the knockout of MoCSN5 decreased the K48-ubiquitination level of MoAtg6 (Fig. 6J), which means that MoCsn5 promotes the K48-ubiquitination of MoAtg6 in M. oryzae.
Previous studies have shown that the K48-ubiquitination of Beclin1 (a homologous protein of Atg6) is a negative pathway for autophagy regulation [50, 51]. K48-ubiquitination promotes proteasome-dependent Beclin1 degradation, thereby effectively downregulating autophagy. Our research indicated that MoCsn5 promotes the K48-ubiquitination of MoAtg6, which promotes MoAtg6 degradation and thus downregulates autophagy in rice blast fungus.
MoCsn5 inhibits autophagy by suppressing the K48-ubiquitination of MoTor
Autophagosome formation is regulated by the TORC1 complex, and active TOR signaling inhibits autophagy through the phosphorylation of Atg proteins [52, 53]. To test whether MoCsn5 regulates MoTor activity, we used rapamycin to validate the relationship between MoCsn5 and MoTor kinase. Compared with 70-15 and the complemented strain Mocsn5-C, the ΔMocsn5 mutant was more sensitive to rapamycin on CM at 25 °C for 9 days (Fig. 7A and B). This finding suggested that the activity of the TORC1 complex in ΔMocsn5 was altered. To verify the regulatory effect of MoCsn5 on MoTor activity, we examined the phosphorylation of the TORC1 activity marker MoRps6. Compared with those in the 70-15 strain, the phosphorylation levels of MoRps6 in the ΔMocsn5 mutant strain were significantly lower after treatment with rapamycin, indicating reduced TORC1 complex activity (Fig. 7C).

MoCsn5 inhibits autophagy by suppressing the K48-ubiquitination of MoTor. A and B Colony morphology and relative growth rate of 70-15, ΔMocsn5 and Mocsn5-C on CM supplemented with 100 ng/mL rapamycin for 9 days. *p < 0.05. C The phosphorylation levels of MoRps6 in the 70-15 and ΔMocsn5 strains were detected by anti-phospho-Rps6 and anti-Rps6 antibodies. The phosphorylation levels of MoRps6 were calculated as phospho-MoRps6/MoRps6. The protein content of actin was used as a control. D Protein levels of MoTor in 70-15, ΔMocsn5 and Mocsn5-C. The strains were cultured in liquid CM at 25 ℃ for 36 h. The protein content of actin was used as a control. Gene knockout and complementation of MoCSN5 were performed in 70-15 expressing MoTor-Flag to ensure consistent transcript levels of MoTor. E Transcript levels of MoTor in 70-15 and ΔMocsn5. The error bars represent the standard deviations. F Ubiquitination levels of MoTor in 70-15, ΔMocsn5 and Mocsn5-C. After lysis, the strains were immunoprecipitated with anti-Flag beads, and a western blot analysis was subsequently performed with an anti-Flag antibody and an anti-ubiquitin antibody. The level of ubiquitinated MoTor was calculated as ubiquitin/MoTor-Flag. G K48-ubiquitination levels of MoTor in 70-15, ΔMocsn5 and Mocsn5-C. After lysis, the strains were immunoprecipitated with anti-Flag beads, and a western blot analysis was subsequently performed with an anti-Flag antibody and an anti-K48-ubiquitin antibody. The K48-ubiquitination levels of MoTor were calculated as K48-ubiquitin/MoTor-Flag
Our results showed that the reduction in the MoTor content in the ΔMocsn5 mutant induced a decrease in TORC1 complex activity (Fig. 7D), which in turn led to increased autophagy. However, no significant difference in the mRNA levels of MoTOR were found between the ΔMocsn5 and 70-15 strains (Fig. 7E). These results suggest that the decrease in the MoTor content in ΔMocsn5 is due to enhanced degradation rather than decreased expression.
Previous studies have shown that the protein content of Tor is regulated by ubiquitination [54]. The ubiquitination level of MoTor in ΔMocsn5 was significantly greater than that in the WT 70-15 (Fig. 7F), suggesting that MoCsn5-mediated ubiquitination regulates the protein level of MoTor. Measurements of the ubiquitination and K48-ubiquitination levels of MoTor in 70-15, ΔMocsn5, and the complemented strain Mocsn5-C also confirmed that the knockout of MoCSN5 increased the K48-ubiquitination level of MoTor (Fig. 7G). These results showed that MoCsn5 stabilized the MoTor content through specific K48-ubiquitination, thereby inhibiting autophagy.
Discussion
Autophagy is an ancient biological process that maintains cell homeostasis and is facilitated by numerous biological stimuli and environmental pressures [33, 55]. Although autophagy is closely related to the pathogenicity of phytopathogenic fungi [7], the specific regulatory mechanisms involved remain to be explored. During this process, numerous autophagy-related proteins that signal the autophagy machinery have been identified. Among them, Atg6/Beclin1/Vps30 is arguably the most studied molecule and is crucial for processes such as the autophagy, inflammation, tumors, and pathogenicity of pathogenic fungi [56,57,58]. As one of the key initiators of autophagy, Atg6 binds to PtdIns3K (class III phosphatidylinositol 3-kinase), thereby mediating the biogenesis and dynamics of subcellular membranes involved in autophagy [58, 59]. In this study, we identified a novel regulator of MoAtg6, MoCsn5, which is also a new autophagy inhibitor. Our data indicated that to inhibit autophagy, Csn5 inhibits K48-ubiquitination by interacting with Atg6, which is crucial for Atg6 degradation, thereby reducing Atg6 protein expression and inhibiting autophagy.
