
Abstract
Ferroptosis is a newly discovered form of cell death that is featured in a wide range of diseases. Exosome therapy is a promising therapeutic option that has attracted much attention due to its low immunogenicity, low toxicity, and ability to penetrate biological barriers. In addition, emerging evidence indicates that exosomes possess the ability to modulate the progression of diverse diseases by regulating ferroptosis in damaged cells. Hence, the mechanism by which cell-derived and noncellular-derived exosomes target ferroptosis in different diseases through the system Xc−/GSH/GPX4 axis, NAD(P)H/FSP1/CoQ10 axis, iron metabolism pathway and lipid metabolism pathway associated with ferroptosis, as well as its applications in liver disease, neurological diseases, lung injury, heart injury, cancer and other diseases, are summarized here. Additionally, the role of exosome-regulated ferroptosis as an emerging repair mechanism for damaged tissues and cells is also discussed, and this is expected to be a promising treatment direction for various diseases in the future.
Video Abstract
Similar content being viewed by others
Background
Ferroptosis is an iron-dependent form of programmed cell death that was defined in 2012. It is characterized by the excessive accumulation of lipid peroxides and reactive oxygen species (ROS) in cells and is distinct from apoptosis, necrosis, and autophagy [1, 2]. As an evolutionarily conserved process, ferroptosis plays a vital role in the developmental processes of diverse organisms, including both animals and plants [3, 4]. Recent studies on neurological diseases have indicated that the presence of iron metabolism imbalance is closely linked to several pathological characteristics of Alzheimer's disease and that excessive iron accumulation within the brain has been identified as a potential cause for the accelerated cognitive deterioration observed in individuals affected by Alzheimer's disease [5]. Regarding cardiovascular diseases, myocardial apoptosis is an important pathological change in heart failure. Furthermore, research has demonstrated that in addition to apoptosis, ferroptosis is also involved in the progression of heart failure [6, 7]. This phenomenon is also found in liver disease. In the course of nonalcoholic steatotic liver disease, ferroptosis may be a predisposing factor of the inflammatory response; moreover, ferroptosis in hepatocytes may precede apoptosis in the early stage of inflammation [8]. Ferroptosis of human bronchial epithelial cells was also observed in an LPS-induced acute lung injury model [9]. Furthermore, ferroptosis has been found to be involved in the pathological process of acute kidney injury mediated by ischaemia–reperfusion injury (IRI) after cardiac surgery due to the iron released from cardiopulmonary bypass. If this condition is not controlled in time, then increases in the cellular labile iron pool (LIP) can occur [10]. In orthopaedic diseases, it has also been confirmed that iron overload is positively correlated with the occurrence and development of osteoporosis and then inhibits the activity of osteoblasts [11]. In addition, the occurrence and development pathways of some cancers are closely related to ferroptosis, and the survival and proliferation of tumour cells are easily affected by ferroptosis [12]. Given that ferroptosis is closely related to the occurrence and development of several diseases, it is essential to develop new strategies against ferroptosis.
Cellular therapy, a novel therapeutic strategy for multiple diseases, has been a research hotspot in recent years. However, cell transplantation therapy still has many limitations, which impedes its application in the clinic [13]. Currently, there is abundant evidence showing that mesenchymal stem cells exert their biological effects by secreting paracrine mediators, especially exosomes [14]. As an important carrier of intercellular communication, exosomes containing a variety of proteins, RNA, and other components play a crucial role in disease diagnosis and treatment by regulating the inflammatory response, angiogenesis and cell protection after injury [15, 16]. Notably, exosomes have been shown to participate in disease progression by regulating ferroptosis [17]. Since the association between exosomes and ferroptosis has been noted, an increasing number of researchers have conducted intensive studies in this field.
This review will focus on the main mechanisms by which exosomes from different sources regulate ferroptosis in different diseases. Subsequently, the application of ferroptosis as an exosome regulatory target in related diseases will be introduced. It is worth noting that exosome-regulated ferroptosis is a "double-edged sword", which suggests the importance for regulating disease progression by reasonable induction or inhibition of ferroptosis (Tables 1 and 2). Additionally, the direction with the most potential for diagnosing and treating different diseases is also provided.
Overview of exosomes
Exosomes are derived from intracellular multivesicular bodies (MVBs) containing intraluminal vesicles, which is a category of extracellular vesicles with a size range of 40 to 160 nm (average ~ 100 nm) in diameter [41]. Exosomes carry biologically active molecules such as proteins, nucleic acids (mRNAs, microRNAs (miRNAs), long noncoding RNAs (lncRNAs), DNA, etc.), lipids, and other components [42]. Exosome-containing RNAs are the main substances regulating cell–cell communication. They can also regulate gene expression in recipient cells through their own degradation and re-expression and then participate in cell cycle progression, cell migration, angiogenesis, or histone modification [43,44,45]. In addition to RNAs, several exosomal proteins and lipids have also been reported. The composition of exosome lipids and proteins may affect their pharmacokinetic properties, and their natural components may play a part in enhancing bioavailability and reducing adverse reactions [41]. There are several surface markers on the exosomal membrane, such as transmembrane proteins (CD63, CD9, CD81), heat shock proteins, TSG101, and Alix, which can regulate processes such as fusion, migration, and adhesion [46,47,48,49]. Recent studies have shown that these exosomes can be secreted by different kinds of cells, including mesenchymal stem cells, immune cells, endothelial and epithelial cells, and various cancer cells [50,51,52]. In addition, noncell-derived exosomes, such as those in body fluids (blood, saliva, etc.) and food-derived exosomes, have also attracted extensive attention [53,54,55,56]. Because its composition, including its surface proteins and lipids, plays a vital role in its function, the selection of exosome sources is critical for treatment [57].
Mechanism and application of exosome-regulated ferroptosis
Ferroptosis is a newly discovered mode of programmed cell death that exists in the pathological processes of various diseases. Therefore, it offers new insights into the development of new cell protection strategies and provides new targets for disease treatment. For example, cancer cells are highly sensitive to ferroptosis [58]. Thus, this sensitivity could be used to induce cancer cell ferroptosis and then eliminate cancer. Ferroptosis is also a new therapeutic target for cardiac diseases [59] and is also being investigated in the treatment of other organs and tissues as well as injuries. Here, the application of ferroptosis as a regulatory point for exosomes in the occurrence and development of different diseases has been presented, and the mechanisms of action and relationship between ferroptosis and exosomes have been revealed.
Exosome-regulated ferroptosis by system Xc-/GSH/GPX4 and its application in treating diseases
The role of system Xc-/GSH/GPX4 in ferroptosis
System Xc- is an amino acid reverse transporter located on the cell membrane; it is mainly composed of the solute carrier family 3 member 2 (SLC3A2) and solute carrier family 7 member 11 (SLC7A11) [60]. System Xc- exchanges extracellular cystine with intracellular glutamic acid in a 1:1 ratio to maintain the balance of REDOX [61]. The ingested cystine undergoes an enzymatic reaction in the cell to form cysteine, which then participates in the biosynthesis of glutathione (GSH), a key factor in regulating ferroptosis. Therefore, the inhibitory effect on the activity of system Xc- affects the synthesis of GSH by inhibiting cystine absorption, which leads to the accumulation of peroxide in vivo that can induce ferroptosis [62]. Studies have shown that regulating the expression of the functional subunit SLC7A11 of system Xc- can affect the activity of system Xc- [63]. The expression of SLC7A11 is regulated by several factors. For example, p53, nuclear factor, erythroid 2-like 2 (NFE2L2), BECN1, BRCA1-associated protein 1 (BAP1), and others can regulate SLC7A11 activity or expression [64,65,66,67].
