
Abstract
Inflammation, although necessary to fight infections, becomes a threat when it exceeds the capability of the immune system to control it. In addition, inflammation is a cause and/or symptom of many different disorders, including metabolic, neurodegenerative, autoimmune and cardiovascular diseases. Comorbidities and advanced age are typical predictors of more severe cases of seasonal viral infection, with COVID-19 a clear example. The primary importance of mitogen-activated protein kinases (MAPKs) in the course of COVID-19 is evident in the mechanisms by which cells are infected with SARS-CoV-2; the cytokine storm that profoundly worsens a patient’s condition; the pathogenesis of diseases, such as diabetes, obesity, and hypertension, that contribute to a worsened prognosis; and post-COVID-19 complications, such as brain fog and thrombosis. An increasing number of reports have revealed that MAPKs are regulated by carbon dioxide (CO2); hence, we reviewed the literature to identify associations between CO2 and MAPKs and possible therapeutic benefits resulting from the elevation of CO2 levels. CO2 regulates key processes leading to and resulting from inflammation, and the therapeutic effects of CO2 (or bicarbonate, HCO3−) have been documented in all of the abovementioned comorbidities and complications of COVID-19 in which MAPKs play roles. The overlapping MAPK and CO2 signalling pathways in the contexts of allergy, apoptosis and cell survival, pulmonary oedema (alveolar fluid resorption), and mechanical ventilation–induced responses in lungs and related to mitochondria are also discussed.
Video Abstract
Similar content being viewed by others

Introduction
The SARS-CoV-2 pandemic highlighted how insufficiently we clinically treat excessive inflammation. Although the specific mechanisms leading to the production of proinflammatory cytokines and the activation of immune system components, as well as the signalling and other effects of proinflammatory cytokines and chemokines, are relatively well understood, the interrelationships among these factors are largely unclear. Due to the many mechanisms common to various pathologies and pathogen infections, including signalling pathways leading to inflammation and activated in response to inflammation, it is worth evaluating lessons learned during the COVID-19 pandemic, making conclusions about the treatments provided, and continuing the intensive search for effective therapies for inflammation.
Mitogen-activated protein kinases (MAPKs) regulate cell proliferation, survival, differentiation, migration, and apoptosis; oncogenesis; and neurodegeneration [1,2,3,4,5]. Signals from cellular receptors are transduced by MAPKs to a wide variety of effector proteins, including transcription factors, which regulate cell functions according to environmental conditions. In this review, we focus on three subfamilies of MAPKs, namely, c-Jun N-terminal kinases (JNKs), extracellular signal-regulated kinases 1 and 2 (ERK1/2) and p38 MAPKs, as they are key players in the regulation of inflammation and play important roles in signalling pathways critical to the course of SARS-CoV-2 infection. In response to a wide variety of chemical and biological agents, these MAPKs not only promote the production of reactive oxygen species (ROS) and proinflammatory cytokines, including interferon-gamma (IFN-γ), interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α) but also regulate cellular responses to a wide range of cytokines [6,7,8,9].
MAPKs have been proposed to be carbon dioxide (CO2) sensors because select MAPKs have been shown to be highly regulated by CO2 in vitro, in human cells and in plants [10,11,12,13]. CO2-dependent regulation of MAPKs has been demonstrated in several animal cell types and tissues (Table 1), but the direct influence of CO2 on MAPK activity has not been previously considered. The effects of CO2 on MAPK activity are very dynamic and depend on the concentration of CO2 and the duration of CO2 exposure. Although the mechanisms of action underlying the effect of CO2 on MAPK functions remain unclear, an emerging pattern indicates that inactive ERK1/2 and plant ERK-type MAPKs are activated, and the functions of all the activated MAPKs studied thus far have been inhibited by increased CO2 levels (Figs. 1 and 2). Importantly, among several groups of proteins proposed to be CO2 sensors, only MAPKs are common to all eukaryotes, with the other eukaryotic CO2 sensors being taxon specific.

Profiles of MAPK activity in response to elevated CO2 based on the data shown in Table 1. a CO2-dependent regulation of baseline ERK1/2 and JNK levels. The blue dashed line represents the extrapolated JNK activity values. b Inactivation of active MAPKs by CO2 at elevated levels

Cellular mechanisms regulating CO2-dependent activation of ERK1/2
In the following parts of this paper, we highlight ERK1/2-dependent processes that are augmented by an increase in CO2 concentration and the harmful effects triggered by MAPK signalling that can be inhibited by elevating CO2 levels. We indicate possible or previously observed consequences of purposely increasing CO2 levels in relation to various aspects of COVID-19 and the most common comorbidities in patients with COVID-19. Since the pathogenesis and therapy of COVID-19 is an extremely broad topic, including the roles of MAPKs and CO2 in these contexts, we do not describe all the possible benefits of increasing CO2 levels in COVID-19 therapy in this article, as these benefits have been discussed in other recently published papers [27,28,29,30]. We focus primarily on the cooperation of the CO2–MAPK signalling module, because the functions of CO2 and MAPK largely overlap (Table 2, Figs. 3 and 4), and it has recently been suggested that MAPKs may be CO2 receptors [10]. Due to text length limitations, we mainly emphasize the benefits of a transient increase in CO2 levels. A broader view of hypercapnia can be found in many recent review papers [31,32,33].

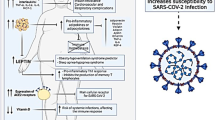
An overview of the involvement of MAPKs and the potential beneficial effects of elevated CO2 levels on the pathogenesis of COVID-19 and comorbidities. Red arrows: confirmed impact of elevated CO2 levels

The physiological and pathological processes regulated by MAPKs and/or CO2. Blue lines: mechanisms regulated by MAPKs; red lines: influence of elevated CO2 levels; black lines: overlapping effects of MAPKs and CO2
Changes in CO2 levels in the pathogenesis of COVID-19
CO2 is an important component of pH regulatory mechanisms in biological systems. In an aqueous environment, dissolved CO2 partially reacts with water to form H2CO3, which dissociates into HCO3− and H+. The balance between H2CO3 and HCO3− levels underlies the most important mechanism of maintaining blood pH; H2CO3 and HCO3− are able to neutralize excess bases and acids, respectively. Additionally, the bicarbonate buffer is highly efficient because the respiratory system efficiently removes CO2 and dissolved inorganic carbon species are distributed on the basis of pH (Bjerrum plot, Fig. 5). Namely, increasing CO2 levels leads to acidification and a shift in the CO2/HCO3− equilibrium towards a further increase in CO2 levels and a decrease in the concentration of HCO3−. These changes result an increased rate of CO2 removal via the lungs and restoration of baseline pH and CO2/HCO3− levels. The transitions between CO2 and HCO3− are catalysed by carbonic anhydrases (CAs), which play important roles in the regulation of pH and CO2 levels [191]. CAs, depending on the isoform, conditions, pH, and CO2/HCO3− ratio, may accelerate the hydration of CO2 or catalyse the reaction in the opposite direction, thereby affecting the flow of CO2 across cell membranes because CO2 can be efficiently transported through membranes via diffusion, while HCO3− transport through membranes requires the action of transporters [192, 193].

Distribution of the species of dissolved inorganic carbon as a function of change in pH (Bjerrum plot)
Numerous reports indicate that both HCO3− [194,195,196] and total CO2 concentrations are lower in patients who die with COVID-19 than in patients who have recovered from COVID-19 [197,198,199]. HCO3− levels lower than 22 mM have been identified as an important risk factor for mechanical ventilation [200] and a predictor of clinical deterioration in patients with nonsevere COVID-19 [201]. The decrease in the total pool of CO2/HCO3− in COVID-19 patients is accompanied by an increased level of lactate. The decrease in CO2/HCO3−, which may result from the intensive removal of CO2 by the lungs during hyperventilation due to a decrease in blood oxygen saturation (SpO2), leads to an increase in the pH of bodily fluids. The increased production of lactate compensates for failure to maintain normal pH. However, the primary cause of the increase in lactate may be an increase in the local glycolysis rate, which is a typical response to infection, inflammation and a decrease in oxygen supply [202]. A reduced rate of aerobic respiration in conjunction with increased glycolytic lactate production leads not only to a decrease in mitochondrial CO2 production but also to a decrease in pH. This acidification forces a shift in the CO2/HCO3− equilibrium, which increases the arterial partial pressure of CO2 (PaCO2), and elevated PaCO2 accelerates the removal of CO2 via the lungs, resulting in a reduction in the total pool of CO2/HCO3−.
Since lactate, in contrast to CO2, cannot be removed easily via gas exchange, an increase in lactate can lead to a permanent decrease in pH and metabolic acidosis. Although metabolic acidosis does not typically occur in the course of acute COVID-19, blood lactate levels were the highest in nonsurvivors and were higher in hospitalized COVID-19 patients than in ambulatory patients [199, 203, 204]. The greatest differences between a group of patients with COVID-19-related acute respiratory distress syndrome (ARDS) who presented with the "hyperinflammatory" phenotype, with a significantly higher mortality rate and a group of those who presented with the "hypoinflammatory" phenotype, was elevated lactate levels and decreased HCO3− levels in patients with the "hyperinflammatory" phenotype; the differences in other markers of inflammation were much less pronounced [205].
Similar to the effect of lactate, permissive and therapeutic hypercapnia leads to a decrease in pH due to an increase in PaCO2 resulting from insufficient CO2 removal through the lungs and the inhalation of CO2, respectively. However, hypercapnia-induced respiratory acidosis does not lead to the many complications attributed to metabolic acidosis and exerts a protective effect in patients with one of many other medical conditions [206,207,208,209].
In addition to studies demonstrating the ability of elevated CO2 to inhibit the proinflammatory response induced by SARS-CoV-2 elements [10], there are also reports based on randomized trials and case studies showing the benefits of using HCO3− in experimental COVID-19 therapies to improve prognosis. Patients with mild COVID-19 who received 14-day nasal NaHCO3 irrigation twice daily showed an eightfold lower risk of hospitalization than the overall population [210]. Supplementing standard COVID-19 therapy with 8.4% NaHCO3 steam inhalation led to an improvement in clinical parameters in patients with mild to moderate symptoms [211]. A positive effect of 10 ml administration of 4.2% NaHCO3 every 6 h was found on mechanically ventilated patients [212].
Opposing effects of elevated CO2 levels
Although the physiological importance of CO2 is well understood, its effects at the molecular and cellular levels are poorly understood, and the broad spectrum of CO2 concentrations has almost never been compared in CO2 signalling studies or for the therapeutic application of CO2. The different effects of specific CO2 concentrations have been reflected in numerous seemingly contradictory results from research groups that reported results based on different CO2 concentrations. Notably, the long-term effect of severe hypercapnia exerted the exact opposite effect of short-term CO2 application, e.g., airway muscles were constricted after long-term (3 or 7 days of 10% CO2 inhalation) treatment with CO2 [213] and were dilated after short-term exposure to elevated CO2 [67]. CO2 dilates airways that are constricted, e.g., by drugs such as serotonin, methacholine, bethanechol and carbachol, or by the occlusion of the pulmonary artery [68, 69]. Importantly, short- but not the long-term effects of hypercapnia are opposite those of hypocapnia, as transient hypercapnia dilates and hypocapnia constricts airways, as found in dog [70], porcine [71] and rat [72] models. In contrast to acute long-term hypercapnia, slightly elevated CO2 levels exert a bronchodilator effect in healthy subjects and in patients with asthma before and after exercise [73, 74]. Consistent with the proposed inhibition of active ERK1/2 induced by elevated CO2, inhibition of ERK1/2 increased airway conductance in patients with asthma [75].
The abovementioned opposing effects of CO2 at the physiological level are analogous to the opposite effects of CO2 on ERK1/2 functions (the activation of inactive ERK1/2 and inhibition of activated ERK1/2). For example, in healthy subjects, elevated PaCO2 levels increased pulmonary artery pressure [214], but inhaled 5% CO2 reduced preexisting pulmonary artery hypertension (PAH) [77]. Importantly, PAH has been associated with high levels of ERK1/2 and p38 activity [80]. A direct comparison indicated that cyclic stretch–induced injury in human bronchial and alveolar epithelial cells was more efficiently inhibited by hypercapnia applied after cell stretching had begun than by preconditioning the cells via induced hypercapnic acidosis [53]. The specific effects of CO2 on MAPK activity and physiology are limited to a relatively narrow range of CO2 concentrations. For example, drastically elevated CO2 (20%, PaCO2 140 mm Hg) did not inhibit ERK1/2 activity [18] unlike lower CO2 levels. Similarly, moderate hypercapnia (PaCO2 of 80–100 mm Hg), conferred better protection from high-pressure ventilation–induced inflammatory injury on rat lungs than PaCO2 > 100 mm Hg [54].
In summary, prolonged exposure to very high levels of CO2 exerts detrimental effects on organisms. However, in the following sections of this review, we focus on the beneficial effects of CO2 and its potential therapeutic use, specifically, the short-term effect of slightly elevated CO2 concentrations.