The COP9 signalosome is an evolutionarily highly conserved multifunctional complex that controls the cell cycle, signal transduction, circadian rhythm, embryonic development, autophagy, and other biological functions in mammals, plants, and fungi [60,61,62]. In previous studies, the regulatory function of CSN subunits in autophagy has been controversial. The impaired fusion between autophagosomes and lysosomes in CR-CSN8KO mice (in which Csn8 was knocked out) is the first line of evidence showing that CSN regulates autophagy [63, 64]. Subsequent studies showed that Csn8 plays an important role in autophagosome maturation. In mammals, Csn3 not only is necessary for autophagosome formation but also promotes autophagosome maturation. Niu et al. showed that Csn3 deficiency inhibits the LC3B-I-to-LC3B-II transformation and promotes the mTOR pathway, which inhibits autophagy in mammalian osteosarcoma [22]. In rice blast fungus, Csn6 deletion leads to enhanced autophagy, which is manifested by an increased number of autophagosomes and an accelerated autophagy rate [26]. In this study, we demonstrated that Csn5 inhibits autophagy in M. oryzae by promoting ubiquitin-dependent Atg6 protein degradation and stabilizing the TOR pathway in autophagosome formation. This difference may be due to the fact that different CSN subunits do not play the same roles in autophagy or to species differences. At present, the role of CSN in autophagy regulation is emerging, but the specific functions of other subunits in autophagy regulation still need to be discovered.
The expression and degradation of proteins are crucial for various aspects of cell development. In eukaryotes, the autophagy‒lysosomal pathway and the ubiquitin‒proteasome system are two highly conserved protein clearance pathways [8, 9, 65, 66]. Although these two pathways are often discussed as separate functional pathways, in recent years, there has been increasing evidence of crosstalk between the ubiquitin‒proteasome system and the autophagy‒lysosomal pathway. Zhang et al. demonstrated that Cand2-regulated ubiquitination and autophagy are essential for pathogenic fungal pathogenicity [67]. We found that MoCsn5 not only regulates the overall ubiquitination level of organisms but also mediates autophagy by directly targeting the ubiquitination of Atg6 and Tor. Using the Csn5 subunit of the key regulatory complex CSN in ubiquitin as an entry point, we revealed a novel mechanism by which ubiquitination regulates autophagy and thus affects pathogenicity in rice blast fungus.
The pathogenicity of pathogenic fungi is not regulated by a single pathway but by the joint regulation of multiple pathways. The Osm1-MAPK pathway modulates the response of pathogenic fungi to hyperosmotic stress [44]. The Pmk1-MAPK pathway is necessary for appressorium-mediated infection of M. oryzae [68, 69]. Although starvation induces autophagy, autophagy is maintained at a basic level even under normal growth conditions, thus controlling the metabolism, development and proliferation of eukaryotes through a comprehensive regulatory pathway [7]. We found that the absence of MoCsn5 downregulated the phosphorylation of MoOsm1 and MoPmk1, reducing the resistance to hyperosmotic stress and the invasion ability of the appressorium in M. oryzae. Studies in mammals have shown that the MAPK signaling pathways (ERK1/2, ERK5, p38, and JNK1/2/3 signaling pathways) are regulated by E3 ubiquitin ligases in cancer [70]. In rectal cancer, Usp14 (ubiquitin-specific peptidase 14) deubiquitinates and stabilizes JNK, thereby promoting MAPK/JNK signaling cascade activation [71]. In fission yeast, the autophagy-related protein Atg1 acts in the Pmk1-MAPK pathway. Pmk1 signaling is suppressed in the Δatg1 strain, and the growth defects caused by overexpression of Pck2, an upstream activator of Pmk1-MAPK, are mitigated by the deletion of ATG1 [72]. We hypothesize that the pleiotropic defects in the growth, development, and pathogenicity of ΔMocsn5 are the result of the dysregulation of different signaling pathways. MoCsn5 may not only regulate autophagy through ubiquitination but also mediate signaling pathways associated with rice blast fungus infection, such as the MoOsm1-MAPK and MoPmk1-MAPK pathways, via ubiquitination and/or autophagy, but the specific regulatory mechanisms involved remain to be investigated, and we believe that these mechanisms will be a very interesting subject.
Taken together, our findings revealed new associations between Csn5 and autophagy, ubiquitination, and pathogenicity in rice blast fungus (Fig. 8). MoCsn5 regulates the protein expression levels of MoAtg6 and MoTor through ubiquitination, thereby inhibiting autophagy. Our results also indicate that MoCsn5 is essential for fungal development, germ tube germination, and appressorium formation in M. oryzae. Although meaningful progress has been made, there are still many questions waiting to be answered. First, are the effects of different CSN subunits on autophagy in different species due to the species or functional specificity of the subunits? Second, does Csn5 regulate the MoOsm1-MAPK and MoPmk1-MAPK pathways through ubiquitination or independently of ubiquitination? Third, as a multifunctional complex, does the CSN complex or its subunits regulate autophagy through other pathways? The regulatory function of the CSN is much more complex than expected. The resolution of these issues could deepen our understanding of CSN-regulated autophagy, which is important for the development of new therapeutic strategies for pathogenic fungi.

