
Abstract
Background
The purinergic receptor P2X7 plays a crucial role in infection, inflammation, and cell death. It is thought that P2X7 receptor stimulation triggers processing and release of the pro-inflammatory cytokine interleukin (IL)-1β by activation of the NLRP3 inflammasome; however, the underlying mechanisms remain poorly understood.
Methods
Modulation of IL-1β secretion was studied in THP-1 macrophages. Adenosine 5’-triphosphate (ATP), BzATP, nigericin and pharmacological inhibitors of P2X receptors, inflammatory caspases and the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein 3 (NLRP3) inflammasome were used to characterize signaling.
Results
In primed macrophages, IL-1β release was increased after P2X7 receptor activation by ATP and 2,3-O-(4-benzoylbenzoyl)-ATP (BzATP). Pharmacological inhibition or genetic knockout of NLRP3 does not completely inhibit IL-1β release in TLR2/1-primed macrophages. Increase in extracellular K+ as well as inhibition of caspase-1 or serine proteases maintained IL-1β release in macrophages stimulated with P2X7 receptor agonists at 50%.
Conclusions
Our findings suggest a previously unrecognized mechanism of P2X7 receptor mediated IL-1β release and highlight the existence of an NLRP3-independent pathway in human macrophages.
Video Abstract
Similar content being viewed by others


Background
IL-1β is a highly potent and proinflammatory cytokine [1]. While IL-1β facilitates antibacterial and antiviral effects, it is also strongly linked to inflammatory and autoimmune diseases [2,3,4]. IL-1β is released following a two-step process in macrophages. The first step, known as priming, is characterized by nuclear translocation of NF-κB which facilitates the expression of pro-IL-1β and NLRP3-associated proteins. The second step, known as activation, leads to NLRP3 inflammasome assembly and the processing of pro-caspase-1 into active caspase-1. Caspase-1, in turn, processes pro-IL-1β into its active form IL-1β [5, 6]. In human macrophages, priming can be initiated by multiple stimuli leading to activation of Toll-like receptors (TLR), Tumor necrosis factor (TNF) receptors, or IL-1 receptors [5, 7], while in human monocytes the priming step is dispensable [8,9,10]. Activation can be achieved by danger associated molecular patterns (DAMPs) such as ATP, particulate matter such as uric acid crystals, and pore-forming toxins such as nigericin. The common denominator connecting these NLRP3 activating stimuli is K+ efflux [11, 12]. However, the precise mechanisms by which the NLRP3 inflammasome responds to a variety of stimuli are poorly understood.
ATP is one of the best studied DAMPs [13, 14]. Extracellular ATP activates cell surface P2X and P2Y receptors. P2X receptors are membrane-bound ion channels that are permeable to calcium, sodium, and potassium [15, 16]. The P2X receptor most involved in inflammation is the P2X7 receptor, whose affinity for ATP is low. However, high ATP concentrations at sites of inflammation or tumors can lead to activation of the receptor [17, 18]. Activation of the P2X7 receptor by ATP or the more potent agonist BzATP [18, 19] serves as an activation signal that leads to IL-1β release mediated by K+ efflux and activation of the NLRP3 inflammasome [17].
To date, inflammatory signaling has often been studied in mouse bone marrow-derived macrophages [11], however, human macrophages have rarely been discussed. In this study, we investigate the underlying mechanism of P2X7 receptor mediated IL-1β release in human macrophages. Our findings are supported by using BzATP, which eliminates the possible confounding effects of high ATP concentrations. Our data suggest the existence of two signaling pathways leading to the release of IL-1β after activation of the P2X7 receptor, with both mechanisms being potentially NLRP3 independent in TLR-primed macrophages. One mechanism is dependent on K+ efflux and requires activation of caspase-1 and serine proteases, while the other mechanism is independent of K+ efflux, caspases, as well as serine, cysteine, and aspartic proteases.
Materials and methods
Cell culture
THP-1 cells (ACC 16, DSMZ-German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany) or THP-1 KO NLRP3 cells (thp-konlrp3z, Invivogen, Toulouse, France) were cultured in RPMI 1640 (11530586, Fisher scientific, Schwerte, Germany) containing 100 U/ml penicillin, 100 μg/ml streptomycin (P4333), 2 mM L-glutamine (G7513, both from Sigma-Aldrich, Taufkirchen, Germany) and 10% heat-inactivated fetal bovine serum (FBS; S0615, Sigma-Aldrich, Taufkirchen, Germany) at a density of 2–8 × 105 cells/ml. Cells were used from passage 4 to 25 and maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Cell lines were regularly tested negative for mycoplasma contamination (VenorGeM Classic Mycoplasma PCR detection kit, 11–8100, Minerva Biolabs, Berlin, Germany). For generating THP-1 derived macrophages, THP-1 monocytes were seeded into 24-well plates at a density of 4 × 105cells/ml in growth medium including 25 ng/ml PMA (phorbol 12-myristate 13-acetate; tlrl-pma, Invivogen, Toulouse, France). After 48 h, adherent cells were carefully washed with PBS (phosphate buffered saline; P04-53500, Pan Biotechne, Aidenbach, Germany) and rested in PMA-free medium for 24 h. PBMCs were isolated from buffy-coat donations (Institute of Experimental Haematology and Transfusion Medicine, University Hospital Bonn) by density gradient centrifugation using Bicoll separation media (BS L6115, Bio&Sell, Nuremberg, Germany). HEK-blue IL-1β cells were cultured in Dulbecco's modified Eagle's medium (DMEM, P04-03500, Pan Biotechne, Aidenbach, Germany) containing 4.5 g/l glucose, 10% (v/v) heat-inactivated fetal bovine serum (FBS; S0615), 2 mM l-glutamine (G7513), 100 U/ml penicillin, 100 μg/ml streptomycin (P4333, all from Sigma-Aldrich, Taufkirchen, Germany), 100 μg/ml normocin, 100 μg/ml zeocin (selective antibiotics, ant-nr-1, ant-zn-1, both from Invivogen, Tolouse, France). Cells were used from passage 3 to 20 and maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. To determine if THP-1 stimulated cells secrete bioactive form of IL-1β a total of 50 μl of THP-1 supernatant was transferred to a 96-well tissue culture–treated plate and mixed with 5 × 104 IL-1β reporter cells in test media (culture media without normocin and zeocin) and incubated for 20 h at 37 °C. After IL-1β receptor stimulation of HEK-Blue cells, NF-κB and AP-1 activation-induced production of secreted embryonic alkaline phosphatase (SEAP) that can be determined with the colorimetric substrate QuantiBlue (rep-qbs, Invivogen, Toulouse, France) by reading the optical density (OD) at 620 nm.