The role of MAPKs in viral infections
MAPKs are activated by viral infection. For example, p38 is activated by hepatitis B virus (HBV), hepatitis C virus (HCV), influenza virus, enterovirus 71, human immunodeficiency virus (HIV) and dengue virus infection [215, 216]. Moreover, MAPKs are involved in many viral infection mechanisms. In addition to induction of a proinflammatory response and regulation of the activity of various types of immune cells during viral infections (e.g., the regulation of CD8+ T-cell apoptosis) [217], ERK, JNK and p38 isoforms have been shown to directly support viral multiplication. First, ERK1/2 may positively influence the entry of SARS-CoV-2 into host cells [218]. Second, the phosphorylation of different host proteins by MAPKs facilitates the replication and translation of many viral proteins [219]. Third, efficient nuclear export of viral ribonucleoprotein complexes depends on the activity of ERK1/2 (e.g., influenza virus ribonucleoproteins [220]), and fourth, phosphorylation of viral proteins by MAPKs facilitates viral complex assembly (e.g., p38α phosphorylates the HCV core protein, leading to its oligomerization [215]). Consist with these findings, p38 and ERK1/2 inhibitors impaired the replication of influenza virus and coronaviruses [221,222,223], including SARS-CoV-2 pseudoviruses, in an in vitro model [215].
MAPKs in the pathogenesis of COVID-19
Angiotensin-converting enzyme 2 (ACE2), the host receptor of SARS-CoV-2, is a negative regulator of MAPK signalling and thus efficiently prevents both the activation of MAPKs and pneumonia caused by exposure to lipopolysaccharide (LPS) [50], bleomycin [224], cigarette smoke [225] or particulate matter 2.5 (PM2.5) [226]. However, during SARS-CoV and SARS-CoV-2 infection, when receptor ACE2 is bound by the viral spike protein, ACE2 function is disrupted, leading to the activation of MAPKs, the production of proinflammatory cytokines and the pathogenesis of pneumonia or even ARDS [227,228,229,230,231,232]. In a model mice, COVID-19-like symptoms, including acute lung injury, were caused by inactive SARS-CoV-2 [233] or SARS-CoV-2 spike protein alone [234]. Moreover, ERK1/2 were activated by both SARS-CoV-2 and the spike protein alone in, e.g., human bronchial epithelial cells [10], human dendritic cells [235] and murine primary macrophages [236]. Activation of p38 by spike was found in Vero E6 cells [237], human peripheral blood mononuclear cells [238], HEK293T cells, BHK21 cells [215], murine alveolar macrophages [239] and microglia [240].
Multiple mechanisms lead to the activation of MAPKs by SARS-CoV-2. One such mechanism is renin-angiotensin system (RAS) dysregulation, as SARS-CoV-2 causes internalization of ACE2 by inhibiting the primary function of ACE2, which is the cleavage of angiotensin (Ang) II to form Ang1-7. As a result, the production of Ang1-7 decreases, and the level of AngII increases, leading to the activation of AngII receptor type 1 (AT1) and downstream MAPKs. In addition to ACE2, other membrane receptors have been shown to interact with SARS-CoV-2 and trigger MAPK-mediated signalling; for example, spike protein activates p38 and ERK1/2 via the receptor CD147 in vivo [241] and in primary human cardiac pericytes [242], respectively. The role of MAPKs in SARS-CoV-2 signalling is multifaceted, as the activation of ERK1/2, JNKs and p38 MAPKs is also triggered by the SARS-CoV-2 nucleocapsid protein [243].
The importance of MAPKs has been confirmed not only in classic signalling studies involving MAPK inhibitors but also in many more-objective high-throughput analyses. Proteomic approaches clearly indicated that MAPKs have been found to be among the most highly activated proteins after SARS-CoV-2 infection, regardless of the experimental approaches, cell and sample types evaluated, or period of SARS-CoV-2 infection [237, 244,245,246]. RNA-seq data revealed that in addition to the regulation of MAPKs by SARS-CoV-2 at the transcriptional level in many cell types [247], the components of the MAPK signalling pathway were also strongly regulated via alternative polyadenylation sites in human peripheral blood mononuclear cells from COVID-19 patients [248].
All types of ACE2-positive immune cells, which are crucial for the pathogenesis of severe COVID-19, can be directly infected by SARS-CoV-2, and as a result of infection, activated MAPK signalling stimulates transcription factors such as NF-κB and AP-1, which trigger the production of proinflammatory cytokines. Despite disputes over whether endothelial cells can be infected by SARS-CoV-2, endothelial cells undoubtedly produce a strong proinflammatory response via MAPKs in patients with severe COVID-19 [249,250,251,252]. Vascular endothelial cells in infected organs recruit monocytes/macrophages and neutrophils to inflammation sites and promote further production of proinflammatory cytokines, leading to a cytokine storm [253, 254]. Uncontrolled activation of macrophages not only leads to the secretion of high levels of IFN‐γ, IP-10, IL-6, IL-17, IL-10/23 and TNF-α but also causes a loss of inflammatory coordination mediated by type-I interferons, which is a hallmark of COVID-19. Importantly, type-I interferon production is inhibited by activated ERK1/2 in macrophages [255]. In addition, MAPKs contribute to a decrease in lymphocyte counts, including lymphocyte necrosis and NK and T-cell exhaustion promoted by IL-6, which is commonly observed in COVID-19 patients [254].
In addition to regulating transcription factors, MAPKs regulate other types of effector proteins. For example, p38 and ERK1/2 phosphorylate and thereby increase the catalytic activity of a disintegrin and metalloprotease 17 (ADAM17). ADAM17, due to its proteolytic activity, can release the ectodomains of a variety of proteins; ADAM17 induces ACE2 shedding and the activation of proinflammatory cytokines and fibrotic factors, leading to enhanced organ dysfunction via increased inflammation and fibrosis [256]. The roles of MAPKs in key processes in the pathogenesis of COVID-19 are very broad, as cellular responses to cytokines leading to severe disease in COVID-19 patients depend on MAPK signalling pathways. More detailed information on MAPK signalling in relation to SARS-CoV-2 infection can be found in recent comprehensive reviews [218, 254, 257].
Hypertension in COVID-19
Hypertension is one of the most common comorbidities that worsens the prognosis of COVID-19. The RAS plays a unique role in regulating blood pressure in patients with COVID-19 due to the direct effect of SARS-CoV-2 on ACE2. SARS-CoV-2 enhances the vasoconstrictive effect of AngII while reducing the amount of Ang1-7, which exert a vasodilating effect. MAPKs constitute a hub for these opposing activities, as both AngII and Ang1-7 signalling is mediated by MAPKs. p38 and ERK1/2 are activated in response to AngII binding by AT1 receptors and induce severe vasoconstriction, increasing blood pressure and heart rate. Moreover, MAPKs have been proposed to be sensors of pressure overload because activation of JNK, p38 and ERK1/2 is proportional to the amount of pressure overload and pressure overload-induced myocardial remodelling in hypertensive patients [258]. In contrast to that of AngII, the activation of Ang1-7 signalling by Ang1-7 binding to the Mas1 receptor leads to inactivation of ERK1/2 via the induction of MAPK phosphatase-1 (MKP-1) in endothelial and vascular smooth muscle cells, which causes not only hypotension but also antiproliferative, antithrombotic and fibrotic effects [259,260,261].
p38 is overactive in the endothelium and adventitia of hypertensive model rodents in contrast to its activation level in normotensive animals. Activation of p38 in response to AngII activity was transient in normotensive rats but sustained in hypertensive rats [262]. Progressive and sustained hypertension induced by AngII, a high-salt and high-fat diet, or monocrotaline treatment in model mice and rats was reversed by p38 inhibitors; similarly, endothelial dysfunction, vascular cell proliferation, cardiac hypertrophy, and enhanced extracellular matrix and collagen deposition leading to vascular remodelling were reversed [81, 83]. Inhibition of overactivated p38 thus prolonged survival and increased endothelium-dependent vascular relaxation [263].
ERK1/2 are activated in vascular smooth muscle cells, arteries and serum of hypertensive patients. ERK1/2 are crucial for AngII- and thrombin-induced smooth muscle cell proliferation and vascular remodelling, leading to hypertension, atherosclerosis, and accelerated cardiovascular damage [264, 265]. Similar to synthetic MAPK inhibitors, aerobic exercise exerts beneficial effects on vascular and endothelial functions, including the inhibition of vascular smooth muscle remodelling, which led to the acquisition of a hypertensive phenotype by promoting the inactivation of overactive p38 and ERK1/2 in spontaneously hypertensive rats [266]. However, basal ERK1/2 activity has been shown to be essential for maintaining endothelial integrity in vivo, and ERK1/2 loss leads to rapid development of hypertension and death within 5 weeks due to widespread endothelial-to-mesenchymal transition and degradation of endothelial cells in various organs [267].
One of the arguments for the use of CO2 in COVID-19 therapy is that the effect of CO2 on the circulatory system is consistent with that of Ang1-7; increased CO2 causes a decrease in vascular resistance and an increase in blood flow to organs [268, 269]. Experimental COVID-19 therapies based on various vasodilators have improved the prognosis. For example, sildenafil shortened the length of hospital stays and reduced the need for invasive mechanical ventilation [270].
In recent years, research on the effects of CO2 on blood pressure has focused on the relationship between CO2 levels and PAH, as end-tidal CO2 (EtCO2) is lower in patients with PAH than in control subjects, and PAH is associated with chronic alveolar hyperventilation. It has been shown that lower EtCO2 or PaCO2 results in shorter survival in patients with PAH [78, 79].
Thrombosis
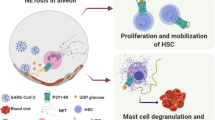
Inflammation of blood vessels in the lungs, heart, brain and other organs is the cause of the most serious complications of severe COVID-19, and COVID-19 is considered a form of inflammatory endotheliitis [249,250,251,252]. Typical pathological changes include thrombosis, which may result from inflammation associated with the induction of tissue factor expression (i.e., factors whose mere presence triggers the production of blood clots). In cells that are in contact with platelets, including both monocytes and endothelial cells, the induction of tissue factor depends on p38. Accordingly, MAPK expression has been associated with platelet activation and thrombosis [88, 89]. However, the frequent incidence of thrombosis and the largeness of the thrombi in vessels of COVID-19 patients are largely due to neutrophil activity not inflammatory processes in endothelial cells. In response to SARS-CoV-2, nucleocapsid or spike proteins, IL-6 or, particularly, IL-8, neutrophils extrude neutrophil extracellular traps (NETs), a web of chromatin-based cytoplasmic materials enriched with antimicrobial agents. NETs promote the accumulation of activated platelets and coagulation factors, forming thrombi [90, 271,272,273].
More NETs have been found in deceased COVID-19 patients than in survivors, and there is a correlation between plasma NET levels and COVID-19 severity [91]. Notably, various studies have consistently shown that the greatest increase in NET production in COVID-19 patients occurs soon after admission to the intensive care unit (ICU). Independent research groups have reported that increased inspiratory airflow and mechanical cell stretch–induced MAPK and NF-κB activation in alveolar macrophages trigger the release of IL-8 and IL-6, which are crucial for NET induction [53, 91, 274].
Treatment with JNK, ERK1/2 or p38 inhibitors abrogates NET formation [90,91,92]. Similarly, elevated CO2 levels inhibit the processes that contribute to the formation of NETs, including bronchoalveolar fluid neutrophil infiltration, NF-κB activation, and the production of IL-6 and IL-8 [53, 85]. Moreover, the effect of CO2 on the generation of NETs has been recognized. The formation of NETs is highly dependent on pH and the CO2/HCO3− ratio; specifically, NET production is induced by high pH and a low HCO3− level and inhibited by a high CO2 level and low pH [86].
Assuming that the inhibition of MAPKs by CO2 is a universal process, the antithrombotic role of CO2 may be even broader than previously recognized, as thrombosis in patients with acute COVID-19 results from the binding of activated platelets to NETs, and MAPKs have been well documented in platelet activation in response to both SARS-CoV-2 and the spike protein [275]. In addition, the role of CO2 in reducing ROS production, as shown in peripheral occlusive arterial disease, points to a universal antithrombosis-inducing effect of CO2 [57]. Accordingly, acidosis promotes a reversible decrease in blood clotting [87].
Obesity and insulin resistance
Hyperinsulinaemia and hyperglycaemia induce an increase in the SARS-CoV-2 load [276], and high glucose levels lead to higher cytokine and ROS production [277] and activation of ERK1/2 in response to the spike protein in endothelial cells [10]. MAPK-dependent modification of blood vessel development programs, including regulation of the expression of intercellular adhesion molecule-1 (ICAM-1), which is responsible for the recruitment of leukocytes to sites of inflammation [278], increases the risk of patients with type 2 diabetes, among others with vascular disease, experiencing more severe COVID-19 [279]. The incidence of cardiovascular complications, including stroke, and death resulting from venous thromboembolism and pulmonary embolism is several fold higher in diabetic patients than in the general population [280, 281].