A proposed regulatory model for the function of MoCsn5 in M. oryzae. As a novel autophagy inhibitor, MoCsn5 participates in the regulation of the pathogenicity of M. oryzae. MoCsn5 inhibits autophagy by regulating the ubiquitination of the autophagy-related proteins MoAtg6 and MoTor, inhibiting the degradation of MoTor and promoting the degradation of MoAtg6. In addition, MoCsn5 affects the phosphorylation of MoPmk1 and MoOsm1, thereby regulating the infection and hypertonic stress response of M. oryzae
Materials and methods
Strains, growth conditions, and quantitative RT‒PCR
The WT strain of M. oryzae, 70-15, was used in this study. All fungal strains were cultured on CM at 25 °C under a 16–18 h light-dark cycle for 8–10 days. For different stress tests, 100 µΜ menadione (VK3), 2 mM hydrogen peroxide (H2O2), 2 µΜ amphotericin B, 1.5 µΜ myriocin, 0.4 M KCl, 0.6 M NaCl and 0.8 M sorbitol were added to solid CM agar plates. The relative growth rates were calculated using the following formula: (the diameter of the strain treated with chemicals)/(the diameter of the untreated strain). Data analysis was performed using ImageJ 1.51j8 and GraphPad Prism 8.0 software. All assays were repeated three times.
To detect the expression of target genes, the 70-15 and ΔMocsn5 strains were cultured in liquid CM for 40 h at 150 rpm at 25 °C. Total RNA was extracted by the TRIzol method and reverse transcribed into cDNA using a reverse transcription kit (TaKaRa, Japan). qRT-PCR was performed using a SYBR Premix Ex Taq (Tli RNaseH Plus) kit (TaKaRa, Japan). All assays were repeated three times. Data analysis was performed using GraphPad Prism 8.0 software. All primers were designed online by Integrated DNA Technologies (https://sg.idtdna.com/pages) and are listed in Table S1.
Gene knockout and complementation assays
In this study, a homologous recombination strategy based on the Agrobacterium tumefaciens-mediated transformation (ATMT) strategy was used for gene deletion [73]. The knockout vector was designed using pKO3A by Lu et al. as described previously [74]. The 1.5-kb upstream fragment (UF) and downstream fragment (DF) of the targeted gene were amplified by PCR with the specific primers CSN-up/down (F) and CSN-up/down (R). The pKO3A vector was digested with XbaI and HindIII. Then, the hygromycin resistance gene (HPH) fragment, UF, and DF were fused to the linearized vector by ligase (Vazyme, P505-d1). The recombination cassette was transferred into 70-15 by ATMT, and the positive transformants with hygromycin resistance were selected using 200 μg/mL hygromycin B (Sangon Biotech, A600230-0001). The long fragment (LF) and short fragment (SF) in mutants were further validated using the primers CSN-Long-F/Long-HPH-R and CSN-SF/SR. The copy number of HPH was verified by quantitative real-time PCR, and the TUBULIN gene was used as a control. Strains complemented with the pKD5-GFP/pKD7-Flag plasmid vector harboring the sulfonylurea resistance gene (SUR)/genetic resistance gene (G418) were used. Western blotting and fluorescence microscopy were used to screen for positive transformants. The complemented strain with a GFP label was also used for subcellular localization analysis. All abovementioned primers are listed in Table S1.
Phenotypic characterization
For phenotypic assays, strains were grown on CM for 9 days, after which the colony diameter and conidia production were measured. For the conidia germination and appressorium formation assays, the conidia were collected and diluted to 5 × 104 conidia/mL and then inoculated on hydrophobic plastic films. After incubation at 22 °C for 4 h and 24 h, the germination and appressorium formation rates were observed, and counted under an optical microscope. For the pathogenicity assays, mycelial plugs and 5 × 104 mL−1 conidial suspensions were incubated on cut leaves of rice and barley. Diseased leaves were imaged 4 days after inoculation. For the appressorial penetration assays, 5 × 104 mL−1 conidial suspensions were added to the isolated barley leaves for 72 h. All the leaves were decolorized in methanol, and appressorium penetration was analyzed under a light microscope. All assays were repeated three times.
Fluorescence observation
pKD8-MoH2B-mCherry was constructed by inserting a fragment of MoH2B genomic DNA without a terminator codon (TAA) into the linearized vector pKD8 digested with BamHI and SamI. The constructed vector was transferred into the complemented strains with MoCsn5-GFP labeled by ATMT. To determine the cellular localization, conidia were collected from 10-day-old CM agar plates and observed under a fluorescence microscope. Conidial suspensions (5 × 104 mL−1) were inoculated on hydrophobic plastic coverslips to induce appressorium formation and observed at 24 hpi. All primers mentioned above are listed in Table S1.
Ubiquitin assays
To assay the ubiquitination level of total proteins, mycelia were grown in liquid CM at 25 °C for 40 h and then transferred to CM supplemented with 20 µΜ MG132 for 0 and 3 h to induce ubiquitinated protein accumulation. At different time points, mycelial proteins were extracted with protein extraction buffer (150 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, 1% Triton X-100, and 1% protease inhibitor mixture).