Cell stimulation
PBMCs as well as THP-1 monocytes and differentiated THP-1 derived macrophages were preincubated with LPS from Escherichia coli 0111:B4 (tlrl-3pelps), Pam2CSK4 (tlrl-pm2s-1), Pam3CSK4 (tlrl-pms-1, all from Invivogen, Toulouse, France) or growth medium for 3 h. Afterwards PBMCs and THP-1 macrophages were stimulated for additional 3 h or THP-1 monocytes for 6 h with 300 μM BzATP (2'/3'-O-(4-Benzoylbenzoyl)adenosine-5'-triphosphate, NU-1620–25), 5 mM ATP (Adenosine 5´-triphosphate, NU-1010-100G, both from Jena Bioscience, Jena, Germany) 10 μM nigericin (4312/10, Tocris Bioscience, Bristol, United Kingdom). In selected experiments, cells were preincubated with the non-competitive P2X7 receptor antagonist A804598 (1 μM, 4473, Tocris Bioscience, Bristol United Kingdom), irreversible P2X7 receptor antagonist oxidized ATP (oxATP, 300 μM, 505758, Merck, Darmstadt, Germany), P2X4 receptor antagonist 5-BDBD (25 μM, SML0450-5MG, Sigma-Aldrich, Taufkirchen, Germany), P2X receptor antagonist PPADS (100 μM, 0625, Tocris Bioscience, Bristol, United Kingdom), NLRP3 inhibitor MCC950 (10 μM, 5479, Tocris Bioscience, Bristol, United Kingdom) or Bay 11–7082 (20 μM, B5556, Sigma-Aldrich, Taufkirchen, Germany), Caspase-1 inhibitor Ac-YVAD-cmk (40 μM, 10014, Biomol, Hamburg, Germany), pan-caspase inhibitor Z-VAD-fmk (40 μM, tlrl-vad, Invivogen, Toulouse, France), serine protease inhibitor AEBSF (300 μM, 50985.100, Biomol, Hamburg, Germany), cysteine protease inhibitor E64 (10 μM, 324890, Merck, Darmstadt, Germany), aspartic protease inhibitor pepstatin A (50 μM, 2936, Carl Roth, Karlsruhe, Germany) 1 h before stimulation. To determine the optimal concentration of caspase inhibitors required to inhibit IL-1β secretion, we incubated increasing concentrations of Ac-YVAD-cmk and Z-VAD-fmk together with nigericin (Figure S1). To examine the dependency of potassium efflux, Pam3CSK4-primed THP-1 macrophages were stimulated in the presence of potassium chloride (75 mM, 6781.3, Carl Roth, Karlsruhe, Germany). To determine the contribution of the NLRP3 inflammasome, THP1 KO NLRP3 macrophages were primed with 1 μg/ml Pam3CSK4 for 3 h followed by stimulation with BzATP, ATP or nigericin for 3 h.
ELISA
After 3 h of stimulation of Pam3CSK4-primed PBMCs or THP-1 macrophages or after 6 h of stimulation of Pam3CSK4-primed THP-1 monocytes cell culture supernatants were collected and analyzed for IL-1β release using commercially available ELISA kits (88–7261-88 from Thermofisher Scientific, Darmstadt, Germany).
LDH
LDH assay was performed according to the manufacturer’s instructions (Thermofisher Scientific, Darmstadt, Germany). The percentage of LDH release was calculated compared to 100% cell lysis control.