MAPKs are involved in various mechanisms leading to obesity, insulin resistance and diabetes, including low-level systemic inflammation; ERK1/2, JNK and p38, which are activated by a high-fat diet, promote the infiltration of monocytes/macrophages into adipose tissue, proinflammatory signalling and dysregulation of immune responses [112, 113]. However, the central role of JNK in the core insulin signalling pathway is the mechanism best understood thus far. Activated JNK (activated, e.g., by hyperglycaemia, free fatty acids, cytokines or ER stress [101,102,103,104,105]) and ERK1/2 phosphorylate insulin receptor substrate-1 (IRS-1) and thus prevent signal transduction after insulin binding to the insulin receptor, leading to alterations in insulin action [106]. The JNK regulation of metabolism is multidimensional. JNK1 knockout protected mice from IRS-1 serine phosphorylation, insulin resistance, fatty liver and diabetes [107, 108]. JNK isoforms are essential regulators in the transition between obesity and type-2 diabetes [104]; JNK1/2 promotes the development of insulin resistance and obesity, whereas JNK3 protects against excessive adiposity [282]. JNK1/2 and p38 promote adipogenesis [283] and regulate lipogenesis, thermogenesis and the browning of white adipose tissue. One of the most important mechanisms among these processes is the modification of mitochondrial function by inhibition of the transcription factor peroxisome proliferator-activated receptor α (PPARα) and uncoupling protein 1 (UCP1) expression in response to, for example, ER stress or a high-fat diet [104, 105, 114, 115].
Direct relationships between insulin resistance, ERK1/2 and p38 activity, and elevated CO2 levels have been found in adipocytes, which become insulin resistant as a result of postsurgical trauma. ERK1/2 and p38 are highly activated after surgery. Overnight incubation of adipocytes in 10% CO2 inactivated ERK1/2 and p38 and restored insulin receptor and IRS-1 sensitivity to insulin [21]. Consistent with CO2-dependent MAPK regulation, subcutaneous injections of CO2 reduced body fat [109]. In addition to the use of transcutaneous CO2 as carboxytherapy in aesthetic medicine [110], the transcutaneous application of CO2 is an efficient treatment for chronic diabetic wounds [284]. Importantly, the major obstacle to wound healing is excessive neutrophil apoptosis caused by the production of NETs [285]; therefore, the beneficial effects of CO2 on diabetic wound healing are consistent with the anti-NETosis effect of CO2, acidification and MAPK inhibitors [86, 90,91,92].
Stroke and ischeamia
Multiple organ failure is the leading cause of COVID-19 mortality. It is a result of, among other causes, organ hypoxia, including hypoxia due to ischaemic stroke, which is a frequent complication of COVID-19. One of the changes observed in the brain in acute stroke patients is a profound reduction in CO2 level [286].
In addition to ischaemia, further organ damage is caused by the restoration of blood flow to organs; this damage is known as ischaemia–reperfusion injury. MAPKs in the brain are activated both in response to hypoxia and several minutes after reperfusion. MAPK inhibition has been shown to ameliorate brain injury due to reduced proinflammatory signalling and cell death. Decreasing MAPK activity enhances myelin regeneration, increases blood‒brain barrier function, and suppresses inflammation [97]. Many different drugs that exert neuroprotective effects during cerebral ischaemia inhibit MAPKs and downstream responses, including impairment of IL-1β, IL-6 and TNF-α production by purpurin and the inhibition of apoptosis by tetramethyl pyrazine mediated via JNK inactivation [98,99,100]. Therefore, blocking the proinflammatory response by inhibiting NF-κB-dependent gene transcription in neurons leads to a reduction in the number of damaged neurons and even to their recovery, reducing the infarct area and death rate of neurons [287].
Therapeutic hypercapnia exerts broadly understood protective effects against reperfusion and oxidative brain injury after ischaemic stroke [136, 286, 288, 289]. In addition to cerebral vasodilation, therapeutic CO2 reduces blood‒brain barrier damage [289] and increases sensorimotor activity and spatial memory after focal cerebral ischaemia–reperfusion [136]. In hypoxic regions, pH is lowered to be as low as 6.0–6.5, which confers neuroprotection [206, 209, 290]. An increase in the concentration of protons may result in energy benefits; i.e., an increase in the number of protons contributes to maintenance of a higher membrane potential in the mitochondria, which leads to more efficient ATP production under conditions of oxygen deficiency. This protection has been demonstrated in fish [291], neurons [292] and neuroendocrine prostate cancer cells, in which acidic pH shifted cellular metabolism towards oxidative phosphorylation [293]. This phenomenon may explain the protective role of CO2 under conditions of hypoxia and the reduction in oxygen consumption by the brain when CO2 levels are increased [184].
Studies on animal models of ischaemic injury have shown that hypercapnic acidosis exerts a protective effect not only on the central nervous system but also on the lung, myocardium, intestine and liver [34, 93,94,95]. For example, an EtCO2 higher than 20 mm Hg at intubation and its increase after resuscitation reduces neurological damage in patients after cardiac arrest [96].
There are known direct relationships between CO2 and MAPKs in protection against ischaemia‒reperfusion-induced injury; for example, the protective role of elevated CO2 levels in the context of ischaemia‒reperfusion-induced retinal injury is mediated via the inhibition of activated p38 [23]. Hypoxia-induced ERK1/2 activity in mice was suppressed by 10 min percutaneous administration of CO2 once per day, which was accompanied a CO2-induced increase in ischaemic blood flow and capillary density [19].
MAPKs and CO2 levels in response to mechanical ventilation
Permissive hypercapnia is included in the treatment guidelines for intubated COVID-19 patients in most countries and institutions worldwide. A comparison of hypercapnic and normocapnic COVID-19 patients (PaCO2 of 47.1 vs. 39.7 mm Hg, respectively) showed no difference in mortality despite worsened health in the hypercapnic patients; i.e., these patients had a higher body mass index (BMI) and higher number of venous thromboembolic events, chronic obstructive pulmonary disease (COPD) or ARDS [294].
In addition to the effect of hypercapnia on patients with COVID-19, there is debate as to whether hypercapnia should be used for intubated patients, as contradictory results have been obtained from different studies [295]. In addition to reports showing the benefits of hypercapnia, there are studies showing reduced survival times for mechanically ventilated patients with severe hypercapnic acidosis [296, 297]. However, a recent meta-analyses indicated that permissive hypercapnia was associated with lower mortality than imposed hypercapnia under protective ventilation conditions [298]. To date, no studies have been conducted in mechanically ventilated patients with a transient increase in CO2 levels (that is, a level sufficient to inhibit the excessive activity of MAPKs in cultured cells [10], corresponding to 5% CO2 administered for 15–20 min several times a day). It should be emphasized that CO2 is a strong inhibitor of both innate and adaptive immune responses, including inhibition of lymphocyte and natural killer cell cytotoxicity, neutrophil and macrophage migration to sites of infection, and the release of proinflammatory cytokines; therefore, the prolonged use of elevated levels of CO2 may lead to weakened immune protection against bacterial infections and sepsis [31, 85], which may worsen the outcome for ICU patients.
The cytokine storm induced by infection with SARS-CoV-2 is enhanced by mechanical ventilation in patients with severe COVID-19. High-pressure mechanical cell stretching exacerbates lung injury, by changing cell histology, increasing lung infiltration with neutrophils, and inducing AEC apoptosis associated with caspase-3 activation [24]. MAPKs are involved in the mechanisms leading to all of these adverse effects in lungs; the most widely recognized of these mechanisms is the role of ERK1/2, which lead to the downstream activation of NF-κB [53], ICAM-1 [54] or ADAM17 [20]. Activation of these signalling pathways as well as ventilation-induced lung injury, AEC apoptosis and increased neutrophil infiltration can be reduced by either elevating the CO2 level or inhibiting MAPK function [20, 24, 53, 54, 56].
Since high levels of oxygen support are used in mechanically ventilated patients with severe COVID-19, it seems that there may be additional benefits from the use of elevated CO2 levels in these patients, as hyperoxia induces profound lung injury, AEC apoptosis, and ROS and proinflammatory cytokine production [59,60,61,62,63,64,65,66, 299, 300], and CO2 inhibits the production of ROS and stimulates the production of antioxidants [57, 58].
MAPKs and CO2 regulate the resorption of alveolar fluid
Symptoms such as shortness of breath, a low SpO2 level and lung failure, as well as organ failure, are largely due to oedema fluid (alveolar lining fluid, ALF) flooding alveolar spaces. Excess ALF markedly reduces the amount of oxygen delivered to erythrocytes. In healthy lungs, membrane transporters trigger vectorial ion transport, followed by osmotic influx water, from the apical surface to the basolateral surface of alveolar epithelial cells (AECs), i.e., from the lumina of the alveoli into the lung interstitium and endothelium. Failure of alveolar fluid clearance (AFC) results from a decrease in the level of membrane transporters needed for the flow of ions. AFC is lowered under hypoxic conditions [301], hyperoxic conditions [302, 303], elevated airway pressure [304], pathogen infection and high levels of proinflammatory cytokines, including IL-1β, IL-8, TNF-α and transforming growth factor β1 (TGF-β1), in ALF [38, 301,302,303,304].
The main mechanism underlying AFC is the transport of Na+ ions across the apical membranes of AECs via epithelial Na+ channels (ENaCs). Activated ERK1/2 in AECs phosphorylate the β and γ subunits of ENaC, leading to enhanced interaction of ENaC with Nedd4, an E3 ubiquitin ligase, and to the endocytosis of the ENaC complex and subsequent downregulation [11, 39,40,41,42]. Thus, Na+ ions accumulate in ALF, leading to an increase in its pH and volume [305]. In addition, ERK1/2 have been indicated to be upstream regulators in the pathway leading to increased degradation of ENaC due to the phosphorylation of Nedd4 by JNK [11]. Inhibition of ERK1/2 or JNK restores the stability of ENaC in the cell membrane, resulting in an increase in AFC. Moreover, in response to IL-1β, p38 inhibits the activity of the α-ENaC gene promoter and the trafficking of ENaC to the apical membranes of type II AECs (ATII) [38]. After absorption into alveoli, Na+ ions are eliminated through the basolateral side of AECs mainly via the action of Na/K-ATPase. Both activated ERK1/2 [12] and JNK [25, 26] inhibit Na/K-ATPase, reducing the AFC rate.
In the studies on ENaC and Na/K-ATPase presented above, elevated CO2 levels (60–120 mm Hg vs. 40 mm Hg of the control) inhibited AFC but were applied under control conditions to activate MAPKs [11, 12, 25, 26, 42]. Under control conditions, inactive ERK1/2 were activated by elevated CO2 levels; therefore, the reduction in AFC rate induced by an increase in CO2 level was expected. However, elevating CO2 levels shows potential therapeutic value for use under pathological conditions in which excessive ALF production and AFC impairment are observed as a result of high ERK1/2 activity stimulated by infection or inflammation. As active ERK1/2 are inactivated by a transient increase in CO2 concentration [10], the therapeutic transient elevation of CO2 in the lungs might lead to an increase in AFC under pathological conditions with elevated MAPK activity. These hypotheses are supported, at least in part, by reports showing that transient (20 min) hypercapnia (10% CO2) increased AFC when ALF production induced by forskolin was also increased [35].
Moreover, AFC regulation by MAPKs is mediated by aquaporin channels (AQPs), through which water flows following an osmotic gradient. The expression of a key aquaporin, AQP5, is downregulated by p38 and JNK, e.g., in human SPC-A1 cells [43] or murine lungs [44]. Generally, events or factors that activate ERK1/2, p38 and JNK (e.g., infection or cytokines) lead to a decrease in the expression of membrane ion or water transporters and an increase in pulmonary oedema via the action of MAPKs. Consistently, research on inflammation induced by pre-B-cell colony-enhancing factor (PBEF) has shown that ERK1/2 downregulated the main transporters responsible for AFC, i.e., ENaC, Na/K-ATPase, and AQP1 [45].
The commonly held view is that the beneficial therapeutic effect of NaHCO3 is due to elevated pH. However, there should be no long-term changes in pH in the lungs after inhalation of NaHCO3, as any elevation in pH should be rapidly neutralized by Na+ influx into the cytoplasm of AECs via ENaC and across the basolateral membrane into bodily fluids. Otherwise, the increased Na+ concentration would be followed by increased secretion of ALF with all the associated negative consequences. In addition, the pH of ALF is regulated by paracellular HCO3− flux across the airway epithelium. At the correct (i.e., slightly acidic) pH of ALF, HCO3− is secreted. In contrast, when the ALF pH is increased (e.g., in response to infection or proinflammatory cytokines, and presumably after NaHCO3 inhalation), HCO3− flow is reversed, limiting pH changes [305,306,307]. These arguments may support the effect of elevated CO2 but not an increase in pH in the alveolar epithelium via the therapeutic use of NaHCO3.