The cysteine-GSH-GPX4 axis is a downstream node of system Xc- and has been considered a major factor in resistance to ferroptosis for many years [68, 69]. Glutathione peroxidase 4 (GPX4) is a key regulator of ferroptosis that inhibits ferroptosis by lowering hydroperoxides and blocking lipoxygenase pathways [70]. GPX4 is essential to preserve tissue homeostasis and avoid cell death during tissue injury [71]. Studies have shown that inactivation of GPX4 with RSL3 (an inducer of ferroptosis) and ML162 (an inhibitor of GPX4) can increase the production of lipid ROS, thus inducing ferroptosis [72, 73]. GPX4 activity and expression are not only regulated by GSH but also controlled by the amino acid selenocysteine in its active centre, which can be embedded into GPX4 through the transporter selenocysteine tRNA to maintain the activity of GPX4 and scavenge lipid ROS, thus inhibiting ferroptosis [4]. Therefore, blocking the GSH synthesis pathway or selenium deficiency leads to suppression of GPX4 activity and induces ferroptosis.
The application of exosome-regulated ferroptosis in treating different diseases through system Xc-/GSH/GPX4
Exosomes play different regulatory roles depending on the cell from which they are derived and the microenvironment in which they reside. Thus, in some diseases, exosomes of their own origin can regulate the ferroptosis of target cells via system Xc-/GSH/GPX4 to promote disease progression. Studies have shown that long-term consumption of a high-fat diet (HFD) causes myocardial damage through myocardial cell ferroptosis [74]. A HFD causes a sustained, low-grade inflammatory response in adipose tissue, which is maintained by inflammatory macrophages [75]. MiR-140-5p has been found to be highly expressed in adipose tissue macrophage-derived exosomes (ATM-Exo) in obesity-induced myocardial injury. Exosomal miR-140-5p targeting SLC7A11 inhibits GSH expression and promotes ferroptosis in cardiomyocytes, exacerbating damage to cardiomyocytes [19]. Similarly, in another myocardial injury model of atrial fibrillation, cardiac fibroblast-derived exosomes (CF-Exo) inhibit SLC7A11 expression and promote cardiac ferroptosis by delivering rich miR-23a-3p [20]. However, it also provides potential therapeutic targets for the treatment of obesity-related cardiomyopathy and for the continued development of atrial fibrillation.
Exogenous administration of mesenchymal stem cell-derived exosomes (MSC-Exos) to regulate ferroptosis in some diseases has also been widely studied. In an acute liver injury (ALI) model induced by carbon tetrachloride (CCL4), Lin et al. found that there was a high concentration of lipid peroxide accumulation in the liver. This accumulation caused the ubiquitination of SLC7A11 and its abnormal expression, leading to ferroptosis in liver cells. However, MSC-Exos can maintain the stability and function of SLC7A11 by increasing the expression of CD44 (regulating SLC7A11) and OTUB1 (deubiquitination), thus activating system Xc − to rescue ALI caused by ferroptosis in hepatocytes [22]. Cerebral ischaemia/reperfusion injury (I/R) causes irreversible damage to the brain and neurons [76]. Therefore, effectively preventing the occurrence of cerebral I/R is an issue that urgently needs to be addressed. By establishing cerebral I/R models in vivo and in vitro, Liu et al. found that Fer-1-loaded bone marrow mesenchymal stem cell-derived extracellular vesicles (BMMSC-EVs/Fer-1) could successfully deliver Fer-1 to neurons, reduce the occurrence of neuronal ferroptosis and inflammation, and alleviate cerebral I/R injury in mice. Further investigation of its mechanism showed that BMMSCs-EVs/Fer-1 could regulate the GPX4/COX2 axis and play a protective role in the brain by upregulating GPX4 and inhibiting COX2 expression [34]. In addition, exosome-regulated ferroptosis also plays an important role in skin injury and wound healing. Zha et al. found that hypoxia-pretreated adipose-derived stem cell (ADSC) exosomes (HExo) could reverse ROS generation by reducing miR-700-5p and increasing GPX4 expression via delivery of circ-Ash1l, thereby improving UV-induced skin damage [39]. Previous studies have shown that exosomes can promote wound healing in diabetic foot ulcers (DFUs), thereby alleviating their symptoms [77]. A recent study found that ferroptosis may play a significant role in this process. The circRNA-itchy E3 ubiquitin protein ligase (circ-ITCH) derived from BMMSC-Exo can recruit TATA-Box binding protein associated factor 15 (TAF15) to activate the Nrf2 signalling pathway, thereby inhibiting ferroptosis of venous endothelial cells, promoting angiogenesis, and accelerating the wound healing process of DFU mice [40].
While cellular ferroptosis may seem to be an unfavourable option, promoting ferroptosis may be beneficial for certain diseases. Increased activated hepatic stellate cells (HSCs) are a key point in the progression of liver fibrosis, and HSCs are considered to be an important target for the treatment of liver fibrosis. Tan et al. found that human umbilical cord mesenchymal stem cell-derived exosomes (HucMSC-Exo) could reduce the expression of xCT/GPX4 by transferring BECN1, thereby inducing ferroptosis and inhibiting the activation of HSCs, providing a new perspective for liver fibrosis treatment [18, 78]. In addition, Guo et al. found that GOT1 protein enrichment in exosomes secreted by pancreatic cancer cells inhibits ferroptosis of cancer cells and promotes tumour development. In terms of mechanism, GOT1 can upregulate the expression of the C–C motif chemokine receptor-2 (CCR2), thereby activating the Nrf2/HO-1 axis to resist ferroptosis in cancer cells [79].
Exosome-regulated ferroptosis by the NAD(P)H/FSP1/CoQ10 system and its application in treating diseases
The role of the NAD(P)H/FSP1/CoQ10 system in ferroptosis
Ferroptosis inhibitor protein 1 (FSP1) is a key protein that protects against ferroptosis. FSP1 knockout cell lines are more sensitive to ferroptosis inducers and can be rescued by overexpression of FSP1 [29, 80]. As a member of the quinone oxidoreductase (NDH-2) family, FSP1 can reduce CoQ10 and reduce ubiquinone (CoQ) to dihydroubiquinone (CoQH2) in the plasma membrane and thereby prevent the accumulation of lipid peroxides. Moreover, Doll et al. found that FSP1 could catalyse the regeneration of COQ10 through NAD(P)H. In addition, FSP1 exerts a protective effect against ferroptosis induced by GPX4 deficiency. Collectively, the NAD(P)H/FSP1/CoQ10 pathway is another antioxidant pathway parallel to the classic glutathione-based GPX4 that works together with GPX4 to inhibit lipid peroxidation and ferroptosis [80, 81].
The application of exosome-regulated ferroptosis in treating different diseases through the NAD(P)H/FSP1/CoQ10 system
Exosomes in the NAD(P)H/FSP1/CoQ10 system mainly regulate the expression of FSP1 through their RNAs to affect ferroptosis of target cells and then participate in disease development. Exosomal miR-4443 has been reported to be a key regulator of oncogenesis and metastasis and a key mediator of drug resistance in tumours [82, 83]. Song et al. found that exosomal miR-4443 was enriched in clinical non-small cell lung carcinoma (NSCLC) resistant to cisplatin and exosomes of cisplatin-resistant NSCLC cell lines compared to cisplatin-sensitive tumour tissues. Through further bioinformatics analysis and luciferase assays, miR-4443 was shown to regulate FSP1 expression in an N6-methyladenosine-dependent manner through its target gene methyltransferase-like 3 (METTL3) and increase the resistance of tumour cells to ferroptosis, promoting resistance to cisplatin in NSCLC [84]. In addition, altered expression of ferroptosis-related markers was observed in an in vivo acute spinal cord injury (ASCI) model [35]. Shao et al. established in vitro models of ASCI mice and hypoxic cell in vivo models and found that MSC-Exos were able to inhibit ROS production by upregulating FSP1 expression. Therefore, damaged neurons are protected from ferroptosis, and the proliferation of hypoxic nerve cells and neurological dysfunction are restored in ASCI mice. Further mechanistic study found that LncGm36569, which was highly expressed in MSC-Exos, could bind to its competing endogenous RNA, miR-5627-5p, to upregulate the expression of its target gene FSP1, thereby inhibiting ferroptosis and promoting the repair of nerve function [85,86,87].