RNA isolation, cDNA synthesis and qRT-PCR
Total RNA isolation was performed using innuPREP RNA Mini Kit 2.0 (845-KS-2040050, AnalytikJena, Jena,Germany) according to the manufacturer’s protocol. Hence, cDNA was synthesized with the help of iScript cDNA Synthesis Kit (1708891, Bio-Rad, Feldkirchen, Germany). Quantitative real-time RT-PCR (qRT-PCR) was performed as previously described [20, 21]. Primers (synthesized by TIB Molbiol, Berlin, Germany or eurofins genomics, Ebersberg, Germany) with the following sequences were used: GAPDH, 5’-CTCTCTGCTCCTCCTGTTCGAC-3’ and 5’-TGAGCGATGTGGCTCGGCT-3’; IL1B, 5’-TGGAGCAACAAGTGGTGT-3’ and 5’-TTGGGATCTACACTCTCCAGC-3’; IL18, 5´-TGCCAACTCTGGCTGCTAAA-3´ and 5´-TTGTTGCGAGAGAAGCGAT-3´, PRTN3, 5´- GCCGGCCACATAACATTTGC-3´and 5´-TACCCGCGTGAAGAAGTCAGG-3´; ELANE, 5´-AACGGCTACGACCCCGTAAA-3´ and 5´-CTGCACGTTGGCGTTGATGG-3´; CTSG, 5´-GCTGAGGCAGGGGAGATCATCG-3´ and 5´-GGGTGTTTTCCCGTCTCTGGA-3´
Fold difference in gene expression was normalized to the housekeeping gene GAPDH showing the most constant expression levels. The reaction mix containing cDNA template, primers and SYBR Green (iTaq Universal SYBR Green Supermix; 172–5125, Bio-Rad) was run under the conditions as previously described.
Statistical analysis
Data are expressed as means + SEM. For multiple comparisons, statistically significant differences were determined by one-way ANOVA followed by a Dunnett´s or Tukey´s post-test and considered significant at *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. For studies of inhibitory effects ATP, BzATP or nigericin induced IL-1β release was set to 100%. All other values were calculated accordingly. Statistical differences were assessed by one-sample t test against 100%. Statistical analysis was performed using GraphPad Prism software.
Results
Pam3CSK4-primed human THP-1 macrophages are a suitable model for studying NLRP3-mediated IL-1β release
We primed peripheral blood mononuclear cells (PBMCs) with the TLR2/1 ligand Pam3CSK4 followed by stimulation with ATP, BzATP, and nigericin. We confirmed that priming was necessary to induce IL-1β release in PBMCs (Fig. 1A). To determine pyroptotic cell death, release of lactate dehydrogenase (LDH) was quantified [22]. ATP, BzATP and nigericin increased LDH release in Pam3CSK4 primed PBMCs by 22%, 7%, and 25%, respectively, compared to unstimulated PBMCs (Fig. 1B). The human THP-1 cell line is a well-established and widely used model for studying activation of the NLRP3 inflammasome [23]. To confirm this, we stimulated both THP-1 monocytes and THP-1 macrophages identically to PBMCs (Fig. 1C, and D). We observed a strong increase in IL-1β release after stimulation with ATP, BzATP or nigericin, although the response was less than that observed in PBMCs.

Priming of macrophages is essential to induce release of bioactive IL-1β. A and B PBMCs were primed with Pam3CSK4 and then stimulated with ATP, BzATP or nigericin for 3 h. Supernatants were analyzed (A) for IL-1β concentration by ELISA and (B) LDH-release. Results are expressed as % of maximal LDH-release. Mean + SEM (n = 3—4). C THP-1 monocytes were primed with Pam3CSK4 and afterwards stimulated with ATP, BzATP and nigericin for 6 h. IL-1β concentration in the supernatants was analyzed by ELISA. Mean + SEM (n = 3—5). D THP-1 macrophages were primed and stimulated as described in (A). IL-1β concentration in the supernatants was analyzed by ELISA. Mean + SEM (n = 3—4). Two-tailed two-sample t test, ns ≥ 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. E THP-1 macrophages were primed with Pam3CSK4. Cells were then stimulated with ATP, BzATP and nigericin for 3 h. Supernatants from stimulated THP-1 macrophages were transferred on HEK-blue IL-1β reporter cells. SEAP production was detected by QUANTI-Blue and optical density was measured at 620 nm. Mean + SEM (n = 4). One-way ANOVA followed by Dunnett´s post-test, ns ≥ 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001
To investigate the bioactivity of the secreted IL-1β, we added the supernatants of stimulated THP-1 macrophages to HEK-blue IL-1β reporter cells (Fig. 1E). Bioactive IL-1β stimulates the IL-1 receptor of HEK-blue cells, which is accompanied by an NF-κB-mediated SEAP release, reflected by a colorimetric shift and higher absorbance of detection media. Supernatants of Pam3CSK4-primed THP-1 macrophages stimulated with ATP and BzATP resulted in a significant increase in absorbance confirming that ATP and BzATP induced the bioactive form of IL-1β. Although nigericin strongly induced IL-1β release in THP-1 macrophages we did not observe a significant increase in absorbance compared to control, which might be explained by cytotoxic effects of remaining nigericin in the supernatant.
P2X7 receptor induced IL-1β secretion is mostly NLRP3 independent
To determine whether ATP mediates the release of IL-1β via the P2X7 receptor or whether other receptors are also involved, we tested various P2X receptor antagonists. BzATP, a more potent P2X7 receptor agonist [18, 19], served as a reference. In THP-1 macrophages, IL-1β release by ATP and BzATP was inhibited in the presence of the P2X7 receptor-selective antagonists A804598 and oxATP (Fig. 2A). The inhibition of ATP-induced cytokine release by the irreversible antagonist oxATP was less pronounced than by the non-competitive reversible antagonist A804598 [24, 25]. Addition of the P2X4 receptor-specific antagonist 5-BDBD [26] did not decrease IL-1β release by ATP and BzATP. PPADS, a selective purinergic P2X receptor antagonist [27, 28], reduced the release of IL-1β by THP-1 macrophages stimulated with ATP and BzATP to 60.6% and 4.2%, respectively. As expected, IL-1β release mediated by the potassium ionophore nigericin was not affected by the addition of P2X receptor antagonists.