Among the most widely used and most effective (though still insufficient) drugs in the treatment of acute COVID-19 is highly concentrated dexamethasone, a corticosteroid that inhibits the activity of MAPKs [308]. In addition to its anti-inflammatory effect, dexamethasone increases the amount of ENaC in AECs by inhibiting ERK1/2 [46, 47] and increases the AFC rate. However, under many pathological conditions where ERK1/2 are activated, decreased sensitivity to glucocorticoids is observed [309, 310]. The molecular mechanism underlying the dexamethasone-dependent regulation of MAPKs is the upregulation of MKP-1 and, as a result, increased inactivation of MAPKs. Transcriptomic data indicate that SARS-CoV-2 infection leads to downregulation of MKP-1, reducing cell sensitivity to corticosteroids [311, 312]. Consequently, in contrast to elevated CO2, dexamethasone was not able to block the activity of ERK1/2 induced by the spike protein in bronchial epithelial cells in the presence of IFN-γ and TNF-α [10].
MAPKs and CO2 in allergy
Among the immune cells with functional SARS-CoV-2-entry machinery, i.e., the expression of ACE2 and TMPRSS2, there are mast cells that handle allergic reactions [313]. Mast cells are stimulated during SARS-CoV-2 infection via ERK1/2, which activate the transcription factors NF-κB and AP-1, leading to the release of a wide variety of proinflammatory factors [120,121,122]. In addition, ERK1/2 stimulate histamine production by regulating the expression of histidine decarboxylase [123, 124]. The histamine signal is received by a wide range of cells in various organs through H1, H2, H3 and H4 receptors, and signalling downstream of each of these receptors is mediated by MAPKs [125,126,127]. Histamine signalling mediated via ERK1/2 also regulates the activity of the mast cells themselves, regulating the production of important molecules such as nerve growth factor (NGF) following activation of the H1 receptor [122] and IL-6, TNF-α, TGF-β1, IL-8, macrophage inflammatory protein-1α (MIP-1α/CCL3), and monocyte chemoattractant protein-1 (MCP-1/CCL2) in response to H4 receptor stimulation [128].
Because of the participation of mast cells in the course of COVID-19, antihistamines are among the most commonly prescribed medications in COVID-19 therapy. The importance of H1 receptor antagonists has been confirmed not only in numerous in silico and in vitro studies but also through its use in the clinic to alleviate the symptoms of COVID-19 in patients. Information on various H1 receptor modulators in the treatment of COVID-19 can be found in a recent review article [314]. In addition, competitive inhibitors of histamine H2 receptors, such as famotidine, are very effective in relieving mild COVID-19 symptoms [315] and in protecting against death and intubation [124, 316], although the exact molecular mechanisms are yet to be elucidated.
The inhibition of overactive MAPKs by elevated CO2 levels in allergic reactions is of particular interest because MAPK/NF-κB-inhibiting drugs (e.g., lidocaine or p38 inhibitors) [129] and elevated CO2 levels are both effective in treating allergy symptoms. In various clinical trials, noninhaled 100% CO2 (flow rate 5–10 ml/s) was effective in the treatment of allergic rhinitis [116]. The effect of a single dose of CO2 administered intranasally for 10–30 s lasted 4 to 6 h, and a 60-s dose lasted 24 h. Similarly, after a 20-s exposure to CO2 prior to allergen exposure, the acute responses to allergen challenge were reduced; for example, there were a significant reduction in sneezing, secretion weight and bilateral rhinorrhoea symptoms. CO2 also led to inhibited histamine release [117]. Accordingly, CO2 inhibited mast cell degranulation and histamine release in vitro [118]. Moreover, a decrease in PaCO2 is one of the most common initial symptoms of anaphylactic reactions [119], suggesting the benefits of using elevated CO2 levels for inhibiting the most severe allergic reactions. Taken together, the evidence suggests that the regulation of CO2-MAPK pathways in the inhibition of allergic reactions is a promising direction for future research.
COVID-19 and smoking
Early in the COVID-19 pandemic, controversial analyses indicated that, contrary to predictions, smoking did not only not worsen the prognosis but also may have protected patients against the development of severe COVID-19 symptoms [317,318,319]. Various studies have pointed to a lower rate of daily smokers presenting with symptomatic COVID-19 [319] and a lower risk of hospitalization, serious illness or death compared to the general population [320,321,322,323,324,325,326]. Interestingly, even in studies that concluded that smoking worsened the prognosis of COVID-19, the proportion of smokers with severe COVID-19 compared to the proportion of smokers in the general population showed that smoking conferred a protective effect [327].
Although the reasons for the potential protective effects of tobacco smoke are unknown, reports of the beneficial effects of smoking have been increasing, so clinical trials have been launched based on the hypothesis that nicotine plays a protective role against the development of COVID-19. Although the arguments presented in this paper may suggest that the effects of smoking considered to be positive may be due to the inhalation of elevated levels of CO2 during smoking; however, this hypothesis should be considered with caution. Notably, as smokers have a much higher risk of cardiovascular and respiratory disease, their milder COVID-19 cases may have be due to the protective effects of the medications they take. On the other hand, numerous comorbidities in this group of patients, compared to the general population, may suggest a very strong protective effect of inhaled tobacco smoke.
MAPKs and CO2 in breathing regulation
Shortness of breath is a typical symptom caused by infection with early variants of SARS-CoV-2; thus, the regulation of breathing plays an important role in the pathogenesis of COVID-19. During wakefulness, CO2 levels are sensed mainly by central chemoreceptors, i.e., the chemosensory neurons in the medulla oblongata sensitive to CO2, which also sense a decrease in pH of cerebrospinal fluid, and carotid body chemoreceptors determine the sensitivity of central chemoreceptors to CO2 [131, 132]. An elevated CO2 level is the primary factor for increasing ventilation and blood flow to the brain. However, prolonged hypercapnia reduces the sensitivity of chemoreceptors to CO2, leading to slower of CO2-induced rapid breathing over time. Similarly, breathing becomes faster as the concentration of inspired CO2 increases up to 9–10%, and a further increase in CO2 concentration leads to a decrease in ventilation.
The mechanics of breathing regulation, especially, CO2 level sensing, are very poorly understood at the molecular level. Mitochondria appear to be crucial for the response to elevated CO2, as CO2 induces the immediate release of ATP from chemosensitive regions of the ventral surface of the medulla oblongata [133]. ATP release is mediated by connexin 26 [328] and potentiates the release of acetylcholine [329]. Studies with model animals indicated that ERK1/2 were crucial for the regulation of respiration, as inhibition of ERK1/2 in brainstem preparations led to impaired breathing responses to CO2-induced acidosis [134].
One of the most important goals in the treatment of severe COVID-19 is an increase in low SpO2 levels. Brief inhalation of CO2 increases SpO2 [184]. The increase in SpO2 was immediate, within ~ 1 min, and 2-min sessions of inhaled 4, 8, or 12% CO2 with nebulized perflubron, a synthetic surfactant, caused SpO2 to increase by 1.7, 1.9 and 2.3 percentage points, respectively. The increase in SpO2 was maintained for 20 min, and subsequent inhalation treatments (twice per day) further stabilized the patient, suggesting a cumulative beneficial effect. Statistically significant increases in SpO2 in patients with cystic fibrosis are maintained 9 days after completion of the 5-day series of CO2 inhalation [330]. In another trial, inhalation of 8% CO2 increased SpO2 in subjects with mild allergic asthma after allergen-induced bronchoconstriction [331].
In addition to increasing SpO2, inhaled CO2 increase the supply of oxygen to tissues because CO2 allows oxygen to be released from haemoglobin (the Bohr effect) [332,333,334]. Thus, the CO2 inhalation treatment led to simultaneous increases in SpO2, better utilization of the oxygen in tissues because of the Bohr effect, and increases in blood flow due to vasodilation, which may support the use of less intense oxygen therapy.
CO2 and sleep
Sleep disturbances are common symptoms of both COVID-19 and post-COVID syndrome. Changes in CO2 levels are associated with sleep onset, wakening, and sleep stages. During sleep, PaCO2 is typically 2–8 mm Hg higher than it is during waking hours, depending on the sleep stage, and the sensitivity of the medulla oblongata chemosensors to CO2 decreases and hypoventilation occurs [154, 155]. Thus, an increase in PaCO2 of 2–8 mm Hg in mechanically ventilated patients may not be considered hypercapnic but a desirable baseline physiological level.
The intensity of neuromotor responses regulating breathing is significantly reduced during sleep compared to that during wakefulness; therefore, only marked increase in hypoxemia or hypercapnia increase ventilation during sleep. Similarly, waking up may be triggered only by a decrease in SpO2 to 70% or an increase in PaCO2 by 15 mm Hg compared to eupnoeic levels. In contrast, the physiological tolerance for decreased PaCO2 during sleep is low, since a decrease in PaCO2 by 3–6 mm Hg during sleep leads to sleep apnoea. Therefore, eupnoeic PaCO2 when awake may not be enough to sustain eupnoeic breathing during sleep [156, 157].
Maintaining waking PaCO2 leads to long-term sleep deprivation in patients in a medically induced coma. In mechanically ventilated patients, REM sleep is absent (and markedly reduced in patients with noninvasive mechanical ventilation) [335,336,337]. REM sleep is associated with an additional increase in PaCO2 of 1–2 mm Hg [158], local increases in low-frequency oscillations and global decreases in high-frequency oscillations in the electroencephalography (EEG) spectrum [338]. CO2 is the determining factor for changes in brain activity; inhaled CO2 leads to an increase in low-frequency power in the EEG spectrum [184]. There is a close connection between ERK1/2 and CO2 in the regulation of REM sleep; active ERK1/2-brain-derived neurotrophic factor (BDNF) signalling in the pedunculopontine tegmentum promotes homeostatic control of REM sleep [339]. Neurotransmitters involved in the regulation of sleep, the circadian rhythm and treatments that prevent major depressive disorder activate the key transcription factor cAMP response element-binding (CREB) via ERK1/2 signalling [159,160,161].
In mechanically ventilated patients, CO2 supplementation is particularly beneficial because mechanical ventilation decreases both pO2 and PaCO2 in the lungs. Inhalation of 1.5–2% CO2 is required to maintain the target EtCO2 of 4.7–4.9% in mechanically ventilated patients [340]. Interestingly, studies in model animals have indicated that the decrease in pO2 and PaCO2 that occurs in mechanically ventilated lungs can be inhibited by ERK1/2, p38 and JNK inhibitors [341].
Regulation of memory by MAPKs, CO2 and mitochondria
The levels of cellular CO2 produced via aerobic oxidation of carbohydrates are higher than those produced via other ATP synthesis pathways. Therefore, CO2 signalling particularly affects organs that consume carbohydrates as their main sources of energy. Therefore, CO2 is an important regulator of brain function, and as much as 20% of all CO2 in the body is generated in the brain, even though the brain represents approximately 2% of human body weight. Hypocapnia occurs in 74% of individuals post-COVID, and patients show neurological symptoms: fatigue, insomnia, depression and post-COVID brain fog [342]. Hypocapnia triggers known negative effects in various neurological diseases and conditions, such as severe traumatic brain injury, and ischaemic or haemorrhagic strokes. Moreover, hypocapnia negatively affects neonatal brain development, and the harmful effects can be reversed by inhaling 5% CO2 [343]. CO2 inhalation enhances the formation of memories and long-term memory [136,137,138,139]. The role of CO2 in the regulation of memory has been indirectly discerned from numerous studies on CAs. CA inhibitors (e.g., acetazolamide) impair [140], and administration of CA activators enhances [141, 142] memory and learning in model animals.
The positive effect of activated CAs on memorization and learning is mediated through the activation of ERK1/2 in the cerebral cortex and hippocampus, among other brain structures, and ERK1/2 are key elements required for the formation, retrieval, consolidation, reconsolidation, and persistence of memory [145,146,147]. Factors that increase cognitive abilities (e.g., amphetamine, methamphetamine, D-phenylalanine, phentermine, mephentermine, chlorphentermine and cocaine- and amphetamine-regulated transcript (CART), and neuropeptide) exert effects via ERK1/2 activation in the hippocampus [148,149,150] and potently activate CAs [141,142,143,144]. In contrast, memory-impairing drugs, such as hypnotic, amnestic and anaesthetic agents (e.g., butylphthalide, ketamine, midazolam, pentobarbital, isoflurane, propofol and scopolamine) reduce ERK1/2 activity in the brain [151, 152]. All naturally occurring mutations in the genes encoding ERK1/2 (MAPK1 and MAPK3) in humans, including mutation in noncoding regions of the genes, lead to cognitive impairment. Moreover, Erk2−/− mice showed a deficit in long-term memory [153].
Notably, the connections between ERK1/2 and mitochondria, which are sites of CO2 production, are important because endogenous CO2 is a natural regulator of neuron function, and mitochondria are critical to neuron function, including memory formation [344]. Local synaptic ATP production must be adjusted to meet high energy demand [345]. Mitochondrial mobility in neurons is essential for the formation of memories, and during learning, the number of mitochondria increases, but the size of mitochondria decreases. These mitochondrial changes promote the formation of multicontact synapses, which increases the information storage capacity of new synapses [346]. In contrast, in the neurons of the ageing brain, mitochondria become elongated as autophagy, fusion and fission of the mitochondria are disrupted [347].