Exosome-regulated ferroptosis by the iron metabolism pathway and its application in treating diseases
The role of the iron metabolism in ferroptosis
Iron is one of the essential trace elements in the body and is essential for cell survival. However, iron metabolism disorders, especially iron overload, promote ROS generation and accumulation through the Fenton reaction, thus triggering ferroptosis [33, 88]. Extracellular iron (Fe3+) can form a complex with circulating transferrin (TF) and bind to a specific membrane transferrin receptor protein-1 (TFR1) in cell endosomes. Subsequently, Fe 3+ in the nucleus is reduced to ferrous iron (Fe 2+) by six-transmembrane epithelial antigen of prostate 3 metalloreductase (STEAP3). Then, mediated by divalent metal transporter 1 (DMT1) or zentz-iron regulatory protein family 8/14 (Zip8/14), Fe 2+ is released into the cytoplasm and stored in LIP or ferritin [89]. Ferritin is the main intracellular iron storage protein complex, consisting of two subunits, ferritin light peptide 1 (FTL1) and ferritin heavy peptide 1 (FTH1) [90]. Moreover, Fe 2+ can also be transported out of cells via ferroprotein (FPN) [91]. Ferritin can be degraded by autophagy and is regulated by nuclear receptor coactivator 4 (NCOA4). Thus, a large amount of free Fe 2+ is released, which increases the accumulation of intracellular iron and ROS and leads to cell ferroptosis [92]. In addition, iron response element binding protein 2 (IREB2), a major regulator of iron metabolism, regulates the expression of TRFC, FTH1, and FTL [93]. Fe 2+ entering cells can transport iron out of cells through DMT1, ferritin MVBs, and exosomes. Inhibition of DMT1 or blocking of MVBs can limit intracellular iron outflow and thus increase the intracellular iron level and induce cell ferroptosis. Notably, Brown et al. demonstrated that extracellular vesicles, especially exosomes, could expel intracellular iron when stimulated by ferroptosis, which is an important mechanism driving ferroptosis resistance [94, 95]. In short, increased iron intake, decreased iron storage, or decreased iron outflow can lead to iron overload, which can lead to ferroptosis.
The application of exosome-regulated ferroptosis in treating different diseases through the iron metabolism pathway
It is known that the maintenance of intracellular iron homeostasis is very important for cell survival and even for normal functioning of the body. As a result, in some diseases, self-derived exosomes can regulate the progression of ferroptosis by regulating proteins involved in iron metabolism, leading to an exacerbation of the disease. Sepsis induces sepsis-associated encephalopathy (SAE) and aggravates the progression of the disease. Through a rat model of sepsis, Wei et al. found that the blood–brain barrier of septic rats was damaged and that the levels of the serum exosome nuclear-enriched transcript 1 (NEAT1), a type of lncRNA, were significantly increased [21, 96]. Previous studies have shown that NEAT1 acts as a ceRNA of miR-9-5p, and its target genes are TFRC and glutamic oxaloacetic transaminase 1 (GOT1), an iron export protein that is essential for iron transport between different cells [97, 98]. NEAT1 sponging miR-9-5p in exosomes promotes the expression of TFRC and GOT1, induces ferroptosis of brain microvascular endothelial cells, leads to SAE and aggravates disease [21]. Similarly, exosomal miR-222 from HBV-infected LO-2 cells inhibits ferroptosis by inhibiting TFRC and promotes the activation of the hepatic stellate cell line LX-2. This leads to liver fibrosis and ultimately accelerates the development of liver failure [23]. In contrast, as an important adipokine, the effect of adipsin on injured cardiomyocytes after myocardial infarction (MI) is surprising. Man et al. found that exosomes derived from pericardial adipose tissue can effectively deliver adipsin to myocardial tissue. They can also interact with iron regulatory protein 2 (IRP2) to upregulate the level of FTH in the peri-infarct area and downregulate the level of transferrin TFR to maintain iron homeostasis, thereby protecting cardiomyocytes against ferroptosis [99, 100].
BMMSC transplantation is an important option for the treatment of a variety of diseases, which can enter damaged tissues and contribute to tissue repair [31]. However, interference with the internal environment and metabolism can affect efficacy, especially since the latter is prone to ferroptosis [101, 102]. Studies have found that modification of BMMSCs (HO-1/BMMSCs) by haem oxygenase-1 (HO-1), an antioxidant enzyme that protects cells from oxidative stress, can prolong their retention time in vivo and optimize their paracrine function, thereby enhancing the enrichment and function of exosomes [103, 104]. Hepatic IRI is an inevitable outcome after a liver transplant. However, steatotic liver, a common type of enlarged standard donor liver, is sensitive to postoperative IRI and poorly tolerated [105, 106]. Hepatic IRI is also closely related to the abnormal expression of iron metabolism-related proteins leading to ferroptosis [107]. Therefore, Li and his team constructed the IRI model of fatty liver and the HR model of fatty liver cells (SHP-HR). They found that the expression of IREB2 was increased in steatotic liver IRI and SHP-HR, which led to ferroptosis of hepatocytes and further aggravated liver tissue damage. HO-1/BMMSC-derived exosomes enriched with miR-29a-3p, which downregulates IREB2, ameliorated liver tissue and hepatocyte injury and inhibited ferroptosis in vitro and in vivo [108]. In the same year, the team used the rat liver transplantation model with severe steatotic donor liver as the donor and the SHP-HR model to verify again that HO-1/BMMSC-derived exosomes were involved in the regulation of hepatocyte ferroptosis. To further investigate its intrinsic mechanism, Song et al. used miRNA sequencing technology to screen upregulated miRNAs in exosomes and analysed the target genes of the upregulated miRNAs. They found that STEAP3 could interact with miR-124-3p. HO-1/BMMSC-derived exosomes enriched with miR-124-3p can reduce the degree of ferroptosis of hepatocytes by inhibiting STEAP3, thereby alleviating liver injury [24]. In conclusion, these studies provide a new therapeutic target for IRI in steatosis donor livers and a new exosome transplantation strategy for exosome-regulated ferroptosis.
In addition to HO-1/BMMSCs, Yi et al. experimentally demonstrated that ADSCs overexpressing miR-19b-3p could efficiently package it into their exosomes. Exosomes can deliver miR-19b-3p to the site of intracerebral haemorrhage to inhibit ferroptosis and reduce cell damage. It has been shown that miR-19b-3p achieves these results by targeting IRP2 [25]. In addition, exosomes themselves are rich in miRNAs. The transplantation of HucMSC-derived exosomes enriched with miR-23a-3p has been shown to improve myocardial infarction. It is worth noting that the expression of DMT1 was significantly higher in mice with acute myocardial infarction at 24 and 48 h than in mice in the control group. Further studies showed that miR-23a-3p could reduce myocardial injury by targeting DMT1 to improve cardiac myocyte ferroptosis [32]. Furthermore, vascular endothelial cell-derived exosomes (EC-Exos) have been identified as promising targeted nanoparticles, especially in the skeletal system [30]. Using Kyoto Encyclopedia of Genes and Gnomes (KEGG) pathway enrichment analysis, Yang et al. found that dexamethasone treatment induced activation of the ferroptosis pathway. EC-Exos could resist glucocorticoid-induced inhibition of osteoblasts by inhibiting ferrophagy-dependent ferroptosis in vitro and in vivo, thereby alleviating osteoporosis [109].