P2X7-mediated IL-1β release is NLRP3 independent. A THP-1 macrophages were primed with Pam3CSK4 and then stimulated with ATP, BzATP or nigericin for 3 h. Antagonists of P2X7 receptor (A-804598, oxATP), P2X4 receptor (5-BDBD) or P2X receptors (PPADS) were added 1 h before stimulation. IL-1β concentration in the supernatants was analyzed by ELISA. ATP, BzATP or nigericin induced IL-1β release was set to 100%. Mean + SEM (n = 3). One-sample t test against 100%, ns ≥ 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. B THP-1 macrophages were primed with Pam3CSK4. Potassium chloride was added together with ATP, BzATP or nigericin for 3 h. IL-1β concentration in the supernatants was analyzed by ELISA. ATP, BzATP or nigericin induced IL-1β release was set to 100%. Mean + SEM (n = 3). One-sample t test against 100%, *P ≤ 0.05, ****P ≤ 0.0001. C THP-1 macrophages were primed with Pam3CSK4 and then stimulated with ATP, BzATP or nigericin for 3 h. NLRP3 inhibitors MCC950 or Bay 11–7082 were added 1 h before stimulation. IL-1β concentration in the supernatants was analyzed by ELISA. ATP, BzATP or nigericin induced IL-1β release was set to 100%. Mean + SEM (n = 3—4). One-sample t test against 100%, ns ≥ 0.05, *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001 (D) NLRP3 KO THP-1 macrophages were primed with Pam3CSK4. Cells were then stimulated with ATP, BzATP or nigericin for 3 h. IL-1β concentration in the supernatants was analyzed by ELISA. Mean + SEM (n = 3). Two-tailed two sample t test, ns ≥ 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001
It has been proposed that whether the P2X7 receptor is activated by ATP or BzATP, or nigericin is used as an ionophore, ultimately K+ efflux from the intracellular space is critical for activation of the NLRP3 inflammasome [11, 12]. To determine the optimal extracellular potassium chloride concentration required to inhibit IL-1β secretion, we incubated increasing potassium concentrations together with nigericin (Figure S2A). A concentration of 75 mM extracellular potassium chloride proved to be nontoxic (Figure S2B). Nigericin-induced IL-1β release by THP-1 macrophages was completely blocked by 75 mM extracellular potassium chloride, whereas IL-1β secretion induced by ATP or BzATP could be reduced only by 50% (Fig. 2B). Assuming that the release of IL-1β is dependent on NLRP3, we tested the specific NLRP3 inflammasome inhibitor MCC950 [29]. MCC950 suppressed IL-1β secretion induced by ATP and nigericin to 24.4% and 0.9% (Fig. 2C). IL-1β release induced by BzATP was not affected, suggesting an NLRP3-independent release of IL-1β. We attempted to verify this result by using the NLRP3 inflammasome inhibitor Bay 11–7082 [30]. In the presence of Bay 11–7082, ATP-, BzATP-, and nigericin-induced IL-1β release was reduced to 50.5%, 53.7%, and 7.8%, respectively (Fig. 2C). The observed differences between MCC950 and the non-specific NLRP3 inhibitor Bay 11–7082 could be explained by interference of Bay 11–7082 with NF-κB signaling [7], therefore being able to reduce IL-1β release beyond direct inhibition of the NLRP3 inflammasome. To further support our results independently of pharmacological inhibition, we used THP-1 macrophages that are deficient for NLRP3. Stimulation with BzATP or ATP following priming resulted in a significant increase in IL-1β release, with IL-1β release being higher by BzATP than ATP. In contrast, nigericin failed to increase IL-1β release in primed NLRP3 knockout THP-1 macrophages (Fig. 2D).
P2X7 receptor induced IL-1β secretion is partially caspase independent
NLRP3 oligomerization leads to self-cleavage of pro-caspase-1 into its active form [31]. Subsequently, active caspase-1 cleaves pro-IL-1β into mature and bioactive IL-1β. To determine to which extent IL-1β secretion depends on caspases, we used the caspase-1 inhibitor Ac-YVAD-cmk and the pan-caspase inhibitor Z-VAD-fmk. Inhibition of caspase-1 activity decreased IL-1β secretion induced by ATP, BzATP and nigericin to 64.8%, 58.4% and 30.3%, respectively, without having toxic effects (Fig. 3A, and S3). The effect of the pan-caspase inhibitor Z-VAD-fmk on IL-1β release mediated by ATP and nigericin was even more pronounced and reduced cytokine levels to 22.6% and 1.6%, respectively. However, BzATP-induced IL-1β release was reduced to 56.4% and showed comparable levels to those observed for the specific caspase-1 inhibitor.