To assay the ubiquitination level of a specific protein, using MoAtg6 as an example, pKD3 with a GFP tag was linearized at the EcoRI/SamI sites and fused with MoATG6 and the native promoter of MoATG6. Recombined plasmids were transferred into 70-15. Knockout of MoCSN5 was performed in 70-15 expressing MoAtg6-GFP, and complementation of MoCSN5 was performed in the resulting strain to ensure consistent transcript levels of MoATG6. These three strains were grown in liquid CM at 25 °C for 40 h. Then, the three strains were extracted and immunoprecipitated with anti-GFP beads and immunoblotted with anti-GFP, anti-ubiquitin, and anti-K48-ubiquitin antibodies. The formula ubiquitin/MoAtg6-GFP was used to calculate the ubiquitination levels of MoAtg6 in the different strains.
Data analysis was performed using ImageJ and GraphPad Prism 8.0 software. All assays were repeated three times. The agents and antibodies used in the abovementioned assays were MG132 (Cell Signaling Technology, 2194S), the anti-GFP affinity beads 4FF (Smart-lifesciences, SA070001), the anti-Flag affinity beads 4FF (Smart-lifesciences, SA042001), the anti-ubiquitin antibody (Cell Signaling Technology, 3936S, 1:1000), the anti-K63-ubiquitin antibody (Cell Signaling Technology, 12930S, 1:1000), the anti-K48-ubiquitin antibody (Cell Signaling Technology, 8081S, 1:1000) and the anti-β-actin antibody (ABclonal, China, AC004, 1:2000). All abovementioned primers are listed in Table S1.
Autophagy assays
To detect the endogenous lipidation of MoAtg8, 70–15 and ΔMocsn5 were grown in liquid CM for 38 h and then transferred to synthetic defined media without amino acids and ammonium sulfate (SD-N) for 3 h and 6 h. The TCA-acetone method was used to extract mycelial protein, and anti-Atg8 and anti-Actin antibodies were used for protein detection by western blotting. Atg8 and Atg8-PE, which have similar molecular weights, were separated by 13.5% SDS‒PAGE with 6 M urea. ImageJ software was used to calculate the gray values of the protein bands.
To monitor autophagic flux, the GFP-MoAtg8 vector constructed in previous studies was transformed into 70-15, ΔMocsn1, ΔMocsn4, ΔMocsn5, or ΔMocsn7a by ATMT [75]. The strains were grown in liquid CM for 38 h before being transferred to liquid SD-N medium for 3 h and 6 h to induce autophagy. At different time points, mycelial proteins were extracted with protein extraction buffer, and after centrifugation at 12000 rpm for 20 min, the supernatant was used for western blotting. SDS-PAGE without urea (12.5%) was used to detect the exogenous insertion protein GFP-MoAtg8/GFP. To observe the localization of GFP-MoAtg8, 7-amino-4-chloromethylcoumarin (CMAC) was used to stain the abovementioned mycelia at 0 h and 3 h, and blue and green fluorescence was observed under a fluorescence microscope. ImageJ software was used to calculate the gray values of the protein bands.
Data analysis was performed using ImageJ and GraphPad Prism 8.0 software. All assays were repeated three times. The agents and antibodies used in the abovementioned assays were anti-β-actin antibody (ABclonal, China, AC004, 1:2000), anti-GFP antibody (Abcam, AB32146, 1:5000), anti-Atg8 antibody (MBL, PM090,1:2000), goat anti-rabbit HRP (FDbio, China, FDR007, 1:8000), goat anti-mouse HRP (FDbio, China, FDM007, 1:8000), protease inhibitor mixture (FDbio, China, FD1001), CMAC (Invitrogen, C2110), urea (Sangon Biotech, A600148-0500), TEMED (Sangon Biotech, A10076-0100), ammonium persulfate (Sangon Biotech, A100486-0025), Acryl/Bis 30% solution (Sangon Biotech, B546017-0500), 1.4 M Tris-HCl (FDbio, FD2084), and 1.5 M Tris-HCl (FDbio, FD2080).
Phosphorylation level assay and western blotting
To assay phosphorylated Pmk1, mycelia were grown in liquid yeast extract-glucose (YEG) media at 25 °C for 40 h. The TCA-acetone method was used for the total protein extraction [76]. To assay phosphorylated Rps6, mycelia were grown in liquid CM at 25 °C for 40 h, and the TCA-acetone method was used for the extraction of total proteins. To assay phosphorylated Osm1 and Ypk1, mycelia were grown in liquid CM at 25 °C for 40 h and then transferred to CM supplemented with 0.6 M NaCl for 0, 30, 60, or 90 min or to CM supplemented with 2 µΜ amphotericin B for 0, 1, or 2 h. The TCA-acetone method was then used for total protein extraction. To detect the protein content of Atg6, mycelia were grown in liquid CM at 25 °C for 40 h. Mycelial proteins were subsequently extracted with protein extraction buffer (150 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, 1% Triton X-100, and 1% protease inhibitor mixture).