The abovementioned processes are regulated by ERK1/2, which activate mitochondrial fission and inhibit fusion [185]. Cycles of fission and fusion help the mitochondrial network adapt to changing metabolic needs and are part of a fusion–fission–mitophagy quality control pathway enabling the removal of dysfunctional mitochondria. The large GTPase dynamin‐related protein 1 (DRP1) is recruited to sites of mitochondrial constriction, where it forms a higher-order ring structure that promotes fission via GTP‐dependent scission. The phosphorylation of DRP1 at Ser616 by ERK1/2 promotes mitochondrial translocation of DRP1 and subsequent mitochondrial fission/fragmentation [181, 186, 187]. ERK1/2 have been identified in mitochondria in several independent studies and are strongly regulated by essential mitochondrial products, i.e., CO2, ATP and H2O2 [348]. Moreover, ERK1/2 regulate mitochondrial biogenesis; in general, they induce mitochondrial biogenesis under control conditions and inhibit it under pathological conditions [188, 189]. ERK1/2 are involved in the regulation of the transition from mitochondrial respiration to glycolysis [190], and it has been proposed that mitochondrial ERK1/2 provide information about mitochondrial energetic and redox status to the nuclear pathways [349].
Mitochondrial dysfunction is closely associated with a variety of neurological disorders and ultimately leads to neuronal apoptosis. Synaptic mitochondrial dysfunction occurs during ageing and correlates with age-related memory loss. Synaptic mitochondria are the primary targets of both amyloid-β [350, 351] and phosphorylated tau [352] toxicity, which contributes to synaptic and memory impairment in Alzheimer's disease. Restoration of mitochondrial function is being intensively developed as a therapeutic strategy for dementia and learning and memory problems [351, 353, 354].
Understanding the relationship between CO2, ERK1/2 and mitochondria may allow for deeper insight into memory mechanisms and the emergence of new therapeutic possibilities. Neurodegenerative disorders are inflammatory in nature and characterized by contradictions in the functioning of ERK1/2; ERK1/2 are essential for the normal function of neurons, but their excessive activity leads to the development of inflammation. Therefore, despite the positive effect of active ERK1/2 on cognition, increased ERK1/2 activity is evident in neurodegenerative disorders. Thus, MAPK inhibition promotes neuroprotection [1, 170,171,172]. However, complete inhibition of ERK1/2 induced by synthetic inhibitors can be problematic, since basal ERK1/2 activity is essential for neuronal survival and memory. Elevated CO2 may be a universal therapeutic alternative that attenuates these drawbacks. CO2, on the one hand, may stimulate insufficiently active ERK1/2 (e.g., in an ageing brain) but, on the other hand, inhibit overactive proinflammatory MAPKs.
In addition to the role of mitochondria in the mechanisms underlying memory discussed here, the understanding of the effects of SARS-CoV-2 on mitochondria, including fusion, fission, mitophagy, metabolic reprogramming, and of the regulation of the immune response and apoptosis, has increased, and these effects are thoroughly discussed in numerous recent reviews [355,356,357].
MAPKs and CO2 in cell survival and apoptosis
Lymphocytes are among the cells whose death contributes most to severe COVID-19, with T-cell apoptosis accounting for T lymphopenia in patients with severe COVID-19 [358]. Negative regulation of apoptosis by ERK1/2 has previously been shown to be required to ensure survival of T and B lymphocytes [182]. ERK1/2 promote cell survival by activating prosurvival BCL-2 proteins (BCL-2, BCL-xL and MCL1) and repressing prodeath protein (BAD, BIM, BMF and PUMA) activity, including the key mechanism underlying the phosphorylation of BIMEL by ERK1/2, thereby preventing homo-oligomerization of BAX, a proapoptotic member of the BCL-2 family responsible for the permeabilization of the mitochondrial outer membrane; loss of potential across the inner mitochondrial membrane and cytochrome c release [181, 183].
In response to a large cellular imbalance, e.g., caused by DNA damage or excessive inflammation, ERK1/2 are hyperactivated and may exert a proapoptotic effect [4, 183]. However, in general, ERK1/2 promote cell survival and proliferation, whereas activation of JNK and p38 may induce apoptosis [359]. Since a slight increase in CO2 concentration (from 5 to 8%) activates ERK1/2 and inhibits p38 and JNK under control conditions [10], the question arises: Does such a slight increase in CO2 levels support the prosurvival activity of ERK1/2 while inhibiting apoptotic p38 and JNK? Indeed, the overlapping functions of CO2 and ERK1/2 include the regulation of apoptosis and longevity, and the lifespan of mammals positively correlates with blood PaCO2 and HCO3− [5, 167, 173, 174]. Moreover, human cells are unable to proliferate without CO2, and elevated CO2 levels support cell proliferation [175, 176]; CO2 has been shown to exert an effect via ERK1/2 in a cell line derived from human small cell lung cancer [14]. Furthermore, hypercapnia induces the expression of anti-apoptotic BCL-2 and BCL-xL and inhibits autophagy in macrophages [178] and ischaemic penumbra astrocytes and neurons [167]. Similarly, CAs [179] and acidification [360], which shifts the CO2/HCO3− equilibrium towards an increase in the level of CO2, activate ERK1/2 and thus delay neutrophil apoptosis, improving neutrophil migration and wound healing. Therefore, the inhibition of CAIX resulted in a reduction in the level of active ERK1/2 and reduced neutrophil viability and mitochondrial function [180]. An extreme case of the prosurvival effect of CO2 involves the accelerated growth of the aquatic uniflagellate phycomycetes Blastocladia ramosa and Blastocladia pringsheimii induced by an increase in CO2 of 5–20% [177].
It is believed that at the time when life was formed, the Earth’s atmosphere and water reservoirs contained much more CO2 than they contain today. To survive, primitive organisms needed to be adapted to the natural environment, i.e., to high concentrations of CO2. The change to the oxidizing atmosphere was followed by endosymbiosis, allowing eukaryotes to maintain high levels of CO2 by producing CO2 inside the cells in mitochondria.
Typical mitochondrial respiration involving oxygen consumption is associated with CO2 production during the entry of pyruvate into the Krebs cycle and two stages of the Krebs cycle. Anaerobic energy production by organisms or cells consuming organic compounds is also associated with increased CO2 levels. However, this production can be mediated by acidification, e.g., by the glycolytic production of lactate, which shifts the CO2/HCO3− equilibrium towards an increased CO2 concentration. There are species of multicellular eukaryotes that tolerate periods of complete oxygen deprivation; e.g., the larvae of oriental fruit flies (Bactrocera dorsalis) can tolerate up to 24 h of anoxia without a significant reduction in survival [361]. In several species of fish adapted to life under completely oxygen-deprived conditions; for example, one of the clearest differences in crucian carp compared to aerobic species is the activity of pyruvate decarboxylase, which produces CO2 independent of the Krebs cycle [362, 363]. In addition, some turtles survive complete anoxic conditions, and upregulation of the prosurvival proteins ERK1/2 and suppression of p38 and JNK underlie neuronal survival [364, 365].
Conclusions and future perspectives
Considering the previously published data presented here, it can be concluded that MAPKs play a central role in regulating cellular responses to changing CO2 levels (Fig. 6). Detailed studies on the regulatory mechanisms MAPK activity by CO2 suggest that reassessing the functioning of MAPK signalling pathways while taking into account the level of CO2, as each MAPK pathway may function differently under altered CO2 levels, is needed. The effect of CO2 on the activity of ERK1/2, JNKs and p38 MAPKs varies depending on its concentration. In addition, individual MAPKs function differently in different signalling pathways. Therefore, it can be expected that relatively narrow ranges of CO2 concentrations will modify MAPK activity in the desired way under specific conditions. Therefore, the basis for future research and the first priority is to determine the effects of specific CO2 concentrations on individual MAPK isoforms in detail; this research should include in vitro studies using recombinant MAPKs, to confirm the direct CO2-sensing ability of MAPKs. We expect that these studies will accelerate CO2 research because MAPKs are involved in many developmental processes and oncogenesis.

Cellular MAPK signalling pathways regulated by CO2. Dashed lines represent secondary or concurrent signalling pathways. Multiple arrows indicate indirect regulation
Advances in our understanding of CO2 signalling have been relatively slow. The first genome-wide proteomic, transcriptomic [366] and genetic [367] analyses of the yeast CO2 response were reported only recently. These data support the hypotheses presented in this paper by showing that the MAPK pathway is critical for CO2 sensing and CO2 signalling in yeast. Hope for progress in CO2 research is offered via the recent development of selective fluorescent CO2 molecular sensors, which are expected to lead to breakthrough insights into biochemical processes [368], and detection methods for carboxylation of the amine group in lysine residues [369].
MAPKs are signalling molecules connecting various aspects of the inflammatory response to viral infection, including SARS-CoV-2 infection, and comorbidity pathogenesis. In addition, CO2 appears to be a molecule that universally counteracts the MAPK-induced proinflammatory response, including in patients with severe course COVID-19 and complications leading to death. The benefits of using elevated CO2 levels in the treatment of various diseases are reflected in numerous completed and ongoing clinical trials established to evaluated CO2 use as a medication. Advances in research on the regulation of CO2-MAPK may significantly increase the number of new therapeutic applications of CO2 because a number of MAPK inhibitors have been approved as potent drugs for the treatment of numerous diseases, including cancers, and a significant number of these inhibitors are under evaluation in different stages of clinical trials. However, many MAPK inhibitors cannot be fully exploited at the most effective concentrations due to their toxicity and resistance mechanisms; for example, the activation of ERK5 can enable cells to bypass RAF-MEK1/2-ERK1/2 inhibitors [370]. CO2, which is safe under controlled conditions [331, 371], may overcome these limitations because it inhibits various ERKs, JNKs and p38 MAPKs when administered in a precise concentration range. Other advantages involve the delivery of CO2 to cells independent of membrane transporters and the possibility of administering very high concentrations of CO2 locally without causing systemic effects and only nominal effects on the cells surrounding the CO2 application site.
In conclusion, understanding the molecular mechanisms underlying CO2-dependent regulation of MAPKs, including the opposing effects of elevated CO2 on active and inactive ERK1/2, is essential to precisely guide the development of therapeutic CO2 applications.
Availability of data and materials
Not applicable.
Abbreviations
- 15-HETE:
-
15-Hydroxyeicosatetraenoic acid
- ACE2:
-
Angiotensin-converting enzyme 2
- ADAM17:
-
A disintegrin and metalloprotease 17
- AEC:
-
Alveolar epithelial cell
- AFC:
-
Alveolar fluid clearance
- Akap1:
-
A-kinase anchoring protein 1
- ALF:
-
Alveolar lining fluid
- Ang:
-
Angiotensin
- AQP:
-
Aquaporin
- ARDS:
-
Acute respiratory distress syndrome
- ASIC:
-
Acid-sensing ion channel
- AT1:
-
Angiotensin II receptor type 1
- ATII cell:
-
Type II alveolar epithelial cell
- BDNF:
-
Brain-derived neurotrophic factor
- BMI:
-
Body mass index
- CA:
-
Carbonic anhydrase
- CART:
-
Cocaine- and amphetamine-regulated transcript
- COPD:
-
Chronic obstructive pulmonary disease
- CREB:
-
cAMP response element-binding
- DRP1:
-
Dynamin‐related protein 1
- EEG:
-
Electroencephalography
- EGFR:
-
Epidermal growth factor receptor
- ENaC:
-
Epithelial Na+ channel
- ER:
-
Endoplasmic reticulum
- ERK1/2:
-
Extracellular signal-regulated kinases 1 and 2
- EtCO2 :
-
End-tidal CO2
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- HO-1:
-
Heme oxygenase-1
- HSF1:
-
Heat shock factor 1
- ICAM-1:
-
Intercellular adhesion molecule-1
- ICU:
-
Intensive care unit
- IFN-γ:
-
Interferon-gamma
- IL:
-
Interleukin
- IRS-1:
-
Insulin receptor substrate 1
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1/CCL2:
-
Monocyte chemoattractant protein-1
- MIP-1α/CCL3:
-
Macrophage inflammatory protein-1α
- MKP-1:
-
MAPK phosphatase 1
- NET:
-
Neutrophil extracellular trap
- NK:
-
Natural killer
- PaCO2 :
-
Arterial partial pressure of CO2
- PAH:
-
Pulmonary artery hypertension
- PBEF:
-
Pre-B-cell colony enhancing factor
- PKC:
-
Protein kinase C
- PKA:
-
Protein kinase A
- PM2.5:
-
Particulate matter 2.5
- PPARα:
-
Peroxisome proliferator-activated receptor α
- PSD-95:
-
Postsynaptic density 95
- ROS:
-
Reactive oxygen species
- SMAD3:
-
Small mothers against decapentaplegic 3
- TGF-β1:
-
Transforming growth factor β1
- TNF-α:
-
Tumour necrosis factor-α
- UCP1:
-
Uncoupling protein 1
- VEGF:
-
Vascular endothelial growth factor
References
Ahmed T, Zulfiqar A, Arguelles S, Rasekhian M, Nabavi SF, Silva AS, et al. Map kinase signaling as therapeutic target for neurodegeneration. Pharmacol Res. 2020;160: 105090.
Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduction. 2015;35:600–4.
Bohush A, Niewiadomska G, Filipek A. Role of mitogen activated protein kinase signaling in Parkinson’s disease. Int J Mol Sci. 2018;19:2973.
Sugiura R, Satoh R, Takasaki T. ERK: a double-edged sword in cancer. ERK-dependent apoptosis as a potential therapeutic strategy for cancer. Cells. 2021;10:2509.
Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. 2009;8:1168–75.
Gajanayaka N, Dong SXM, Ali H, Iqbal S, Mookerjee A, Lawton DA, et al. TLR-4 agonist induces IFN-γ production selectively in proinflammatory human M1 macrophages through the PI3K-mTOR- and JNK-MAPK-Activated p70S6K Pathway. J Immunol. 2021;207:2310–24.
Zibara K, Zeidan A, Bjeije H, Kassem N, Badran B, El-Zein N. ROS mediates interferon gamma induced phosphorylation of Src, through the Raf/ERK pathway, in MCF-7 human breast cancer cell line. J Cell Commun Signal. 2017;11:57–67.
Lucas RM, Luo L, Stow JL. ERK1/2 in immune signalling. Biochem Soc Trans. 2022;50:1341–52.
Ronkina N, Gaestel M. MAPK-activated protein kinases: servant or partner? Annu Rev Biochem. 2022;91:505–40.
Galganska H, Jarmuszkiewicz W, Galganski L. Carbon dioxide inhibits COVID-19-type proinflammatory responses through extracellular signal-regulated kinases 1 and 2, novel carbon dioxide sensors. Cell Mol Life Sci. 2021;78:8229–42.
Gwoździńska P, Buchbinder BA, Mayer K, Herold S, Morty RE, Seeger W, et al. Hypercapnia Impairs ENaC Cell surface stability by promoting phosphorylation, polyubiquitination and endocytosis of β-ENaC in a human alveolar epithelial cell line. Front Immunol. 2017;8:591.
Welch LC, Lecuona E, Briva A, Trejo HE, Dada LA, Sznajder JI. Extracellular signal-regulated kinase (ERK) participates in the hypercapnia-induced Na. K-ATPase downregulation FEBS Lett. 2010;584:3985–9.
Gałgańska H, Gałgański Ł. Mitogen-activated protein kinases are carbon dioxide receptors in plants. bioRxiv. 2020;2020.05.09.086116. Available from: http://biorxiv.org/content/early/2020/05/10/2020.05.09.086116.abstract. Accesed 12 May 2020.
Merryman JI, Park PG, Schuller HM. Carbon dioxide, an important messenger molecule for small cell lung cancer. Chest. 1997;112:779–84.
Kuo NT, Agani FH, Haxhiu MA, Chang CH. A possible role for protein kinase C in CO2/H+-induced c-fos mRNA expression in PC12 cells. Respir Physiol. 1998;111:127–35.
Xu Y-J, Elimban V, Dhalla NS. Suppression of phosphorylated MAPK and caspase 3 by carbon dioxide. Mol Cell Biochem. 2017;436:23–8.
Kurihara J, Katsura K, Siesjö BK, Wieloch T. Hyperglycemia and hypercapnia differently affect post-ischemic changes in protein kinases and protein phosphorylation in the rat cingulate cortex. Brain Res. 2004;995:218–25.
Wang N, Gates KL, Trejo H, Favoreto S, Schleimer RP, Sznajder JI, et al. Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J. 2010;24:2178–90.
Izumi Y, Yamaguchi T, Yamazaki T, Yamashita N, Nakamura Y, Shiota M, et al. Percutaneous carbon dioxide treatment using a gas mist generator enhances the collateral blood flow in the ischemic hindlimb. J Atheroscler Thromb. 2015;22:38–51.
Otulakowski G, Engelberts D, Gusarova GA, Bhattacharya J, Post M, Kavanagh BP. Hypercapnia attenuates ventilator-induced lung injury via a disintegrin and metalloprotease-17. J Physiol. 2014;592:4507–21.
Danielsson A, Ost A, Lystedt E, Kjolhede P, Gustavsson J, Nystrom FH, et al. Insulin resistance in human adipocytes occurs downstream of IRS1 after surgical cell isolation but at the level of phosphorylation of IRS1 in type 2 diabetes. FEBS J. 2005;272:141–51.
Ko MJ, Mulia GE, van Rijn RM. Commonly used anesthesia/euthanasia methods for brain collection differentially impact MAPK activity in male and female C57BL/6 Mice. Front Cell Neurosci. 2019;13:96.
Lin L-T, Chen J-T, Tai M-C, Chen Y-H, Chen C-L, Pao S-I, et al. Protective effects of hypercapnic acidosis on Ischemia-reperfusion-induced retinal injury. PLoS ONE. 2019;14: e0211185.
Yang WC, Song CY, Wang N, Zhang LL, Yue ZY, Cui XG, et al. Hypercapnic acidosis confers antioxidant and anti-apoptosis effects against ventilator-induced lung injury. Lab Invest. 2013;93:1339–49.
Dada LA, Trejo Bittar HE, Welch LC, Vagin O, Deiss-Yehiely N, Kelly AM, et al. High CO2 Leads to Na, K-ATPase Endocytosis via c-Jun Amino-Terminal Kinase-Induced LMO7b Phosphorylation. Mol Cell Biol. 2015;35:3962–73.
Vadász I, Dada LA, Briva A, Helenius IT, Sharabi K, Welch LC, et al. Evolutionary conserved role of c-Jun-N-terminal kinase in CO2-induced epithelial dysfunction. PLoS ONE. 2012;7: e46696.
Cicconetti F, Sestili P, Madiai V, Albertini MC, Campanella L, Coppari S, et al. Extracellular pH, osmolarity, temperature and humidity could discourage SARS-CoV-2 cell docking and propagation via intercellular signaling pathways. PeerJ. 2021;9: e12227.
Rashedi J, Mahdavi Poor B, Asgharzadeh M. Sodium bicarbonate nebulized therapy in patients with confirmed COVID-19. Adv Pharm Bull. 2021;11:397–8.
Badhe RV, Nipate SS. The use of negative oxygen ion clusters [O(2)(-)(H(2)O)(n)] and bicarbonate ions [HCO(3)(-)] as the supportive treatment of COVID-19 infections: a possibility. Med Hypotheses. 2021;154: 110658.
El-Betany AMMM, Behiry EM, Gumbleton M, Harding KG. Humidified warmed CO(2) treatment therapy strategies can save lives with mitigation and suppression of SARS-CoV-2 infection: an evidence review. Front Med (Lausanne). 2020;7: 594295.
Masterson C, Horie S, McCarthy SD, Gonzalez H, Byrnes D, Brady J, et al. Hypercapnia in the critically ill: insights from the bench to the bedside. Interface Focus. 2021;11:20200032.
Petran J, Ansems K, Rossaint R, Marx G, Kalvelage C, Kopp R, et al. Effects of hypercapnia versus normocapnia during general anesthesia on outcomes: a systematic review and meta-analysis. Braz J Anesthesiol. 2022;72:398–406.
Shigemura M, Welch LC, Sznajder JI. Hypercapnia regulates gene expression and tissue function. Front Physiol. 2020;11: 598122.
Laffey JG, Tanaka M, Engelberts D, Luo X, Yuan S, Keith Tanswell A, et al. therapeutic hypercapnia reduces pulmonary and systemic injury following in vivo lung reperfusion. Am J Respir Crit Care Med. 2000;162:2287–94.
Turner MJ, Saint-Criq V, Patel W, Ibrahim SH, Verdon B, Ward C, et al. Hypercapnia modulates cAMP signalling and cystic fibrosis transmembrane conductance regulator-dependent anion and fluid secretion in airway epithelia. J Physiol. 2016;594:1643–61.
Tang S-E, Wu S-Y, Chu S-J, Tzeng Y-S, Peng C-K, Lan C-C, et al. Pre-treatment with ten-minute carbon dioxide inhalation prevents lipopolysaccharide-induced lung injury in mice via down-regulation of toll-like receptor 4 expression. Int J Mol Sci. 2019;20:6293.
Wu S-Y, Li M-H, Ko F-C, Wu G-C, Huang K-L, Chu S-J. Protective effect of hypercapnic acidosis in ischemia-reperfusion lung injury is attributable to upregulation of heme Oxygenase-1. PLoS ONE. 2013;8: e74742.
Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, et al. Interleukin-1beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J Biol Chem. 2005;280:18579–89.
Lazrak A, Chen L, Jurkuvenaite A, Doran SF, Liu G, Li Q, et al. Regulation of alveolar epithelial Na+ channels by ERK1/2 in chlorine-breathing mice. Am J Respir Cell Mol Biol. 2012;46:342–54.
Hallows KR, Bhalla V, Oyster NM, Wijngaarden MA, Lee JK, Li H, et al. Phosphopeptide screen uncovers novel phosphorylation sites of Nedd4-2 that potentiate its inhibition of the epithelial Na+ channel. J Biol Chem. 2010;285:21671–8.
Booth RE, Stockand JD. Targeted degradation of ENaC in response to PKC activation of the ERK1/2 cascade. Am J Physiol Renal Physiol. 2003;284:F938–47.
Shi H, Asher C, Chigaev A, Yung Y, Reuveny E, Seger R, et al. Interactions of beta and gamma ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem. 2002;277:13539–47.
Shen Y, Chen Z, Wang Y, Song Z, Zhang Z, Jin M, et al. Aquaporin 5 expression inhibited by LPS via p38/JNK signaling pathways in SPC-A1 cells. Respir Physiol Neurobiol. 2010;171:212–7.
Ba F, Zhou X, Zhang Y, Wu C, Xu S, Wu L, et al. Lipoxin A4 ameliorates alveolar fluid clearance disturbance in lipopolysaccharide-induced lung injury via aquaporin 5 and MAPK signaling pathway. J Thorac Dis. 2019;11:3599–608.
Xu W, Zhou J, You M, Lu C, Yang W, Gong Y, et al. Pre-B-cell colony enhancing factor regulates the alveolar epithelial sodium-water transport system through the ERK and AKT pathways. Am J Transl Res. 2019;11:5824–35.
Wang HC, Zentner MD, Deng HT, Kim KJ, Wu R, Yang PC, et al. Oxidative stress disrupts glucocorticoid hormone-dependent transcription of the amiloride-sensitive epithelial sodium channel alpha-subunit in lung epithelial cells through ERK-dependent and thioredoxin-sensitive pathways. J Biol Chem. 2000;275:8600–9.
Jiang L, Wang J, Su C, Qian W, Chen J, Zhu B, et al. α-ENaC, a therapeutic target of dexamethasone on hydrogen sulfide induced acute pulmonary edema. Environ Toxicol Pharmacol. 2014;38:616–24.
Laffey JG, Honan D, Hopkins N, Hyvelin J-M, Boylan JF, McLoughlin P. Hypercapnic acidosis attenuates endotoxin-induced acute lung injury. Am J Respir Crit Care Med. 2004;169:46–56.
Lang CJ, Dong P, Hosszu EK, Doyle IR. Effect of CO 2 on LPS-induced cytokine responses in rat alveolar macrophages. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2005;289:L96-103.
Li Y, Zeng Z, Li Y, Huang W, Zhou M, Zhang X, et al. Angiotensin-converting enzyme inhibition attenuates lipopolysaccharide-induced lung injury by regulating the balance between angiotensin-converting enzyme and angiotensin-converting enzyme 2 and inhibiting mitogen-activated protein kinase activation. Shock. 2015;43:395–404.
Li Y, Zeng Z, Cao Y, Liu Y, Ping F, Liang M, et al. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci Rep. 2016;6:27911.
Schuh K, Pahl A. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochem Pharmacol. 2009;77:1827–34.
Horie S, Ansari B, Masterson C, Devaney J, Scully M, O’Toole D, et al. Hypercapnic acidosis attenuates pulmonary epithelial stretch-induced injury via inhibition of the canonical NF-κB pathway. Intensive Care Med Exp. 2016;4:8.
Yang W, Yue Z, Cui X, Guo Y, Zhang L, Zhou H, et al. Comparison of the effects of moderate and severe hypercapnic acidosis on ventilation-induced lung injury. BMC Anesthesiol. 2015;15:67.
Contreras M, Ansari B, Curley G, Higgins BD, Hassett P, O’Toole D, et al. Hypercapnic acidosis attenuates ventilation-induced lung injury by a nuclear factor-κB–dependent mechanism. Crit Care Med. 2012;40:2622–30.
Kim S-H, Li M, Pyeon T-H, So K-Y, Kwak S-H. The volatile anesthetic sevoflurane attenuates ventilator-induced lung injury through inhibition of ERK1/2 and Akt signal transduction. Korean J Anesthesiol. 2015;68:62.