Exosome-regulated ferroptosis by the lipid metabolism pathway and its application in treating diseases
The role of the lipid metabolism in ferroptosis
The heart of ferroptosis is the accumulation of iron-dependent lipid peroxides, so lipid metabolism is critical to the occurrence and development of ferroptosis. It has been shown that ROS can react with polyunsaturated fatty acids (PUFAs) on lipid membranes to cause lipid peroxidation, which is one of the necessary stages for ferroptosis to eventually occur. Free PUFAs are substrates for the synthesis of lipid signal transduction mediators. They are esterified by acyl-CoA synthase long chain family member 4 (ACSL4) and incorporated into membrane phospholipids (PLs) with the help of recombinant lysophosphatidylcholine acyltransferase 3 (LPCAT3). ACSL4 is known to be positively correlated with susceptibility to ferroptosis, and the loss of LPCAT3 protects cells from damage caused by ferroptosis [37]. In turn, ferroptosis signals can be transmitted under lipid oxygenase (LOX) peroxide [110, 111]. Among different kinds of LOXs, arachidonate 15-lipoxygenase (ALOX15) is believed to play a core role in lipid peroxidation and ferroptosis [112]. Therefore, the combined action of ACSL4, LPCAT3, and ALOX15 can produce overwhelming lipid peroxidation in the cell, leading to the occurrence of cellular ferroptosis. In addition, phosphatidylethanolamine (PE), containing arachidonic acid (AA) and its derivative adrenal acid – a type of oxidized PL – has been shown to be the phospholipid most likely to induce ferroptosis [113].
The application of exosome-regulated ferroptosis in treating different diseases through the lipid metabolism pathway
Lipid metabolism plays an important role in tumorigenesis, invasion, metastasis, and many other tumour processes [114]. It has been shown that exosomes can regulate the function of target cells by secreting their contents into the tumour microenvironment and thus affect the occurrence and development of tumours [115, 116]. Additionally, the regulatory effect of exosomes on tumours is also associated with ferroptosis [117]. As a result, many studies have focused on ferroptosis and lipid metabolism to investigate the regulatory effects of exosomes in cancer cells. Using mass spectrometry of clinical samples from gastric cancer, Zhang and his team found that the levels of ALOX15 decreased, but the levels of ubiquitin-specific protease 7 (USP7) and heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) increased significantly in tumour tissue. By establishing relevant cell models, such as primary stromal cells and cancer cells, they found that cancer-associated fibroblast (CAF)-derived exosomes can secrete miR-552, a potential inhibitor of ALOX15, and hnRNPA1 as well as USP7 can increase the secretion and expression of miR-552 in exosomes. Cisplatin and paclitaxel promote the secretion of miR-552 from CAFs through the USP7/hnRNPA1 axis. This leads to the inhibition of ALOX15 and a reduction in lipid ROS accumulation in cancer cells, ultimately leading to a reduction in chemotherapy sensitivity [118]. Similarly, in 2021, the same team discovered that exosomal lncFERO (exo-lncFERO) secreted by gastric cancer cells could regulate the tumour-inducing properties of gastric cancer stem cells (GCSCs) by regulating ferroptosis of GCSCs through lipid ROS [26]. Recent studies have found that lipid metabolism pathways in GCSCs are significantly upregulated compared to those in differentiated GC cells, and stearoyl-coa-desaturase (SCD1), an enzyme that regulates the production of tumour lipid ROS, has the highest expression level [27, 119, 120]. Based on this, in vitro and in vivo experimental data provided by Zhang and his team demonstrated that exo-lncFERO can interact with SCD1 in GCSCs and recruit hnRNPA1 to promote the expression of SCD1. This leads to the dysregulation of PUFA levels in GCSCs and subsequent inhibition of ferroptosis. In addition, hnRNPA1 also enhanced the secretion of exo-lncFERO by GC cells, indicating that the exo-lncFERO/hnRNPA1/SCD1 axis can control the tumorigenic properties and resistance to chemotherapy of GCSCs [26]. Previous evidence suggests that obesity is strongly associated with cancer progression and poor prognosis [121, 122]. Zhang et al. found that microsomal triglyceride transfer protein (MTTP) expression was increased in plasma exosomes of colorectal cancer (CRC) patients with a high body fat rate through clinical studies. This increased expression reversed the occurrence of ferroptosis in CRC cells and made them resistant to oxaliplatin chemotherapy. Using transfection of KD-MTTP lentivirus into the abdominal fat of obese mice, a mechanistic study in colorectal cancer organoids found that adipocyte-derived exosomes could reduce the level of PUFA by upregulating GPX4 and xCT through the MTTP/PRAP1/ZEB1 axis, thereby inhibiting the production of lipid ROS and the incidence of ferroptosis [123]. In addition, the anti-ferroptosis properties of lung adenocarcinoma (LUAD) are also regulated by exosomes on lipid metabolism. By analysing plasma and tissue samples from healthy individuals and LUAD patients, Wang et al. found that plasma-derived exosomes from LUAD patients reduced lipid peroxidation. Further analysis of the LUAD mouse model showed that this phenomenon may be attributed to the increased expression of circRNA_101093 (cir93) in LUAD cells by exosomes. Cir93 can interact with fatty acid binding protein 3, thereby enhancing the transport of AA and its reaction with taurine and reducing the overall level of AA in the cell. Additionally, NAT, the reaction product of taurine and AA, can also inhibit the infiltration of AA into the plasma membrane by inhibiting ACSL4, LPCAT3, etc., and finally increase the ferroptosis resistance of LUAD [28]. In addition to its role in cancer, exosome regulation of ferroptosis via the lipid metabolic pathway is equally important in AKI. Sun et al. established a mouse model of AKI induced by IRI. It was demonstrated that human urine-derived stem cell-derived exosomes (USC-Exos) enriched with the lncRNA taurine-upregulated gene 1 (TUG1) effectively alleviated cell damage and ferroptosis. The potential mechanism may be related to the interaction between RNA binding protein serine/arginine splicing factor 1 (SRSF1) and ACSL4 mRNA stability [36].
Conclusions
Research has indicated that the modulation of ferroptosis could offer a novel approach for the treatment of specific ailments. As regenerative medicine continues to advance, exosome-regulated ferroptosis in disease treatment has garnered increasing scholarly interest. Simultaneously, we have observed that the application of exosome-regulated ferroptosis is less relevant in the context of inflammatory diseases and other fields. Inflammation, as a pivotal pathological process underlying numerous diseases, typically arises from detrimental stimuli such as infection and tissue damage, thereby initiating a complex biological response [38]. Intriguingly, ferroptosis exhibits a close association with inflammation. Studies have demonstrated that intracellular iron overload can promote M1 macrophage polarization and amplify inflammatory responses [124]. Furthermore, during the course of inflammation, disruption in iron homeostasis occurs alongside excessive tissue and cellular damage [125]. In brief, the interaction between ferroptosis and inflammation in inflammatory diseases has the potential to exacerbate the progression of the disease. Consequently, employing a combination of anti-ferroptosis and anti-inflammatory therapies may disrupt this detrimental cycle and potentially achieve synergistic effects (1 + 1 > 2). Currently available evidence suggests that exosomes can alleviate inflammatory bowel disease through inhibition of ferroptosis; however, further investigations are warranted to elucidate the precise mechanisms involved [126].