Data analysis was performed using ImageJ and GraphPad Prism 8.0 software. All assays were repeated three times. The agents and antibodies used in the abovementioned assays were anti-phospho-p44/42 MAPK antibody (Cell Signal Technology, 4370S, 1:500), anti-ERK1/2 MAPK antibody (Santa Cruz Biotechnology, sc-514302, 1:500), anti-phospho-MoYpk1 (S619) antibody and anti-MoYpk1 antibody (prepared by ABclonal Biotechnology Co., Ltd.), anti-phospho-MoRps6 antibody and anti-MoRps6 antibody (prepared by ABclonal Biotechnology Co., Ltd.), anti-Beclin1 antibody (ABclonal, China, A21191, 1:1000), and anti-β-actin antibody (ABclonal, China, AC004, 1:2000).
Yeast two-hybrid assays, pulldown assays, and coimmunoprecipitation assays
In this study, the yeast strain Y2HGold was used, and pGADT7-T and pGBKT7-53 were used as positive controls. The coding DNA sequences (CDSs) of MoCSN5 and MoATG14 were amplified and fused with the bait vector pGBKT7 and the prey vector pGADT7, respectively. After sequencing, a pair of bait and prey vectors were cotransformed into the yeast Y2HGold strain according to the Matchmaker Gold Yeast Two-Hybrid System (Clontech, Mountain View, CA, USA), and the cell growth on SD-Leu-Trp-His-Ade media and SD-Leu-Trp media was observed. All abovementioned primers are listed in Table S1.
For pull-down assays, the full-length cDNA of MoAtg14 was amplified and inserted into the vector pGEX4T-1 to obtain the GST-MoAtg14 plasmid. The full-length cDNA of MoCsn5 was amplified and inserted into the vector pET35a to obtain the His-MoCsn5 plasmid. These plasmids were subsequently transformed into E. coli strain BL21(DE3) for expression. The bacterial cells were lysed to obtain bacterial lysates according to a previously described method [77, 78]. The extracted protein was subjected to SDS-PAGE and Coomassie brilliant blue staining to ensure protein expression. The soluble GST or GST fusion protein was incubated with 55 µl of glutathione agarose beads with GST tags (Smart-Lifesciences Biotech, SA042001) at 4 °C for 2 h. Then, the agarose beads were washed five times with washing buffer and incubated with the supernatant containing His-MoCsn5 protein for 2 h. The proteins were eluted with elution buffer and detected by immunoblotting with an anti-His antibody (Huabio, R1207-2, 1:2000) and an anti-GST antibody (Huabio, EM80701, 1:2000). All abovementioned primers are listed in Table S1.
For coimmunoprecipitation assays, taking MoAtg14 and MoCsn5 as examples, the pKD5-MoAtg14-GFP vector was constructed by inserting a fragment of MoATG14 genomic DNA without a terminator codon (TAA) into the linearized vector pKD5 digested with BamHI and SamI. Then the constructed empty GFP and MoAtg14-GFP vectors with SUR resistance were transferred into the complemented strains with the MoCsn5-3 × Flag label (G418 resistance). The positive transformants were screened by PCR, western blot, and fluorescence microscopy. Total proteins were extracted from the mycelia of the positive transformants coexpressing GFP and MoCsn5-3 × Flag, MoAtg14-GFP, and MoCsn5-3 × Flag with protein extraction buffer. After centrifugation at 12,000 rpm for 20 min, the supernatant of the lysates was incubated with GFP beads at 4 °C for 4 h. Moreover, anti-GFP antibody and anti-Flag antibody were used to detect the whole protein and the eluent of affinity beads eluted with acidic amino acids, respectively. All abovementioned primers are listed in Table S1.
Availability of data and materials
Dataset used or analyzed during the current study are available from the corresponding author on reasonable request.
References
Dean R, Van Kan JA, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, Foster GD. The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol. 2012;13:414–30.
Wilson RA. Magnaporthe oryzae. Trends Microbiol. 2021;29:663–4.
Zhang H, Zheng X, Zhang Z. The Magnaporthe grisea species complex and plant pathogenesis. Mol Plant Pathol. 2016;17:796–804.
Foster AJ, Talbot NJ. Getting a grip on blast. Nat Microbiol. 2020;5:1457–8.
Talbot NJ. Appressoria. Curr Biol. 2019;29:R144–6.
Turrà D, Segorbe D, Di Pietro A. Protein kinases in plant-pathogenic fungi: conserved regulators of infection. Annu Rev Phytopathol. 2014;52:267–88.
Zhu XM, Li L, Wu M, Liang S, Shi HB, Liu XH, Lin FC. Current opinions on autophagy in pathogenicity of fungi. Virulence. 2019;10:481–9.
Wang X, Su H, Ranek MJ. Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol. 2008;45:11–27.
Pearce C, Hayden RE, Bunce CM, Khanim FL. Analysis of the role of COP9 Signalosome (CSN) subunits in K562; the first link between CSN and autophagy. BMC Cell Biol. 2009;10:31.
Fudal I, Collemare J, Böhnert HU, Melayah D, Lebrun MH. expression of Magnaporthe grisea avirulence gene ACE1 is connected to the initiation of appressorium-mediated penetration. Eukaryot Cell. 2007;6:546–54.
Zhao X, Xu JR. A highly conserved MAPK-docking site in Mst7 is essential for Pmk1 activation in Magnaporthe grisea. Mol Microbiol. 2007;63:881–94.