Influence of caspases, serine proteases, cysteine proteases and aspartic proteases on IL-1β release. A THP-1 macrophages were primed with Pam3CSK4 and then stimulated with ATP, BzATP or nigericin for 3 h. Inhibitors of Caspase-1 (Ac-YVAD-cmk) or pan-caspase (Z-VAD-fmk), serine protease (AEBSF), cysteine protease (E64), aspartic protease (pepstatin A) were added 1 h before stimulation. IL-1β concentration in the supernatants was analyzed by ELISA. ATP, BzATP or nigericin induced IL-1β release was set to 100%. Mean + SEM (n = 3—4). One-sample t test against 100%, ns ≥ 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. B THP-1 macrophages were primed with Pam3CSK4 and then stimulated with BzATP in the absence or presence of KCl for 3 h. Pan-caspase inhibitor (Z-VAD-fmk) or serine protease inhibitor (AEBSF) was added 1 h before stimulation. IL-1β concentration in the supernatants was analyzed by ELISA. BzATP induced IL-1β release was set to 100%. Mean + SEM (n = 3—7). One-way ANOVA followed by Tukey´s post-test, ns ≥ 0.05 (C) THP-1 macrophages were primed with Pam3CSK4 and then stimulated with ATP, BzATP or nigericin for 3 h. Gene expression of PRTN3 and ELANE and CTSG was normalized to GAPDH. Mean + SEM (n = 3)
To elucidate the involvement of other proteases in IL-1β secretion, we used inhibitors of serine-, cysteine-, and aspartic proteases. In the presence of the serine protease inhibitor AEBSF we observed a decrease in IL-1β secretion mediated by ATP, BzATP and nigericin (Fig. 3A). When AEBSF was used in combination with a pan-caspase inhibitor, IL-1β secretion by ATP and BzATP was further reduced, whereas IL-1β release induced by nigericin was completely blocked. IL-1β levels secreted by ATP-, BzATP- and nigericin-stimulated macrophages remained unchanged in the presence of the cysteine protease inhibitor E64 or the aspartic protease inhibitor pepstatin A. In contrast, the combination of E64 or pepstatin A with Z-VAD-fmk reduced IL-1β release mediated by ATP, BzATP, and nigericin. No toxic effects were observed (Figure S3). To determine whether caspase-dependent IL-1β release by BzATP was related to potassium efflux or whether these signaling cascades occur separately, we inhibited potassium efflux and caspases simultaneously. A synergistic effect could be ruled out, as the combination of potassium chloride and pan-caspase inhibitor Z-VAD-fmk reduced BzATP-induced IL-1β release to levels similar to the addition of potassium chloride alone (Fig. 3B). This trend was repeated when using the serine protease inhibitor AEBSF (Fig. 3B). Combining AEBSF with Z-VAD-fmk could not reduce BzATP-mediated IL-1β release any further than AEBSF alone. This observation suggests that potassium efflux, serine proteases, and caspases operate in the same signaling pathway. Gene expression levels of the serine proteases proteinase 3 (PRTN3), neutrophil elastase (ELANE), or cathepsin G (CTSG), which have been shown to alternatively process IL-1β compared to caspase-1 [32], were not affected by stimulation with ATP, BzATP or nigericin (Fig. 3C).
Priming with different TLR ligands affects signal transduction of P2X7 mediated IL-1β release
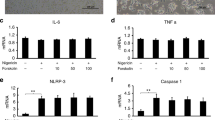
Not only TLR2/1 ligands can serve as priming signal for NLRP3 inflammasome activation. Therefore, we also used the TLR2/6 ligand Pam2CSK4 and the TLR4 ligand LPS and observed increased IL-1β release after stimulation with ATP, BzATP, or nigericin (Fig. 4A). After demonstrating that priming is essential to induce IL-1β release in THP-1 macrophages, we wanted to further characterize the role of different TLR signaling pathways. The relative gene expressions of IL1B and IL18 did not show a significant difference between TLR2 and TLR4 signaling (Fig. 4B). In macrophages stimulated with nigericin, inhibition of the NLRP3 inflammasome resulted in a comparable decrease in IL-1β release, regardless of which TLR ligand was used for priming (Fig. 4C). Furthermore, ATP-induced IL-1β release consistently showed partial independence from NLRP3 regardless of the TLR ligand used for priming. However, the extent of NLRP3 independence was different. Although incubation with MCC950 did not result in significant differences in IL-1β release when priming with TLR2/1 or TLR2/6 agonists, priming with a TLR4 agonist led to a significantly lower level of inhibition. Opposite results were observed in BzATP-stimulated cells. As demonstrated above (Fig. 2C), cytokine levels induced by BzATP remained unchanged in the presence of the NLRP3 inflammasome inhibitor MCC950 when priming was performed by TLR2/1 ligand Pam3CSK4. In contrast, inhibition of NLRP3 reduced BzATP-induced IL-1β cytokine levels to 44.6% and 57.0% in LPS- and Pam2CSK4-primed cells, respectively, indicating that TLR signaling differentially modulates signal transduction of P2X7-mediated IL-1β release (Fig. 4C).