Furthermore, ferroptotic cells can elicit proinflammatory damage-associated molecular patterns (DAMPs) and trigger immune responses [127]. The interplay between ferroptosis and immune cells has garnered significant attention, particularly in the context of antitumour effects. Ferroptosis in tumour cells leads to the release of various immune-stimulatory signals, which can facilitate dendritic cell maturation, enhance macrophage phagocytosis of ferroptotic cells, and further augment CD8+ Tcells infiltration into tumours [128]. Moreover, exosome delivery carrier characteristics offer additional possibilities for combating tumour drug resistance by leveraging ferroptosis. Consequently, the combination of exosome-mediated ferroptosis and tumour immune activation has become a new antitumour immunotherapy strategy. Cai et al. developed a nanoreactor Cu a ZIF-8 coated surface layer loaded with the ferroptosis agonist erastin to activate cancer cell ferroptosis while influencing TAM phenotypic approach. This approach also increased IFNγ secretion by CD8+T cells and further promoted erastin-induced ferroptosis mediated by the nanoreactor [129]. Additionally, Wang et al. harnessed exosome properties along with ferroptosis to construct mixed nanoparticles comprising a biocompatible oleic acid-Fe3O4 core attached with oxaliplatin polymers and Prominin2 siRNA. These nanoparticles inhibited iron efflux from tumour cells while enhancing their susceptibility to undergo ferroptosis and activating their immunogenicity against metastasis [130].
Overall, this review provides a comprehensive analysis, surpassing prior studies, to systematically elucidate that exosomes can regulate ferroptosis through four main pathways: system Xc-/GSH/GPX4, system NAD(P)H/FSP1/CoQ10, iron metabolism and lipid metabolism (Figs. 1 and 2). Based on the new insight that reasonable induction or inhibition of ferroptosis can regulate disease progression, we believe that ferroptosis may be the most promising candidate for exosome influence on the progression of different diseases. However, there remain several questions that necessitate attention prior to the broader implementation of exosome-regulated ferroptosis. Additional investigations are imperative to ascertain the safety, dosage response, and potential adverse effects of exosomes. However, the understanding of the upstream and downstream mechanisms involved in exosome-mediated ferroptosis, as well as its potential therapeutic applications, remains limited due to a lack of investigations. Additionally, the present study mostly focused on the regulation of nucleic acid molecules in exosomes, neglecting a comprehensive exploration of the impact of protein and lipid molecules on ferroptosis. Moreover, much of the current research is based on animal models and cell experiments, but there is a lack of connection between these basic studies and clinical practice.

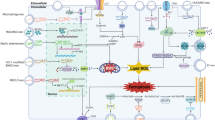
Mechanisms of exosome-regulated ferroptosis through “defence pathways”. The system Xc-/GSH/GPX4 and NAD(P)H/FSP1/CoQ10 axes serve as two “defence pathways” to protect cells from ferroptosis. In addition, the activation of the Nrf2 antioxidant pathway cooperated with GPX4 to inhibit lipid peroxidation and ferroptosis. Exosomes derived from different cells can directly or indirectly regulate the above pathways through their functional components to regulate the occurrence and development of ferroptosis in cells. This diagram illustrates the pathway by which exosomes regulate ferroptosis by inhibiting or promoting the key targets SLC7A11, GPX4, FSP1, and Nrf2. SLC7A11: solute carrier family 7 member 11; GPX4: glutathione peroxidase 4; FSP1: ferroptosis suppressor protein 1; Nrf2: nuclear factor E2-related factor 2

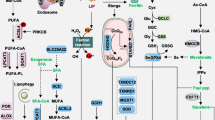
Mechanisms of exosome-regulated ferroptosis through the “offensive pathway”. Iron metabolism and lipid metabolism pathways serve as two “offensive pathways” that can promote ferroptosis by promoting the accumulation of intracellular iron and lipid peroxidation. Exosomes derived from different cells can directly or indirectly inhibit iron metabolism and lipid metabolism pathways to inhibit the occurrence of intracellular ferroptosis. The key targets of exosomes in regulating lipid metabolism pathways included TFR1, IRP2, IREB2, STEAP3, and DMT1. The key targets of exosomes in regulating lipid metabolism pathways included PUFA, ACSL4, and ALOX15. TFR1: transferrin receptor 1; IRP2: iron regulatory protein 2; IREB2: iron response element-binding protein 2; STEAP3: six-transmembrane epithelial antigen of prostate 3 metalloreductase; DMT1: divalent metal transporter 1; PUFA: polyunsaturated fatty acid; ACSL4: acyl-CoA synthetase long-chain family member 4; ALOX15: arachidonate 15-lipoxygenase
Consequently, conducting further comprehensive investigations into the correlation between exosomes and ferroptosis, in conjunction with the management of inflammation, immunity, and other related aspects, will broaden the potential applications of exosome-mediated regulation of ferroptosis across various disease domains. Additionally, this will offer a more defined trajectory and a more robust theoretical foundation for the clinical exploration of exosome-mediated regulation of ferroptosis. Furthermore, a promising direction for future research is to investigate whether exosomes that regulate other emerging forms of cell death, such as cuproptosis, are also involved in disease progression [131]. In any case, the exploration of exosome-regulated ferroptosis may provide benefits in disease intervention and treatment.
Availability of data and materials
Not applicable.