Wang ZH, Shen ZF, Wang JY, Cai YY, Li L, Liao J, Lu JP, Zhu XM, Lin FC, Liu XH. MoCbp7, a novel calcineurin b subunit-binding protein, is involved in the calcium signaling pathway and regulates fungal development, virulence, and er homeostasis in Magnaporthe oryzae. Int J Mol Sci. 2023;24:9297.
Kim Y, Islam N, Moss BJ, Nandakumar MP, Marten MR. Autophagy induced by rapamycin and carbon-starvation have distinct proteome profiles in Aspergillus nidulans. Biotechnol Bioeng. 2011;108:2705–15.
Reiner DJ, Lundquist EA. Small GTPases. WormBook. 2018;2018:1–65.
Qin N, Xu D, Li J, Deng XW. COP9 signalosome: discovery, conservation, activity, and function. J Integr Plant Biol. 2020;62:90–103.
Wei N, Tsuge T, Serino G, Dohmae N, Takio K, Matsui M, Deng XW. The COP9 complex is conserved between plants and mammals and is related to the 26S proteasome regulatory complex. Curr Biol. 1998;8:919–22.
Wei N, Serino G, Deng XW. The COP9 signalosome: more than a protease. Trends Biochem Sci. 2008;33:592–600.
Rozen S, Füzesi-Levi MG, Ben-Nissan G, Mizrachi L, Gabashvili A, Levin Y, Ben-Dor S, Eisenstein M, Sharon M. CSNAP is a stoichiometric subunit of the COP9 signalosome. Cell Rep. 2015;13:585–98.
Wolf DA, Zhou C, Wee S. The COP9 signalosome: an assembly and maintenance platform for cullin ubiquitin ligases? Nat Cell Biol. 2003;5:1029–33.
Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20.
Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, Wei N, Shevchenko A, Deshaies RJ. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–5.
Niu J, Yan T, Guo W, Wang W, Ren T, Huang Y, Zhao Z, Yu Y, Chen C, Huang Q, et al. The COPS3-FOXO3 positive feedback loop regulates autophagy to promote cisplatin resistance in osteosarcoma. Autophagy. 2023;19:1693–710.
Lee MH, Zhao R, Phan L, Yeung SC. Roles of COP9 signalosome in cancer. Cell Cycle. 2011;10:3057–66.
Kato JY, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14:1209–25.
Jiang H, Zhang Y, Wang W, Cao X, Xu H, Liu H, Qi J, Jiang C, Wang C. FgCsn12 is involved in the regulation of ascosporogenesis in the wheat scab fungus Fusarium graminearum. Int J Mol Sci. 2022;23:10445.
Shen ZF, Li L, Wang JY, Zhang YR, Wang ZH, Liang S, Zhu XM, Lu JP, Lin FC, Liu XH. A subunit of the COP9 signalosome, MoCsn6, is involved in fungal development, pathogenicity, and autophagy in rice blast fungus. Microbiol Spectr. 2022;10:e0202022.
Chen A, Ren Y, Han X, Liu C, Zhou Y, Xu C, Qi H, Ma Z, Chen Y. The COP9 signalosome complex regulates fungal development and virulence in the wheat scab fungus Fusarium graminearum. Front Microbiol. 2023;14:1179676.
Wang M, Yang X, Ruan R, Fu H, Li H. Csn5 is required for the conidiogenesis and pathogenesis of the Alternaria alternata tangerine pathotype. Front Microbiol. 2018;9:508.
Mou YN, Ren K, Tong SM, Ying SH, Feng MG. Essential role of COP9 signalosome subunit 5 (Csn5) in insect pathogenicity and asexual development of Beauveria bassiana. J Fungi (Basel). 2021;7:642.
Shen Z-F, Li L, Zhu X-M, Liu X-H, Klionsky DJ, Lin F-C. Current opinions on mitophagy in fungi. Autophagy. 2022;19:747–57.
Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–73.
Zhu XM, Li L, Bao JD, Wang JY, Liang S, Zhao LL, Huang CL, Yan JY, Cai YY, Wu XY, et al. MoVast2 combined with MoVast1 regulates lipid homeostasis and autophagy in Magnaporthe oryzae. Autophagy. 2023;1–19:2353.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010;30:1049–58.
Nakatogawa H, Ishii J, Asai E, Ohsumi Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy. 2012;8:177–86.
Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9.
Jin M, Klionsky DJ. Transcriptional regulation of ATG9 by the Pho23-Rpd3 complex modulates the frequency of autophagosome formation. Autophagy. 2014;10:1681–2.
Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–56.
Mittler R. ROS are good. Trends Plant Sci. 2017;22:11–9.
Li P, Zhao L, Qi F, Htwe N, Li Q, Zhang D, Lin F, Shang-Guan K, Liang Y. The receptor-like cytoplasmic kinase RIPK regulates broad-spectrum ROS signaling in multiple layers of plant immune system. Mol Plant. 2021;14:1652–67.
Jacob S, Foster AJ, Yemelin A, Thines E. High osmolarity glycerol (HOG) signalling in Magnaporthe oryzae: Identification of MoYPD1 and its role in osmoregulation, fungicide action, and pathogenicity. Fungal Biol. 2015;119:580–94.
van Dam TJ, Zwartkruis FJ, Bos JL, Snel B. Evolution of the TOR pathway. J Mol Evol. 2011;73:209–20.