Influence of priming with different TLR ligands on IL-1β release. A THP-1 macrophages were primed with LPS or Pam2CSK4. Cells were then stimulated with ATP, BzATP or nigericin for 3 h. IL-1β concentration in the supernatants was analyzed by ELISA. Mean + SEM (n = 3). Two-tailed two-sample t test, ns ≥ 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. B THP-1 macrophages were primed with Pam3CSK4, Pam2CSK4 or LPS. Gene expression of IL18 or IL1B was normalized to GAPDH. Mean + SEM (n = 3). One-way ANOVA followed by Tukey´s post-test, ns ≥ 0.05. C THP-1 macrophages were primed with Pam3CSK4, Pam2CSK4 or LPS and then stimulated with ATP, BzATP or nigericin. NLRP3 inhibitor (MCC950) was added 1 h before stimulation. IL-1β concentration in the supernatants was analyzed by ELISA. ATP, BzATP or nigericin induced IL-1β release was set to 100%. Mean + SEM (n = 3—5). One-way ANOVA followed by Tukey´s post-test, ns ≥ 0.05, *P ≤ 0.05, **P ≤ 0.01
Discussion
Although our findings support the importance of the NLRP3 inflammasome in P2X7 receptor-mediated IL-1β release [17], we propose an NLRP3-independent pathway in human macrophages. This previously unrecognized mechanism underlines the value of inhibiting the P2X7 receptor in inflammatory diseases [33,34,35] and highlights species-specific differences for IL-1β secretion between mouse and human macrophages. We demonstrated that stimulation with the P2X7 receptor ligand ATP led to NLRP3-independent IL-1β release. In Pam3CSK4-primed human THP-1 macrophages lacking NLRP3, stimulation with ATP led to a significant increase in IL-1β release. These effects were even more pronounced for BzATP, a synthetic ATP derivative and a more potent P2X7 receptor ligand [18].
A limitation of the present study is the use of the THP-1 cell line, which may deviate from primary cells. THP-1 cells are widely used to study the NLRP3 inflammasome [36], however, future studies should include human primary macrophages to confirm our findings. Yet, our results are in line with previous studies showing that the NLRP3 inhibitor MCC950 only partially inhibits IL-1β release in primary human macrophages, while the responses are completely dependent on NLRP3 in mouse macrophages [29, 37, 38].
We confirmed full NLRP3-dependency of nigericin induced IL-1β release [11], however, the contribution of NLRP3 was dispensable for ATP and BzATP. Thus, we aimed to investigate the role of the P2X7 receptor in IL-1β secretion, although it is believed to do so in a NLRP3-dependent manner [17]. Inhibitors of P2X and selective inhibitors of P2X7 receptors blocked BzATP but not nigericin induced IL-1β release indicating that NLRP3-independent IL-1β secretion is fully attributable to the P2X7 receptor. This finding is supported by results obtained with the selective P2X7 inhibitor A438079 in LPS-primed mouse macrophages [39]. Although we did not include P2X7 knockout cells in our study, we focused on various pharmacological inhibitors of P2X receptors with different selectivity and mode of action.
High extracellular potassium concentrations inhibit NLRP3 activation and IL-1β release [11, 12]. Our results suggest that P2X7 receptor-mediated IL-1β release is only partially dependent on K+ efflux, which differs from the previous concept established in mouse macrophages [11]. Our findings are consistent with results obtained in human macrophages [37], although a link to the P2X7 receptor and NLRP3 independence was not established previously. Our findings do not refute potassium efflux being the common trigger of NLRP3 inflammasome activation [11], but further underline the existence of a P2X7 receptor-facilitated NLRP3-independent IL-1β releasing pathway.
Caspase-1 is a central player in inflammasome-induced IL-1β release [5]. Although other caspases, such as caspase-8, can process IL-1β [40], only caspase-1 is involved in BzATP-induced IL-1β release. ATP and nigericin activate additional caspases, as pan-caspase inhibition further inhibited IL-1β release. While nigericin-induced IL-1β release is fully caspase-dependent, the P2X7 receptor agonists ATP and BzATP trigger IL-1β release partially caspase independent. This contradicts the notion that P2X7 receptor activation leads to IL-1β release via caspase-1 only [37, 41, 42], and once again emphasizes the differences of important signaling pathways between human and mouse macrophages. Furthermore, our results prove that K+ efflux is associated with caspase-1 for P2X7 receptor-dependent IL-1β release.
Caspase and NLRP3 independent mechanisms for IL-1β processing have been described, including serine proteases that are involved in caspase-1-independent IL-1β release [43, 44]. A significant reduction of IL-1β release was achieved in response to all stimuli tested by inhibition of serine protease. Serine protease inhibition reduces IL-1β release by human macrophages under acidic stress [37]. We show that serine proteases are also involved in both the NLRP3-dependent and -independent release of IL-1β. Simultaneous inhibition of caspases and serine proteases led to a similar reduction in IL-1β secretion compared to caspase inhibition alone, indicating that serin proteases are only involved in the caspase/IL-1β axis. Because a substantial level of IL-1β release remained after simultaneous inhibition of caspase and serine protease, we checked the involvement of cysteine and aspartic proteases. Aspartic protease inhibition reduced IL-1β release by human macrophages if stimulated with lactic acid [37]. We did not observe a reduction in IL-1β release with inhibition of either class of proteases.
Lastly, priming with TLR agonists differentially modulates NLRP3 dependency of P2X7 mediated IL-1β release. Priming with the TLR2/1 ligand Pam3CSK4 led to completely NLRP3-independent IL-1β release in BzATP-stimulated macrophages, while priming with TLR2/6 and TLR4 ligands led to some degree of NLRP3 dependency. Therefore, it would be interesting to determine the underlying mechanisms in the NLRP3 priming pathway triggered by TLR ligands.