Abbreviations
- ROS:
-
reactive oxygen species
- IRI:
-
ischaemia–reperfusion injury
- LIP:
-
labile iron pool
- MVBs:
-
multivesicular bodies
- miRNA:
-
microRNA
- lncRNA:
-
long non-coding RNA
- SLC3A2:
-
Solute carrier family 3 member 2
- SLC7A11:
-
Solute carrier family 7 member 11
- GSH:
-
glutathione
- NFE2L2:
-
erythroid 2-like 2
- BAP1:
-
BRCA1-associated protein 1
- GPX4:
-
glutathione peroxidase 4
- HFD:
-
high-fat diet
- ATM-Exo:
-
adipose tissue macrophage-derived exosomes
- CF-Exo:
-
cardiac fibroblast derived exosomes
- MSC-Exo:
-
mesenchymal stem cell-derived exosomes
- ALI:
-
acute liver injury
- CCL4:
-
carbon tetrachloride
- I/R:
-
ischemia/reperfusion
- BMMSC-EVs/Fer-1:
-
Fer-1 loaded bone marrow mesenchymal stem cell-derived extracellular vesicles
- ADSC:
-
adipose-derived stem cell
- HExo:
-
hypoxia-pretreated adipose-derived stem cells exosomes
- DFU:
-
diabetic foot ulcer
- circ-ITCH:
-
circRNA-itchy E3 ubiquitin protein ligase
- TAF15:
-
TATA-Box binding protein associated factor 15
- HSCs:
-
hepatic stellate cells
- HucMSC-Exo:
-
human umbilical cord mesenchymal stem cell-derived exosomes
- CCR2:
-
C–C motif chemokine receptor-2
- FSP1:
-
ferroptosis inhibitor protein 1
- NSCLC:
-
non-small cell lung carcinoma
- METTL3:
-
methyltransferase-like 3
- ASCI:
-
acute spinal cord injury
- Fe3+ :
-
extracellular iron
- TF:
-
transferrin
- TFR1:
-
transferrin receptor protein-1
- Fe2+ :
-
ferrous iron
- STEAP3:
-
six-transmembrane epithelial antigen of prostate 3 metalloreductase
- DMT1:
-
divalent Metal Transporter 1
- Zip8/14:
-
zentz-iron regulatory protein family 8/14
- FTL1:
-
ferritin light peptide 1
- FTH1:
-
ferritin heavy peptide 1
- FPN:
-
ferroprotein
- IREB2:
-
iron response element binding protein 2
- SAE:
-
sepsis associated encephalopathy
- NEAT1:
-
nuclear-enriched transcript 1
- GOT1:
-
glutamic oxaloacetic transaminase 1
- MI:
-
myocardial infarction
- IRP2:
-
iron regulatory protein 2
- HO-1/BMMSCs:
-
modification of BMMSCs by heme oxygenase-1
- SHP-HR:
-
HR model of fatty liver cells
- EC-Exo:
-
endothelial cell-derived exosomes
- KEGG:
-
Kyoto Encyclopedia of Genes and Gnomes
- PUFAs:
-
polyunsaturated fatty acids
- ACSL4:
-
acyl-CoA synthase long chain family member 4
- PLs:
-
phospholipids
- LPCAT3:
-
Lysophosphatidylcholine Acyltransferase 3
- LOXs:
-
lipid oxygenase
- ALOX15:
-
arachidonate 15-lipoxygenase
- PE:
-
phosphatidylethanolamine
- AA:
-
arachidonic acid
- USP7:
-
ubiquitin-specific protease 7
- hnRNPA1:
-
heterogeneous nuclear ribonucleoprotein A1
- CAFs:
-
cancer-associated fibroblasts
- exo-lncFERO:
-
exosome lncFERO
- GCSCs:
-
gastric cancer stem cells
- SCD1:
-
stearoyl-Coa-desaturase
- MTTP:
-
microsomal triglyceride transfer protein
- CRC:
-
colorectal cancer
- LUAD:
-
lung adenocarcinoma
- cir93:
-
circRNA_101093
- USC-Exo:
-
human urine-derived stem cells-derived exosomes
- TUG1:
-
taurine-upregulated gene 1
- 1SRSF1:
-
Serine/arginine splicing factor 1
References
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Distéfano AM, Martin MV, Córdoba JP, Bellido AM, D’Ippólito S, Colman SL, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. 2017;216:463–76.
Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172:409–22.
Gleason A, Bush AI. Iron and ferroptosis as therapeutic targets in Alzheimer’s disease. Neurotherapeutics. 2021;18:252–64.
Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314:H659–68.
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–80.
Qi J, Kim JW, Zhou Z, Lim CW, Kim B. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am J Pathol. 2020;190:68–81.
Liu P, Feng Y, Li H, Chen X, Wang G, Xu S, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett. 2020;25:10.
Choi N, Whitlock R, Klassen J, Zappitelli M, Arora RC, Rigatto C, et al. Early intraoperative iron-binding proteins are associated with acute kidney injury after cardiac surgery. J Thorac Cardiovasc Surg. 2019;157:287–97.
Xiao W, Beibei F, Guangsi S, Yu J, Wen Z, Xi H, et al. Iron overload increases osteoclastogenesis and aggravates the effects of ovariectomy on bone mass. J Endocrinol. 2015;226:121–34.
Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–14.
Trohatou O, Roubelakis MG. Mesenchymal stem/stromal cells in regenerative medicine: past, present, and future. Cell Reprogram. 2017;19:217–24.
Phinney DG, Pittenger MF. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells. 2017;35:851–8.
El Andaloussi S, Mäger I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–57.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
Sanwlani R, Gangoda L. Role of extracellular vesicles in cell death and inflammation. Cells. 2021;10:2663.
Tan Y, Huang Y, Mei R, Mao F, Yang D, Liu J, et al. HucMSC-derived exosomes delivered BECN1 induces ferroptosis of hepatic stellate cells via regulating the xCT/GPX4 axis. Cell Death Dis. 2022;13:319.
Zhao X, Si L, Bian J, Pan C, Guo W, Qin P, et al. Adipose tissue macrophage-derived exosomes induce ferroptosis via glutathione synthesis inhibition by targeting SLC7A11 in obesity-induced cardiac injury. Free Radic Biol Med. 2022;182:232–45.
Liu D, Yang M, Yao Y, He S, Wang Y, Cao Z, et al. Cardiac fibroblasts promote ferroptosis in atrial fibrillation by secreting exo-miR-23a-3p targeting SLC7A11. Oxid Med Cell Longev. 2022;2022:3961495.
Wei XB, Jiang WQ, Zeng JH, Huang LQ, Ding HG, Jing YW, et al. Exosome-derived lncRNA NEAT1 exacerbates sepsis-associated encephalopathy by promoting ferroptosis through regulating miR-9-5p/TFRC and GOT1 Axis. Mol Neurobiol. 2022;59:1954–69.
Lin F, Chen W, Zhou J, Zhu J, Yao Q, Feng B, et al. Mesenchymal stem cells protect against ferroptosis via exosome-mediated stabilization of SLC7A11 in acute liver injury. Cell Death Dis. 2022;13:271.
Zhang Q, Qu Y, Zhang Q, Li F, Li B, Li Z, et al. Exosomes derived from hepatitis B virus-infected hepatocytes promote liver fibrosis via miR-222/TFRC axis. Cell Biol Toxicol. 2022;39:467–81.
Wu L, Tian X, Zuo H, Zheng W, Li X, Yuan M, et al. miR-124-3p delivered by exosomes from heme oxygenase-1 modified bone marrow mesenchymal stem cells inhibits ferroptosis to attenuate ischemia-reperfusion injury in steatotic grafts. J Nanobiotechnology. 2022;20:196.
Yi X, Tang X. Exosomes from miR-19b-3p-modified ADSCs inhibit ferroptosis in intracerebral hemorrhage mice. Front Cell Dev Biol. 2021;9:661317.
Zhang H, Wang M, He Y, Deng T, Liu R, Wang W, et al. Chemotoxicity-induced exosomal lncFERO regulates ferroptosis and stemness in gastric cancer stem cells. Cell Death Dis. 2021;12:1116.
Gao Y, Li J, Xi H, Cui J, Zhang K, Zhang J, et al. Stearoyl-CoA-desaturase-1 regulates gastric cancer stem-like properties and promotes tumour metastasis via HIPPO/YAP pathway. Br J Cancer. 2020;122:1837–47.
Zhang X, Xu Y, Ma L, Yu K, Niu Y, Xu X, et al. Essential roles of exosome and circrna_101093 on ferroptosis desensitization in lung adenocarcinoma. Cancer Commun (Lond). 2022;42:287–313.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Song H, Li X, Zhao Z, Qian J, Wang Y, Cui J, et al. Reversal of osteoporotic activity by endothelial cell-secreted bone targeting and biocompatible exosomes. Nano Lett. 2019;19:3040–8.
Feng Y, Xu Q, Yang Y, Shi W, Meng W, Zhang H, et al. The therapeutic effects of bone marrow-derived mesenchymal stromal cells in the acute lung injury induced by sulfur mustard. Stem Cell Res Ther. 2019;10:90.
Song Y, Wang B, Zhu X, Hu J, Sun J, Xuan J, et al. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol Toxicol. 2021;37:51–64.
Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–90.
Liu J, Zhou Y, Xie C, Li C, Ma L, Zhang Y. Anti-ferroptotic effects of bone marrow mesenchymal stem cell-derived extracellular vesicles loaded with Ferrostatin-1 in cerebral ischemia-reperfusion injury associate with the GPX4/COX-2 Axis. Neurochem Res. 2023;48:502–18.