Dogliotti G, Galliera E, Iorio E, De Bernardi Di Valserra M, Solimene U, Corsi MM. Effect of immersion in CO2-enriched water on free radical release and total antioxidant status in peripheral arterial occlusive disease. Int Angiol. 2011;30:12–7.
Bolevich S, Kogan AH, Zivkovic V, Djuric D, Novikov AA, Vorobyev SI, et al. Protective role of carbon dioxide (CO2) in generation of reactive oxygen species. Mol Cell Biochem. 2016;411:317–30.
Parinandi NL, Kleinberg MA, Usatyuk PV, Cummings RJ, Pennathur A, Cardounel AJ, et al. Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L26-38.
Wang X, Lv S, Sun J, Zhang M, Zhang L, Sun Y, et al. Caffeine reduces oxidative stress to protect against hyperoxia-induced lung injury via the adenosine A2A receptor/cAMP/PKA/Src/ERK1/2/p38MAPK pathway. Redox Rep. 2022;27:270–8.
Wang C, Cheng T, Lu Q, Li W, Liu B, Yue L, et al. Oxygen therapy accelerates apoptosis induced by selenium compounds via regulating Nrf2/MAPK signaling pathway in hepatocellular carcinoma. Pharmacol Res. 2023;187: 106624.
Jiang J, Wang J, Li C, Mo L, Huang D. Hyperoxia induces alveolar epithelial cell apoptosis by regulating mitochondrial function through small mothers against decapentaplegic 3 (SMAD3) and extracellular signal-regulated kinase 1/2 (ERK1/2). Bioengineered. 2022;13:242–52.
Liang Z, Zhang X, Liu Y, Wu Q, You C. SEMA3A protects against hyperoxia-induced lung injury in a bronchopulmonary dysplasia model of newborn rat by inhibiting ERK pathway. Allergol Immunopathol (Madr). 2021;49:8–15.
Sidramagowda Patil S, Soundararajan R, Fukumoto J, Breitzig M, Hernández-Cuervo H, Alleyn M, et al. Mitochondrial protein Akap1 deletion exacerbates endoplasmic reticulum stress in mice exposed to hyperoxia. Front Pharmacol. 2022;13: 762840.
Menon RT, Thapa S, Shrestha AK, Barrios R, Shivanna B. Extracellular signal-regulated kinase 1 alone is dispensable for hyperoxia-mediated alveolar and pulmonary vascular simplification in neonatal mice. Antioxidants. 2022;11:1130.
Hu J, Wu Z, Wang H, Geng H, Huo J, Zhu X, et al. Vitamin D ameliorates apoptosis and inflammation by targeting the mitochondrial and MEK1/2-ERK1/2 pathways in hyperoxia-induced bronchopulmonary dysplasia. J Inflamm Res. 2022;15:4891–906.
El Mays TY, Choudhury P, Leigh R, Koumoundouros E, Van der Velden J, Shrestha G, et al. Nebulized perflubron and carbon dioxide rapidly dilate constricted airways in an ovine model of allergic asthma. Respir Res. 2014;15:98.
Shigemura M, Sznajder JI. Elevated CO(2) modulates airway contractility. Interface Focus. 2021;11:20200021.
Duckles SP, Rayner MD, Nadel JA. Effects of CO2 and pH on drug-induced contractions of airway smooth muscle. J Pharmacol Exp Ther. 1974;190:472–81.
Kikuchi R, Kikuchi K, Hildebrandt J, Yanai M, Sekizawa K, Sasaki H. Dependence of collateral and small airway resistances of CO2 and volume in dog lobes. Respir Physiol. 1995;100:245–52.
Lindeman KS, Croxton TL, Lande B, Hirshman CA. Hypocapnia-induced contraction of porcine airway smooth muscle. Eur Respir J. 1998;12:1046–52.
El Mays TY, Saifeddine M, Choudhury P, Hollenberg MD, Green FHY. Carbon dioxide enhances substance P-induced epithelium-dependent bronchial smooth muscle relaxation in Sprague-Dawley rats. Can J Physiol Pharmacol. 2011;89:513–20.
van den Elshout FJ, van Herwaarden CL, Folgering HT. Effects of hypercapnia and hypocapnia on respiratory resistance in normal and asthmatic subjects. Thorax. 1991;46:28–32.
Fisher HK, Hansen TA. Site of action of inhaled 6 per cent carbon dioxide in the lungs of asthmatic subjects before and after exercise. Am Rev Respir Dis. 1976;114:861–70.
Park S-Y, Kang M-J, Jin N, Lee SY, Lee YY, Jo S, et al. House dust mite-induced Akt-ERK1/2-C/EBP beta pathway triggers CCL20-mediated inflammation and epithelial-mesenchymal transition for airway remodeling. FASEB J. 2022;36: e22452.
Shah SD, Nayak AP, Sharma P, Villalba DR, Addya S, Huang W, et al. Targeted inhibition of select ERK1/2 functions mitigates pathological features of asthma in mice. Am J Respir Cell Mol Biol. 2022;68:23–38.
Chuang I-C, Yang R-C, Chou S-H, Huang L-R, Tsai T-N, Dong H-P, et al. Effect of carbon dioxide inhalation on pulmonary hypertension induced by increased blood flow and hypoxia. Kaohsiung J Med Sci. 2011;27:336–43.
Welch CE, Brittain EL, Newman AL, Robbins IM, Pugh ME, Newman JH, et al. End-tidal carbon dioxide as a prognostic feature in pulmonary arterial hypertension. Ann Am Thorac Soc. 2017;14:896–902.
Harbaum L, Fuge J, Kamp JC, Hennigs JK, Simon M, Sinning C, et al. Blood carbon dioxide tension and risk in pulmonary arterial hypertension. Int J Cardiol. 2020;318:131–7.
Yu X, Li T, Liu X, Yu H, Hao Z, Chen Y, et al. Modulation of pulmonary vascular remodeling in hypoxia: role of 15-LOX-2/15-HETE-MAPKs pathway. Cell Physiol Biochem. 2015;35:2079–97.
Church AC, Martin DH, Wadsworth R, Bryson G, Fisher AJ, Welsh DJ, et al. The reversal of pulmonary vascular remodeling through inhibition of p38 MAPK-alpha: a potential novel anti-inflammatory strategy in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2015;309:L333–47.
Ooi H, Cadogan E, Sweeney M, Howell K, O’Regan RG, McLoughlin P. Chronic hypercapnia inhibits hypoxic pulmonary vascular remodeling. American Journal of Physiology-Heart and Circulatory Physiology. 2000;278:H331–8.
Bao W, Behm DJ, Nerurkar SS, Ao Z, Bentley R, Mirabile RC, et al. Effects of p38 MAPK Inhibitor on angiotensin II-dependent hypertension, organ damage, and superoxide anion production. J Cardiovasc Pharmacol. 2007;49:362–8.
Lu J, Shimpo H, Shimamoto A, Chong AJ, Hampton CR, Spring DJ, et al. Specific inhibition of p38 mitogen-activated protein kinase with FR167653 attenuates vascular proliferation in monocrotaline-induced pulmonary hypertension in rats. J Thorac Cardiovasc Surg. 2004;128:850–9.
Marhong J, Fan E. Carbon dioxide in the critically ill: too much or too little of a good thing? Respir Care. 2014;59:1597–605.
Maueröder C, Mahajan A, Paulus S, Gößwein S, Hahn J, Kienhöfer D, et al. Ménage-à-Trois: the ratio of bicarbonate to CO(2) and the pH regulate the capacity of neutrophils to form NETs. Front Immunol. 2016;7:583.
Engström M, Schött U, Romner B, Reinstrup P. Acidosis impairs the coagulation: a thromboelastographic study. J Trauma. 2006;61:624–8.
Bohgaki M, Atsumi T, Yamashita Y, Yasuda S, Sakai Y, Furusaki A, et al. The p38 mitogen-activated protein kinase (MAPK) pathway mediates induction of the tissue factor gene in monocytes stimulated with human monoclonal anti-beta2Glycoprotein I antibodies. Int Immunol. 2004;16:1633–41.
Lee M-K, Lee Y, Huh J-W, Chen H, Wu W, Ha U-H. The Pseudomonas aeruginosa HSP90-like protein HtpG regulates IL-8 expression through NF-κB/p38 MAPK and CYLD signaling triggered by TLR4 and CD91. Microbes Infect. 2020;22:558–66.
Youn Y-J, Lee Y-B, Kim S-H, Jin HK, Bae J-S, Hong C-W. Nucleocapsid and spike proteins of SARS-CoV-2 drive neutrophil extracellular trap formation. Immune Netw. 2021;21: e16.
Middleton EA, He X-Y, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136:1169–79.
An Z, Li J, Yu J, Wang X, Gao H, Zhang W, et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-κB signaling in macrophages. Cell Cycle. 2019;18:2928–38.
Vannucci RC, Towfighi J, Heitjan DF, Brucklacher RM. Carbon dioxide protects the perinatal brain from hypoxic-ischemic damage: an experimental study in the immature rat. Pediatrics. 1995;95:868–74.
Nomura F, Aoki M, Forbess JM, Mayer JEJ. Effects of hypercarbic acidotic reperfusion on recovery of myocardial function after cardioplegic ischemia in neonatal lambs. Circulation. 1994;90:II321-7.
Li A, Quan Y, Guo Y, Li W, Cui X. Effects of therapeutic hypercapnia on inflammation and apoptosis after hepatic ischemia-reperfusion injury in rats. Chin Med J (Engl). 2010;123:2254–8.
Baldi E, Caputo ML, Klersy C, Benvenuti C, Contri E, Palo A, et al. End-tidal carbon dioxide (ETCO2) at intubation and its increase after 10 minutes resuscitation predicts survival with good neurological outcome in out-of-hospital cardiac arrest patients. Resuscitation. 2022;181:197–207.
Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, et al. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98:11569–74.
Kim W, Kwon HJ, Jung HY, Hahn KR, Yoon YS, Hwang IK, et al. neuroprotective effects of purpurin against ischemic damage via MAPKs, bax, and oxidative stress cascades in the gerbil hippocampus. Mol Neurobiol. 2022;59:2580–92.
Appunni S, Gupta D, Rubens M, Ramamoorthy V, Singh HN, Swarup V. Deregulated protein kinases: friend and foe in ischemic stroke. Mol Neurobiol. 2021;58:6471–89.
Zhong M, Ma W, Zhang X, Wang Y, Gao X. Tetramethyl pyrazine protects hippocampal neurons against anoxia/reoxygenation injury through inhibiting apoptosis mediated by JNK/MARK signal pathway. Med Sci Monit. 2016;22:5082–90.
Engin A. The pathogenesis of obesity-associated adipose tissue inflammation. Adv Exp Med Biol. 2017;960:221–45.
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6.
Yung JHM, Giacca A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells. 2020;9:706.
Vernia S, Cavanagh-Kyros J, Barrett T, Tournier C, Davis RJ. Fibroblast growth factor 21 mediates glycemic regulation by hepatic JNK. Cell Rep. 2016;14:2273–80.
Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, et al. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014;20:512–25.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7.
Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–6.
Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2006;103:10741–6.
Alam M, Voravutinon N, Reynolds KA, Poon E. Safety of subcutaneous infiltration of carbon dioxide (Carboxytherapy) for abdominal fat reduction: a pilot study. Dermatol Surg. 2020;46:1249–50.
Kołodziejczak A, Podgórna K, Rotsztejn H. Is carboxytherapy a good alternative method in the removal of various skin defects? Dermatol Ther. 2018;31: e12699.
Yamazaki T, Ushikoshi-Nakayama R, Shakya S, Omagari D, Matsumoto N, Nukuzuma C, et al. The effects of bathing in neutral bicarbonate ion water. Sci Rep. 2021;11:21789.
Zhang X, Liu Z, Li W, Kang Y, Xu Z, Li X, et al. MAPKs/AP-1, not NF-κB, is responsible for MCP-1 production in TNF-α-activated adipocytes. Adipocyte. 2022;11:477–86.
Cui N, Li H, Dun Y, Ripley-Gonzalez JW, You B, Li D, et al. Exercise inhibits JNK pathway activation and lipotoxicity via macrophage migration inhibitory factor in nonalcoholic fatty liver disease. Front Endocrinol (Lausanne). 2022;13: 961231.
Lv S, Zhou Y, Chen J, Yuan H, Zhang Z-N, Luan B. Hepatic ER stress suppresses adipose browning through ATF4-CIRP-ANGPTL3 cascade. Cell Rep. 2022;40: 111422.
Zhang S, Cao H, Li Y, Jing Y, Liu S, Ye C, et al. Metabolic benefits of inhibition of p38α in white adipose tissue in obesity. PLoS Biol. 2018;16: e2004225.
Casale TB, Korenblat PE, Meltzer EO, Yen K, Bhatnagar A. Nasal carbon dioxide for the symptomatic treatment of perennial allergic rhinitis. Ann Allergy Asthma Immunol. 2011;107:364–70.
Baroody FM, Gavanescu L, Wang JH, DeTineo M, Naclerio RM. The effect of intranasal carbon dioxide on the acute response to nasal challenge with allergen. Allergy Asthma Proc. 2011;32:206–12.