Muir A, Roelants FM, Timmons G, Leskoske KL, Thorner J. Down-regulation of TORC2-Ypk1 signaling promotes MAPK-independent survival under hyperosmotic stress. Elife. 2015;4:e09336.
Dixon KP, Xu JR, Smirnoff N, Talbot NJ. Independent signaling pathways regulate cellular turgor during hyperosmotic stress and appressorium-mediated plant infection by Magnaporthe grisea. Plant Cell. 1999;11:2045–58.
Nair U, Yen WL, Mari M, Cao Y, Xie Z, Baba M, Reggiori F, Klionsky DJ. A role for Atg8-PE deconjugation in autophagosome biogenesis. Autophagy. 2012;8:780–93.
Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 2007;12:209–18.
Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast Saccharomyces cerevisiae. FEBS Lett. 2007;581:2156–61.
Obara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes–Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell. 2006;17:1527–39.
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99.
Xu C, Feng K, Zhao X, Huang S, Cheng Y, Qian L, Wang Y, Sun H, Jin M, Chuang TH, Zhang Y. Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy. 2014;10:2239–50.
Abrahamsen H, Stenmark H, Platta HW. Ubiquitination and phosphorylation of Beclin 1 and its binding partners: tuning class III phosphatidylinositol 3-kinase activity and tumor suppression. FEBS Lett. 2012;586:1584–91.
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003.
Zheng XF, Florentino D, Chen J, Crabtree GR, Schreiber SL. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell. 1995;82:121–30.
Jiang Y. Regulation of TORC1 by ubiquitin through non-covalent binding. Curr Genet. 2016;62:553–5.
Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95.
Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–34.
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–80.
Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17:839–49.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39.
Braus GH, Irniger S, Bayram O. Fungal development and the COP9 signalosome. Curr Opin Microbiol. 2010;13:672–6.
Hetfeld BK, Bech-Otschir D, Dubiel W. Purification method of the COP9 signalosome from human erythrocytes. Methods Enzymol. 2005;398:481–91.
Kwok SF, Solano R, Tsuge T, Chamovitz DA, Ecker JR, Matsui M, Deng XW. Arabidopsis homologs of a c-Jun coactivator are present both in monomeric form and in the COP9 complex, and their abundance is differentially affected by the pleiotropic cop/det/fus mutations. Plant Cell. 1998;10:1779–90.
Liu J, Su H, Wang X. The COP9 signalosome coerces autophagy and the ubiquitin-proteasome system to police the heart. Autophagy. 2016;12:601–2.
Su H, Li F, Ranek MJ, Wei N, Wang X. COP9 signalosome regulates autophagosome maturation. Circulation. 2011;124:2117–28.
Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–22.
Li Y, Li S, Wu H. Ubiquitination-proteasome system (UPS) and autophagy two main protein degradation machineries in response to cell stress. Cells. 2022;11:851.
Zhang YR, Wei YY, Wu MH, Liu MY, Liang S, Zhu XM, et al. Cand2 inhibits CRL-mediated ubiquitination and suppresses autophagy to facilitate pathogenicity of phytopathogenic fungi. Plant Commun. 2024;5(2):100720.
Hamel LP, Nicole MC, Duplessis S, Ellis BE. Mitogen-activated protein kinase signaling in plant-interacting fungi: distinct messages from conserved messengers. Plant Cell. 2012;24:1327–51.
Xu JR, Hamer JE. MAP kinase and cAMP signaling regulate infection structure formation and pathogenic growth in the rice blast fungus Magnaporthe grisea. Genes Dev. 1996;10:2696–706.
Park HB, Baek KH. E3 ligases and deubiquitinating enzymes regulating the MAPK signaling pathway in cancers. Biochim Biophys Acta Rev Cancer. 2022;1877:188736.
Du XH, Ke SB, Liang XY, Gao J, Xie XX, Qi LZ, Liu XY, Xu GY, Zhang XD, Du RL, Li SZ. USP14 promotes colorectal cancer progression by targeting JNK for stabilization. Cell Death Dis. 2023;14:56.
Takasaki T, Utsumi R, Shimada E, Bamba A, Hagihara K, Satoh R, Sugiura R. Atg1, a key regulator of autophagy, functions to promote MAPK activation and cell death upon calcium overload in fission yeast. Microb Cell. 2023;10:133–40.
Vijn I, Govers F. Agrobacterium tumefaciens mediated transformation of the oomycete plant pathogen Phytophthora infestans. Mol Plant Pathol. 2003;4:459–67.
Lu J, Cao H, Zhang L, Huang P, Lin F. Systematic analysis of Zn2Cys6 transcription factors required for development and pathogenicity by high-throughput gene knockout in the rice blast fungus. Plos Pathog. 2014;10:e1004432.
Zhu XM, Liang S, Shi HB, Lu JP, Dong B, Liao QS, Lin FC, Liu XH. VPS9 domain-containing proteins are essential for autophagy and endocytosis in Pyricularia oryzae. Environ Microbiol. 2018;20:1516–30.