Zhou H, Yin C, Zhang Z, Tang H, Shen W, Zha X, et al. Proanthocyanidin promotes functional recovery of spinal cord injury via inhibiting ferroptosis. J Chem Neuroanat. 2020;107: 101807.
Sun Z, Wu J, Bi Q, Wang W. Exosomal lncRNA TUG1 derived from human urine-derived stem cells attenuates renal ischemia/reperfusion injury by interacting with SRSF1 to regulate ASCL4-mediated ferroptosis. Stem Cell Res Ther. 2022;13:297.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Wallach D, Kang TB, Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol. 2014;14:51–9.
Zha J, Pan Y, Liu X, Zhu H, Liu Y, Zeng W. Exosomes from hypoxia-pretreated adipose-derived stem cells attenuate ultraviolet light-induced skin injury via delivery of circ-Ash1l. Photodermatol Photoimmunol Photomed. 2022;39:107–15.
Chen J, Li X, Liu H, Zhong D, Yin K, Li Y, et al. Bone marrow stromal cell-derived exosomal circular RNA improves diabetic foot ulcer wound healing by activating the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway and inhibiting ferroptosis. Diabet Med. 2022;40:e15031.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977.
Pathan M, Fonseka P, Chitti SV, Kang T, Sanwlani R, Van Deun J, et al. Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019;47:D516–9.
Isola AL, Chen S. Exosomes: the messengers of health and disease. Curr Neuropharmacol. 2017;15:157–65.
Baglio SR, Rooijers K, Koppers-Lalic D, Verweij FJ, Pérez Lanzón M, Zini N, et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res Ther. 2015;6:127.
Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126:1208–15.
Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913.
Edgar JR, Eden ER, Futter CE. Hrs- and CD63-dependent competing mechanisms make different sized endosomal intraluminal vesicles. Traffic. 2014;15:197–211.
Chen L, Chen R, Kemper S, Brigstock DR. Pathways of production and delivery of hepatocyte exosomes. J Cell Commun Signal. 2018;12:343–57.
Farooqi AA, Desai NN, Qureshi MZ, Librelotto D, Gasparri ML, Bishayee A, et al. Exosome biogenesis, bioactivities and functions as new delivery systems of natural compounds. Biotechnol Adv. 2018;36:328–34.
Shanmuganathan M, Vughs J, Noseda M, Emanueli C. Exosomes: basic biology and technological advancements suggesting their potential as ischemic heart disease therapeutics. Front Physiol. 2018;9:1159.
Wang Y, Shen Y, Liu H, Yin J, Zhang XT, Gong AY, et al. Induction of inflammatory responses in splenocytes by exosomes released from intestinal epithelial cells following Cryptosporidium parvum infection. Infect Immun. 2019;87:e00705–00718.
Calvo V, Izquierdo M. Inducible polarized secretion of exosomes in T and B lymphocytes. Int J Mol Sci. 2020;21:2631.
Baranyai T, Herczeg K, Onódi Z, Voszka I, Módos K, Marton N, et al. Isolation of exosomes from blood plasma: qualitative and quantitative comparison of ultracentrifugation and size exclusion chromatography methods. PLoS ONE. 2015;10:e0145686.
Xiao GY, Cheng CC, Chiang YS, Cheng WT, Liu IH, Wu SC. Exosomal miR-10a derived from amniotic fluid stem cells preserves ovarian follicles after chemotherapy. Sci Rep. 2016;6:23120.
Merchant ML, Rood IM, Deegens J, Klein JB. Isolation and characterization of urinary extracellular vesicles: implications for biomarker discovery. Nat Rev Nephrol. 2017;13:731–49.
Hock A, Miyake H, Li B, Lee C, Ermini L, Koike Y, et al. Breast milk-derived exosomes promote intestinal epithelial cell growth. J Pediatr Surg. 2017;52:755–9.
Ocansey D, Zhang L, Wang Y, Yan Y, Qian H, Zhang X, et al. Exosome-mediated effects and applications in inflammatory bowel disease. Biol Rev Camb Philos Soc. 2020;95:1287–307.
Wu L, Zhou J, Zhou W, Huang XF, Chen Q, Wang W, et al. Sorafenib blocks the activation of the HIF-2α/VEGFA/EphA2 pathway, and inhibits the rapid growth of residual liver cancer following high-intensity focused ultrasound therapy in vivo. Pathol Res Pract. 2021;220:153270.
Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. 2021;11:3052–9.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–8.
Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids. 2012;42:231–46.
Bridges RJ, Natale NR, Patel SA. System xc- cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20–34.
Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu R, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging (Albany NY). 2020;12:12943–59.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181–92.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X(c)(-) activity. Curr Biol. 2018;28:2388–99.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Xu X, Zhang X, Wei C, Zheng D, Lu X, Yang Y, et al. Targeting SLC7A11 specifically suppresses the progression of colorectal cancer stem cells via inducing ferroptosis. Eur J Pharm Sci. 2020;152: 105450.
Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–52.
Mao L, Zhao T, Song Y, Lin L, Fan X, Cui B, et al. The emerging role of ferroptosis in non-cancer liver diseases: hype or increasing hope. Cell Death Dis. 2020;11:518.
Wortmann M, Schneider M, Pircher J, Hellfritsch J, Aichler M, Vegi N, et al. Combined deficiency in glutathione peroxidase 4 and vitamin E causes multiorgan thrombus formation and early death in mice. Circ Res. 2013;113:408–17.
Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol. 2018;9:1371.
Moosmayer D, Hilpmann A, Hoffmann J, Schnirch L, Zimmermann K, Badock V, et al. Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallogr D Struct Biol. 2021;77:237–48.
Pei Z, Liu Y, Liu S, Jin W, Luo Y, Sun M, et al. FUNDC1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through ACSL4-mediated ferroptosis. Metabolism. 2021;122:154840.
Thomas D, Apovian C. Macrophage functions in lean and obese adipose tissue. Metabolism. 2017;72:120–43.
Campbell B, Khatri P. Stroke. Lancet. 2020;396:129–42.
Huang C, Luo W, Wang Q, Ye Y, Fan J, Lin L, et al. Human mesenchymal stem cells promote ischemic repairment and angiogenesis of diabetic foot through exosome miRNA-21-5p. Stem Cell Res. 2021;52:102235.
Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27–42.
Guo Y, Chen T, Liang X, Gou S, Xiong J, Cui J, et al. Tumor cell derived exosomal GOT1 suppresses tumor cell ferroptosis to accelerate pancreatic cancer progression by activating Nrf2/HO-1 Axis via upregulating CCR2 expression. Cells. 2022;11:3893.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Zhang X, Sui S, Wang L, Li H, Zhang L, Xu S, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol. 2020;235:3425–37.
Li M, Zhang X, Ding X, Zheng Y, Du H, Li H, et al. Long noncoding RNA LINC00460 promotes cell progression by sponging miR-4443 in head and neck squamous cell carcinoma. Cell Transplant. 2020;29:963689720927405.
Ebrahimi SO, Reiisi S. Downregulation of miR-4443 and miR-5195-3p in ovarian cancer tissue contributes to metastasis and tumorigenesis. Arch Gynecol Obstet. 2019;299:1453–8.
Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021;276:119399.
Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17:272–83.
Yang Y, Tai W, Lu N, Li T, Liu Y, Wu W, et al. lncRNA ZFAS1 promotes lung fibroblast-to-myofibroblast transition and ferroptosis via functioning as a ceRNA through miR-150-5p/SLC38A1 axis. Aging (Albany NY). 2020;12:9085–102.