Conclusions
In summary, we propose an independent pathway of NLRP3 and caspase for IL-1β release following activation of the P2X7 receptor in human macrophages (Fig. 5). In this pathway, cleavage of pro-IL-1β is not caused by caspase-1, cysteine protease, or aspartic proteases, indicating an unknown mechanism of cleavage of pro-IL-1β to its active form. Our findings have significance for understanding P2X7 receptor-mediated IL-1β release, which will facilitate the development of new therapeutic strategies aimed at modulating inflammatory responses.

Proposed mechanism for NLRP3-independent IL-1β release by human macrophages after P2X7 receptor activation. Activation of TLR2/1 by Pam3CSK4 leads to NF-κB mediated production of pro-IL-1β and components of NLRP3 inflammasome. Nigericin-mediated K+ efflux leads to NLRP3 inflammasome oligomerization causing serine protease- and caspase-dependent IL-1β release. In contrast, P2X7 receptor stimulation by BzATP potentially initiates two distinct IL-1β releasing mechanisms that are both NLRP3-independent. One mechanism relies on K+ efflux and requires activation of caspase-1 and serine proteases. The other mechanism is independent of K+ efflux, caspases, and serine-, cysteine-, and aspartic proteases, suggesting the involvement of other mechanisms for proteolytic processing of IL-1β
Availability of data and materials
All data generated and analyzed during this study are included in the manuscript.
Abbreviations
- ATP:
-
Adenosine 5’-triphosphate
- BzATP:
-
2′(3′)-O-(4-benzoylbenzoyl) adenosine 5′-triphosphate
- IL:
-
Interleukin
- LDH:
-
Lactate dehydrogenase
- NLRP3:
-
Nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein 3
- TLR:
-
Toll-like receptor
References
Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147.
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. https://doi.org/10.1146/annurev.immunol.021908.132612.
Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, Behar SM. IL-1β promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol. 2013;190:4196–204. https://doi.org/10.4049/jimmunol.1202688.
Orzalli MH, Smith A, Jurado KA, Iwasaki A, Garlick JA, Kagan JC. An antiviral branch of the IL-1 signaling pathway restricts immune-evasive virus replication. Mol Cell. 2018;71:825-840.e6. https://doi.org/10.1016/j.molcel.2018.07.009.
Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20133328.
Burns K, Martinon F, Tschopp J. New insights into the mechanism of IL-1beta maturation. Curr Opin Immunol. 2003;15:26–30. https://doi.org/10.1016/s0952-7915(02)00017-1.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–91. https://doi.org/10.4049/jimmunol.0901363.
Netea MG, Nold-Petry CA, Nold MF, Joosten LAB, Opitz B, van der Meer JHM, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113:2324–35. https://doi.org/10.1182/blood-2008-03-146720.
Viganò E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun. 2015;6:8761. https://doi.org/10.1038/ncomms9761.
Gritsenko A, Yu S, Martin-Sanchez F, Diaz-Del-Olmo I, Nichols E-M, Davis DM, et al. Priming is dispensable for NLRP3 inflammasome activation in human monocytes in vitro. Front Immunol. 2020;11:565924. https://doi.org/10.3389/fimmu.2020.565924.
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53. https://doi.org/10.1016/j.immuni.2013.05.016.
Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–9. https://doi.org/10.1038/sj.cdd.4402195.
Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major DAMPs. Apoptosis. 2021;26:152–62. https://doi.org/10.1007/s10495-021-01663-3.
Vénéreau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. 2015;6:422. https://doi.org/10.3389/fimmu.2015.00422.
Valera S, Hussy N, Evans RJ, Adami N, North RA, Surprenant A, Buell G. A new class of ligand-gated ion channel defined by P2x receptor for extracellular ATP. Nature. 1994;371:516–9. https://doi.org/10.1038/371516a0.
Hattori M, Gouaux E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature. 2012;485:207–12. https://doi.org/10.1038/nature11010.
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity. 2017;47:15–31. https://doi.org/10.1016/j.immuni.2017.06.020.
Donnelly-Roberts DL, Namovic MT, Han P, Jarvis MF. Mammalian P2X7 receptor pharmacology: comparison of recombinant mouse, rat and human P2X7 receptors. Br J Pharmacol. 2009;157:1203–14. https://doi.org/10.1111/j.1476-5381.2009.00233.x.
Müller CE, Namasivayam V. Recommended tool compounds and drugs for blocking P2X and P2Y receptors. Purinergic Signal. 2021;17:633–48. https://doi.org/10.1007/s11302-021-09813-7.
Müller G, Lübow C, Weindl G. Lysosomotropic beta blockers induce oxidative stress and IL23A production in Langerhans cells. Autophagy. 2020;16:1380–95. https://doi.org/10.1080/15548627.2019.1686728.
Bockstiegel J, Wurnig SL, Engelhardt J, Enns J, Hansen FK, Weindl G. Pharmacological inhibition of HDAC6 suppresses NLRP3 inflammasome-mediated IL-1β release. Biochem Pharmacol. 2023;215: 115693. https://doi.org/10.1016/j.bcp.2023.115693.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. https://doi.org/10.1038/nrmicro2070.
Sha W, Mitoma H, Hanabuchi S, Bao M, Weng L, Sugimoto N, et al. Human NLRP3 inflammasome senses multiple types of bacterial RNAs. Proc Natl Acad Sci U S A. 2014;111:16059–64. https://doi.org/10.1073/pnas.1412487111.