Qu Y, Wang J, Huang P, Liu X, Lu J, Lin FC. PoRal2 is involved in appressorium formation and virulence via Pmk1 MAPK pathways in the rice blast fungus Pyricularia oryzae. Front Plant Sci. 2021;12:702368.
Li L, Zhu XM, Shi HB, Feng XX, Liu XH, Lin FC. MoFap7, a ribosome assembly factor, is required for fungal development and plant colonization of Magnaporthe oryzae. Virulence. 2019;10:1047–63.
Cai YY, Wang JY, Wu XY, Liang S, Zhu XM, Li L, Lu JP, Liu XH, Lin FC. MoOpy2 is essential for fungal development, pathogenicity, and autophagy in Magnaporthe oryzae. Environ Microbiol. 2022;24:1653–71.
Acknowledgements
We express our gratitude to all those who helped with this study.
Funding
This study was supported by the National Key Research and Development Program of China (2023YFD1400200), the National Natural Science Foundation of China (32370208) and the Key Research and Development Project of Zhejiang Province, China (2021C02010).
Author information
Authors and Affiliations
Contributions
Zi-Fang Shen: Investigation, Methodology, Data curation, Writing-original draft. Lin Li, Jing-Yi Wang and Jian Liao: Methodology, Data curation. Yun-Ran Zhang, Xue-Ming Zhu, Zi-He Wang, Jian-Ping Lu and Xiao-Hong Liu: Investigation, Methodology. Fu-Cheng Lin: Conceptualization, Funding acquisition, Writing-review & editing, Supervision. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethics approval or consent was required since this study did not include patient samples, human subjects or animal experiments.
Consent for publication
This study did not involve human subjects. All co-authors have seen and approved the final draft.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Verification of deletion mutants by PCR and quantitative real-time PCR. (A) Rice blast fungus knockout model. (B) PCR detection was performed on the null mutants, and the recombinant fragment (LF) was cloned from the positive strain but not from the WT strain. The 500-bp characteristic fragment (SF) of the target gene was cloned in the WT strain but not in the mutants. TUBULIN was used as a positive control. (C) The copy number of the resistance gene HPH in the deletion mutants was verified by quantitative real-time PCR. Figure S2. The targeted gene destruction of CSN subunits decreased the growth, sporulation, and pathogenicity of M. oryzae. (A) Colony morphology of 70-15, ΔMocsn1, Mocsn1-C, ΔMocsn2, Mocsn2-C, ΔMocsn3, Mocsn3-C, ΔMocsn4, Mocsn4-C, ΔMocsn7a, and Mocsn7a-C. The strains were grown on CM and MM plates for 9 days. (B) Statistical analysis of the colony growth diameter. The data were analyzed using GraphPad Prism 8.0 software. The error bars represent the standard deviations. ***P < 0.001. (C) Conidiophores of 70-15, ΔMocsn1, Mocsn1-C, ΔMocsn2, Mocsn2-C, ΔMocsn3, Mocsn3-C, ΔMocsn4, Mocsn4-C, ΔMocsn7a, and Mocsn7a-C. The strains were cultivated in an incubator at 25 ℃ for 9 days and observed under an optical microscope. (D) Disease spots of detached barley leaves inoculated with mycelial plugs from 70-15, ΔMocsn1, Mocsn1-C, ΔMocsn2, Mocsn2-C, ΔMocsn3, Mocsn3-C, ΔMocsn4, Mocsn4-C, ΔMocsn7a, and Mocsn7a-C. Leaves were cultured at 25 ℃ for 4 days after inoculation. Figure S3. CSN subunits are involved in the regulation of autophagy in M. oryzae. (A) Yeast two-hybrid assays were used to detect the interactions between CSN subunits and Atg-related proteins. Pairs of pGBKT7-53 and PGADT7-T were used as positive controls. Yeast transformants carrying the indicated constructs were plated onto selective plates supplemented without Leu/Trp or Leu/Trp/His/Ade for growth assays. (B) Autophagic flux analysis of GFP-MoAtg8 in 70-15 and ΔMocsn1.Total GFP-MoAtg8 and free GFP were detected by western blotting. (C) Autophagic flux analysis of GFP-MoAtg8 in 70-15, ΔMocsn4 andΔMocsn7a. Figure S4. Identification of the Csn5 protein in rice blast fungus. (A) Comparison of Csn5 amino acid sequences in different eukaryotes via DNAMAN 8 software. The amino acid sequences used for comparison were from Magnaporthe oryzae (XP_003712833.1), Fusarium graminearum (XP_011318525.1), Mus musculus (NP_038743.1), Caenorhabditis elegans (NP_500841.1), Aspergillus fumigatus (XP_755961.2), Homo sapiens (NP_006828.2) and Neurospora crassa (XP_956786.1). (B) Phylogenetic trees of the Csn5 proteins constructed using MEGA 11. (C) Conserved MPN domain of Csn5 in different eukaryotes was identified using IBS 1.0.1 software.
Additional file 2: Table S1.
Primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shen, ZF., Li, L., Wang, JY. et al. Csn5 inhibits autophagy by regulating the ubiquitination of Atg6 and Tor to mediate the pathogenicity of Magnaporthe oryzae. Cell Commun Signal 22, 222 (2024). https://doi.org/10.1186/s12964-024-01598-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01598-7