Shao C, Chen Y, Yang T, Zhao H, Li D. Mesenchymal stem cell derived exosomes suppress neuronal cell ferroptosis via lncGm36569/miR-5627-5p/FSP1 Axis in acute spinal cord injury. Stem Cell Rev Rep. 2022;18:1127–42.
Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17.
Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41:274–86.
Chen P, Li FM, Zhou YF, Qian C, Li J, Jiang LR, et al. Effects of alpha-lipoic acid on expression of iron transport and storage proteins in BV-2 microglia cells. Pharmacol Rep. 2017;69:1–5.
Coffey R, Ganz T. Iron homeostasis: an anthropocentric perspective. J Biol Chem. 2017;292:12727–34.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–8.
Gammella E, Recalcati S, Rybinska I, Buratti P, Cairo G. Iron-induced damage in cardiomyopathy: oxidative-dependent and independent mechanisms. Oxid Med Cell Longev. 2015;2015:230182.
Turcu AL, Versini A, Khene N, Gaillet C, Cañeque T, Müller S, et al. DMT1 inhibitors kill cancer stem cells by blocking lysosomal iron translocation. Chemistry. 2020;26:7369–73.
Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51:575–86.
Wu H, Liu A. Long non-coding RNA NEAT1 regulates ferroptosis sensitivity in non-small-cell lung cancer. J Int Med Res. 2021;49:300060521996183.
Xie Q, Lin S, Zheng M, Cai Q, Tu Y. Long noncoding RNA NEAT1 promotes the growth of cervical cancer cells via sponging miR-9-5p. Biochem Cell Biol. 2019;97:100–8.
Shen Y, Li X, Zhao B, Xue Y, Wang S, Chen X, et al. Iron metabolism gene expression and prognostic features of hepatocellular carcinoma. J Cell Biochem. 2018;119:9178–204.
Cook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, et al. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science. 1987;237:402–5.
Man W, Song X, Xiong Z, Gu J, Lin J, Gu X, et al. Exosomes derived from pericardial adipose tissues attenuate cardiac remodeling following myocardial infarction by adipsin-regulated iron homeostasis. Front Cardiovasc Med. 2022;9:1003282.
Yamada N, Karasawa T, Wakiya T, Sadatomo A, Ito H, Kamata R, et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: potential role of ferroptosis. Am J Transplant. 2020;20:1606–18.
Liu J, Ren Z, Yang L, Zhu L, Li Y, Bie C, et al. The NSUN5-FTH1/FTL pathway mediates ferroptosis in bone marrow-derived mesenchymal stem cells. Cell Death Discov. 2022;8:99.
Cao H, Yang L, Hou B, Sun D, Lin L, Song HL, et al. Heme oxygenase-1-modified bone marrow mesenchymal stem cells combined with normothermic machine perfusion to protect donation after circulatory death liver grafts. Stem Cell Res Ther. 2020;11:218.
Sun D, Cao H, Yang L, Lin L, Hou B, Zheng W, et al. MiR-200b in heme oxygenase-1-modified bone marrow mesenchymal stem cell-derived exosomes alleviates inflammatory injury of intestinal epithelial cells by targeting high mobility group box 3. Cell Death Dis. 2020;11:480.
Álvarez-Mercado AI, Gulfo J, Romero Gómez M, Jiménez-Castro MB, Gracia-Sancho J, Peralta C. Use of steatotic grafts in liver transplantation: current status. Liver Transpl. 2019;25:771–86.
Wang X, Walkey CJ, Maretti-Mira AC, Wang L, Johnson DL, DeLeve LD. Susceptibility of rat steatotic liver to ischemia-reperfusion is treatable with liver-selective matrix metalloproteinase inhibition. Hepatology. 2020;72:1771–85.
Corradini E, Buzzetti E, Dongiovanni P, Scarlini S, Caleffi A, Pelusi S, et al. Ceruloplasmin gene variants are associated with hyperferritinemia and increased liver iron in patients with NAFLD. J Hepatol. 2021;75:506–13.
Li X, Wu L, Tian X, Zheng W, Yuan M, Tian X, et al. miR-29a-3p in exosomes from heme oxygenase-1 modified bone marrow mesenchymal stem cells alleviates steatotic liver ischemia-reperfusion injury in rats by suppressing ferroptosis via iron responsive element binding protein 2. Oxid Med Cell Longev. 2022;2022:6520789.
Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B, Jiang LS, et al. Exosomes derived from vascular endothelial cells antagonize glucocorticoid-induced osteoporosis by inhibiting ferritinophagy with resultant limited ferroptosis of osteoblasts. J Cell Physiol. 2021;236:6691–705.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–4975.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 2020;31:62–76.
Sung BH, von Lersner A, Guerrero J, Krystofiak ES, Inman D, Pelletier R, et al. A live cell reporter of exosome secretion and uptake reveals pathfinding behavior of migrating cells. Nat Commun. 2020;11:2092.
Wu CY, Du SL, Zhang J, Liang AL, Liu YJ. Exosomes and breast cancer: a comprehensive review of novel therapeutic strategies from diagnosis to treatment. Cancer Gene Ther. 2017;24:6–12.
Shi Y, Qiu B, Huang L, Lin J, Li Y, Ze Y, et al. Exosomes and ferroptosis: roles in tumour regulation and new cancer therapies. PeerJ. 2022;10:e13238.
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. 2020;19:43.
Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019;79:5355–66.
Luis G, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, et al. Tumor resistance to ferroptosis driven by stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and fatty acid biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 2021;43:102006.
Lee J, You JH, Kim MS, Roh JL. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol. 2020;37:101697.
Song M, Giovannucci E. Preventable incidence and mortality of carcinoma associated with lifestyle factors among white adults in the United States. JAMA Oncol. 2016;2:1154–61.
Zhang Q, Deng T, Zhang H, Zuo D, Zhu Q, Bai M, et al. Adipocyte-derived exosomal MTTP suppresses ferroptosis and promotes chemoresistance in colorectal cancer. Adv Sci (Weinh). 2022;9:e2203357.
Zhou Y, Que KT, Zhang Z, Yi ZJ, Zhao PX, You Y, et al. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/ACETYL-p53 pathway. Cancer Med. 2018;7:4012–22.
Dar HH, Anthonymuthu TS, Ponomareva LA, Souryavong AB, Shurin GV, Kapralov AO, et al. A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: INOS/NO (•) sabotage of theft-ferroptosis. Redox Biol. 2021;45:102045.
Zhu Y, Qin H, Sun C, Shao B, Li G, Qin Y, et al. Endometrial regenerative cell-derived exosomes attenuate experimental colitis through downregulation of intestine ferroptosis. Stem Cells Int. 2022;2022:3014123.
Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14:759–67.
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4.
Li K, Xu K, He Y, Yang Y, Tan M, Mao Y, et al. Oxygen self-generating nanoreactor mediated ferroptosis activation and immunotherapy in triple-negative breast cancer. ACS Nano. 2023;17:4667–87.
Wang Y, Chen Q, Song H, Zhang Y, Chen H, Liu P, et al. A triple therapeutic strategy with antiexosomal iron efflux for enhanced ferroptosis therapy and immunotherapy. Small. 2022;18:e2201704.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 82270565; 82270574; 82070545; 82100574). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
X.C., Z.L., Z.H. and K.J. conceived the study and revised the manuscript; L.L. and Y.Y. wrote and revised the manuscript, constructed, and revised the figures; the rest of authors revised the manuscript. All authors approved the final manuscript and agreed to be responsible for this review.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, L., Ye, Y., Lin, R. et al. Ferroptosis: a promising candidate for exosome-mediated regulation in different diseases. Cell Commun Signal 22, 6 (2024). https://doi.org/10.1186/s12964-023-01369-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01369-w