Karasawa A, Kawate T. Structural basis for subtype-specific inhibition of the P2X7 receptor. Elife. 2016. https://doi.org/10.7554/eLife.22153.
Murgia M, Hanau S, Pizzo P, Rippa M, Di Virgilio F. Oxidized ATP. An irreversible inhibitor of the macrophage purinergic P2Z receptor. J Biol Chem. 1993;268:8199–203.
Balázs B, Dankó T, Kovács G, Köles L, Hediger MA, Zsembery A. Investigation of the inhibitory effects of the benzodiazepine derivative, 5-BDBD on P2X4 purinergic receptors by two complementary methods. Cell Physiol Biochem. 2013;32:11–24. https://doi.org/10.1159/000350119.
Michel AD, Clay WC, Ng SW, Roman S, Thompson K, Condreay JP, et al. Identification of regions of the P2X(7) receptor that contribute to human and rat species differences in antagonist effects. Br J Pharmacol. 2008;155:738–51. https://doi.org/10.1038/bjp.2008.306.
Huo H, Fryatt AG, Farmer LK, Schmid R, Evans RJ. Mapping the binding site of the P2X receptor antagonist PPADS reveals the importance of orthosteric site charge and the cysteine-rich head region. J Biol Chem. 2018;293:12820–31. https://doi.org/10.1074/jbc.RA118.003737.
Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55. https://doi.org/10.1038/nm.3806.
Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu J-W, et al. Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285:9792–802. https://doi.org/10.1074/jbc.M109.082305.
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–206. https://doi.org/10.1016/j.cell.2014.02.008.
Provoost S, Maes T, Pauwels NS, Vanden Berghe T, Vandenabeele P, Lambrecht BN, et al. NLRP3/caspase-1-independent IL-1beta production mediates diesel exhaust particle-induced pulmonary inflammation. J Immunol. 2011;187:3331–7. https://doi.org/10.4049/jimmunol.1004062.
Ruiz-Ruiz C, Calzaferri F, García AG. P2X7 receptor antagonism as a potential therapy in amyotrophic lateral sclerosis. Front Mol Neurosci. 2020;13:93. https://doi.org/10.3389/fnmol.2020.00093.
Romagnoli R, Baraldi PG, Cruz-Lopez O, Lopez-Cara C, Preti D, Borea PA, Gessi S. The P2X7 receptor as a therapeutic target. Expert Opin Ther Targets. 2008;12:647–61. https://doi.org/10.1517/14728222.12.5.647.
Calzaferri F, Ruiz-Ruiz C, de Diego AMG, de Pascual R, Méndez-López I, Cano-Abad MF, et al. The purinergic P2X7 receptor as a potential drug target to combat neuroinflammation in neurodegenerative diseases. Med Res Rev. 2020;40:2427–65. https://doi.org/10.1002/med.21710.
Chanput W, Mes JJ, Wichers HJ. THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol. 2014;23:37–45. https://doi.org/10.1016/j.intimp.2014.08.002.
Mizushina Y, Karasawa T, Aizawa K, Kimura H, Watanabe S, Kamata R, et al. Inflammasome-independent and atypical processing of IL-1β contributes to acid aspiration-induced acute lung injury. J Immunol. 2019;203:236–46. https://doi.org/10.4049/jimmunol.1900168.
Schaale K, Peters KM, Murthy AM, Fritzsche AK, Phan M-D, Totsika M, et al. Strain- and host species-specific inflammasome activation, IL-1β release, and cell death in macrophages infected with uropathogenic Escherichia coli. Mucosal Immunol. 2016;9:124–36. https://doi.org/10.1038/mi.2015.44.
Barberà-Cremades M, Baroja-Mazo A, Gomez AI, Machado F, Di Virgilio F, Pelegrín P. P2X7 receptor-stimulation causes fever via PGE2 and IL-1β release. FASEB J. 2012;26:2951–62. https://doi.org/10.1096/fj.12-205765.
Afonina IS, Müller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. 2015;42:991–1004. https://doi.org/10.1016/j.immuni.2015.06.003.
Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, Di Virgilio F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol. 1997;159:1451–8.
Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286:C1100–8. https://doi.org/10.1152/ajpcell.00494.2003.
Hazuda DJ, Strickler J, Kueppers F, Simon PL, Young PR. Processing of precursor interleukin 1 beta and inflammatory disease. J Biol Chem. 1990;265:6318–22.
Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, et al. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci U S A. 1999;96:6261–6. https://doi.org/10.1073/pnas.96.11.6261.
Acknowledgements
Figure 5 was created with Biorender.com.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
J.B.: Methodology, Investigation, Writing &; original draft, Writing &; review &; editing, Visualization. J.E.: Methodology, Investigation, Writing &; original draft, Writing &; review &; editing, G.W.: Conceptualization, Writing &; original draft, Writing &; review &; editing, Supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Studies with human blood were approved by the ethics committee of the University Clinic Bonn (315/22) and written informed consent was obtained from all healthy donors.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary figures S1-S3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bockstiegel, J., Engelhardt, J. & Weindl, G. P2X7 receptor activation leads to NLRP3-independent IL-1β release by human macrophages. Cell Commun Signal 21, 335 (2023). https://doi.org/10.1186/s12964-023-01356-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01356-1