Strider JW, Masterson CG, Durham PL. Treatment of mast cells with carbon dioxide suppresses degranulation via a novel mechanism involving repression of increased intracellular calcium levels. Allergy. 2011;66:341–50.
Gouel-Chéron A, de Chaisemartin L, Jönsson F, Nicaise-Roland P, Granger V, Sabahov A, et al. Low end-tidal CO2 as a real-time severity marker of intra-anaesthetic acute hypersensitivity reactions. Br J Anaesth. 2017;119:908–17.
Conti P, Caraffa A, Tetè G, Gallenga CE, Ross R, Kritas SK, et al. Mast cells activated by SARS-CoV-2 release histamine which increases IL-1 levels causing cytokine storm and inflammatory reaction in COVID-19. J Biol Regul Homeost Agents. 2020;34:1629–32.
Matsubara M, Tamura T, Ohmori K, Hasegawa K. Histamine H1 receptor antagonist blocks histamine-induced proinflammatory cytokine production through inhibition of Ca2+-dependent protein kinase C, Raf/MEK/ERK and IKK/I kappa B/NF-kappa B signal cascades. Biochem Pharmacol. 2005;69:433–49.
Thangam EB, Jemima EA, Singh H, Baig MS, Khan M, Mathias CB, et al. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: the hunt for new therapeutic targets. Front Immunol. 2018;9:1873.
Colucci R, Fleming JV, Xavier R, Wang TC. L-histidine decarboxylase decreases its own transcription through downregulation of ERK activity. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1081-91.
Monczor F, Fernandez N. Current knowledge and perspectives on histamine H1 and H2 receptor pharmacology: functional selectivity, receptor crosstalk, and repositioning of classic histaminergic ligands. Mol Pharmacol. 2016;90:640–8.
Ishola AA, Joshi T, Abdulai SI, Tijjani H, Pundir H, Chandra S. Molecular basis for the repurposing of histamine H2-receptor antagonist to treat COVID-19. J Biomol Struct Dyn. 2022;40:5785–802.
Esbenshade TA, Browman KE, Bitner RS, Strakhova M, Cowart MD, Brioni JD. The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br J Pharmacol. 2008;154:1166–81.
Beermann S, Vauth M, Hein R, Seifert R, Neumann D. Distinct signalling pathways of murine histamine H1- and H4-receptors expressed at comparable levels in HEK293 cells. PLoS ONE. 2014;9: e107481.
Jemima EA, Prema A, Thangam EB. Functional characterization of histamine H4 receptor on human mast cells. Mol Immunol. 2014;62:19–28.
Xiang J, Yang Z, Zhou Q. Lidocaine relieves murine allergic rhinitis by regulating the NF-κB and p38 MAPK pathways. Exp Ther Med. 2022;23:193.
Nardelli LM, Rzezinski A, Silva JD, Maron-Gutierrez T, Ornellas DS, Henriques I, et al. Effects of acute hypercapnia with and without acidosis on lung inflammation and apoptosis in experimental acute lung injury. Respir Physiol Neurobiol. 2015;205:1–6.
Blain GM, Smith CA, Henderson KS, Dempsey JA. Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO(2). J Physiol. 2010;588:2455–71.
Shimokawa N, Dikic I, Sugama S, Koibuchi N. Molecular responses to acidosis of central chemosensitive neurons in brain. Cell Signal. 2005;17:799–808.
Gourine AV, Llaudet E, Dale N, Spyer KM. ATP is a mediator of chemosensory transduction in the central nervous system. Nature. 2005;436:108–11.
Laouafa S, Perrin-Terrin A-S, Jeton F, Elliot-Portal E, Tam R, Bodineau L, et al. Pharmacological, but not genetic, alteration of neural Epo modifies the CO(2)/H(+) central chemosensitivity in postnatal mice. Respir Physiol Neurobiol. 2017;242:73–9.
Iturri P, Joseph V, Rodrigo G, Bairam A, Soliz J. Inhibition of protein kinases AKT and ERK1/2 reduce the carotid body chemoreceptor response to hypoxia in adult rats. Adv Exp Med Biol. 2015;860:269–77.
Tao T, Zhao M, Yang W, Bo Y, Li W. Neuroprotective effects of therapeutic hypercapnia on spatial memory and sensorimotor impairment via anti-apoptotic mechanisms after focal cerebral ischemia/reperfusion. Neurosci Lett. 2014;573:1–6.
Taugher RJ, Wunsch AM, Wang GZ, Chan AC, Dlouhy BJ, Wemmie JA. Post-acquisition CO(2) inhalation enhances fear memory and depends on ASIC1A. Front Behav Neurosci. 2021;15: 767426.
Ziemann AE, Allen JE, Dahdaleh NS, Drebot II, Coryell MW, Wunsch AM, et al. The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell. 2009;139:1012–21.
Du J, Reznikov LR, Price MP, Zha X, Lu Y, Moninger TO, et al. Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proceedings of the National Academy of Sciences U S A. 2014;111:8961–6.
Yang M-T, Chien W-L, Lu D-H, Liou H-C, Fu W-M. Acetazolamide impairs fear memory consolidation in rodents. Neuropharmacology. 2013;67:412–8.
Blandina P, Provensi G, Passsani MB, Capasso C, Supuran CT. Carbonic anhydrase modulation of emotional memory. implications for the treatment of cognitive disorders. J Enzyme Inhib Med Chem. 2020;35:1206–14.
Schmidt SD, Costa A, Rani B, Godfried Nachtigall E, Passani MB, Carta F, et al. The role of carbonic anhydrases in extinction of contextual fear memory. Proc Natl Acad Sci U S A. 2020;117:16000–8.
Canto de Souza L, Provensi G, Vullo D, Carta F, Scozzafava A, Costa A, et al. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology. 2017;118:148–56.
Angeli A, Vaiano F, Mari F, Bertol E, Supuran CT. Psychoactive substances belonging to the amphetamine class potently activate brain carbonic anhydrase isoforms VA, VB, VII, and XII. J Enzyme Inhib Med Chem. 2017;32:1253–9.
Albert-Gascó H, Ros-Bernal F, Castillo-Gómez E, Olucha-Bordonau FE. MAP/ERK signaling in developing cognitive and emotional function and its effect on pathological and neurodegenerative processes. Int J Mol Sci. 2020;21:4471.
Medina JH, Viola H. ERK1/2: a key cellular component for the formation, retrieval, reconsolidation and persistence of memory. Front Mol Neurosci. 2018;11:361.
Kelly A, Laroche S, Davis S. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J Neurosci. 2003;23:5354–60.
Kim SW, Ha NY, Kim KI, Park JK, Lee YH. Memory-improving effect of formulation-MSS by activation of hippocampal MAPK/ERK signaling pathway in rats. BMB Rep Korea (South). 2008;41:242–7.
Zhi W-H, Zeng Y-Y, Lu Z-H, Qu W-J, Chen W-X, Chen L, et al. Simvastatin exerts antiamnesic effect in Aβ25-35 -injected mice. CNS Neurosci Ther. 2014;20:218–26.
Bharne AP, Borkar CD, Bodakuntla S, Lahiri M, Subhedar NK, Kokare DM. Pro-cognitive action of CART is mediated via ERK in the hippocampus. Hippocampus. 2016;26:1313–27.
Salort G, Álvaro-Bartolomé M, García-Sevilla JA. Pentobarbital and other anesthetic agents induce opposite regulations of MAP kinases p-MEK and p-ERK, and upregulate p-FADD/FADD neuroplastic index in brain during hypnotic states in mice. Neurochem Int. 2019;122:59–72.
Zhou J, Yang W-S, Suo D-Q, Li Y, Peng L, Xu L-X, et al. Moringa oleifera seed extract alleviates scopolamine-induced learning and memory impairment in mice. Front Pharmacol. 2018;9:389.
Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, et al. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–76.
Sowho M, Amatoury J, Kirkness JP, Patil SP. Sleep and respiratory physiology in adults. Clin Chest Med. 2014;35:469–81.
Newton K, Malik V, Lee-Chiong T. Sleep and breathing. Clin Chest Med. 2014;35:451–6.
Dempsey JA, Smith CA, Przybylowski T, Chenuel B, Xie A, Nakayama H, et al. The ventilatory responsiveness to CO(2) below eupnoea as a determinant of ventilatory stability in sleep. J Physiol. 2004;560:1–11.
Malhotra A, Owens RL. What is central sleep apnea? Respir Care. 2010;55:1168–78.
Malik V, Smith D, Lee-Chiong T Jr. Respiratory physiology during sleep. Sleep Med Clin. 2012;7:497–505.
Xia Z, Storm D. Role of circadian rhythm and REM sleep for memory consolidation. Neurosci Res. 2017;118:13–20.
Wang X-L, Yuan K, Zhang W, Li S-X, Gao GF, Lu L. Regulation of circadian genes by the MAPK pathway: implications for rapid antidepressant action. Neurosci Bull. 2020;36:66–76.
Ballester Roig M, Leduc T, Areal C, Mongrain V. Cellular effects of rhynchophylline and relevance to sleep regulation. Clocks Sleep. 2021;3:312–41.
Szollosi I, Jones M, Morrell MJ, Helfet K, Coats AJS, Simonds AK. Effect of CO<sub>2</sub> inhalation on central sleep apnea and arousals from sleep. Respiration. 2004;71:493–8.
Lin C-C, Wang Y-P, Chiu C-H, Sun Y-K, Lin M-W, Tzeng I-S. Molecular signalling involved in upper airway remodelling is enhanced in patients with obstructive sleep apnoea. J Laryngol Otol. 2022;136:1096–104.
Huang Q, Wang P, Liu H, Li M, Yue Y, Xu P. Inhibition of ERK1/2 regulates cognitive function by decreasing expression levels of PSD-95 in the hippocampus of CIH rats. Eur J Neurosci. 2022;55:1471–82.
Feinstein JS, Gould D, Khalsa SS. Amygdala-driven apnea and the chemoreceptive origin of anxiety. Biol Psychol. 2022;170: 108305.
de Carvalho CR, Lopes MW, Constantino LC, Hoeller AA, de Melo HM, Guarnieri R, et al. The ERK phosphorylation levels in the amygdala predict anxiety symptoms in humans and MEK/ERK inhibition dissociates innate and learned defensive behaviors in rats. Mol Psychiatry. 2021;26:7257–69.
Tregub P, Malinovskaya N, Hilazheva E, Morgun A, Kulikov V. Permissive hypercapnia and hypercapnic hypoxia inhibit signaling pathways of neuronal apoptosis in ischemic/hypoxic rats. Mol Biol Rep. 2023;50:2317–33.
Nadeev AD, Kritskaya KA, Fedotova EI, Berezhnov AV. «One Small Step for Mouse»: High CO2 inhalation as a new therapeutic strategy for Parkinson’s Disease. Biomedicines. 2022;10:2832.
Tao T, Liu Y, Zhang J, Xu Y, Li W, Zhao M. Therapeutic hypercapnia improves functional recovery and attenuates injury via antiapoptotic mechanisms in a rat focal cerebral ischemia/reperfusion model. Brain Res. 2013;1533:52–62.
Sahu R, Upadhayay S, Mehan S. Inhibition of extracellular regulated kinase (ERK)-1/2 signaling pathway in the prevention of ALS: target inhibitors and influences on neurological dysfunctions. Eur J Cell Biol. 2021;100: 151179.
Miningou N, Blackwell KT. The road to ERK activation: Do neurons take alternate routes? Cell Signal. 2020;68: 109541.
Asih PR, Prikas E, Stefanoska K, Tan ARP, Ahel HI, Ittner A. Functions of p38 MAP Kinases in the Central Nervous System. Front Mol Neurosci. 2020;13: 570586.
Tolstun DA, Knyazer A, Tushynska TV, Dubiley TA, Bezrukov VV, Fraifeld VE, et al. Metabolic remodelling of mice by hypoxic-hypercapnic environment: imitating the naked mole-rat. Biogerontology. 2020;21:143–53.
Muradian K. “Pull and push back” concepts of longevity and life span extension. Biogerontology. 2013;14:687–91.
Schuller HM. Carbon dioxide potentiates the mitogenic effects of nicotine and its carcinogenic derivative, NNK, in normal and neoplastic neuroendocrine lung cells via stimulation of autocrine and protein kinase C-dependent mitogenic pathways. Neurotoxicology. 1994;15:877–86.
Tsuji T, Aoshiba K, Itoh M, Nakamura H, Yamaguchi K. Hypercapnia accelerates wound healing in endothelial cell monolayers exposed to hypoxia. Open Respir Med J. 2013;7:6–12.
Held AA, Emerson R, Fuller MS, Gleason FH. Blastocladia and aqualinderella: fermentative water molds with high carbon dioxide optima. Science. 1969;165:706–8.
Casalino-Matsuda SM, Nair A, Beitel GJ, Gates KL, Sporn PHS. Hypercapnia inhibits autophagy and bacterial killing in human macrophages by increasing expression of Bcl-2 and Bcl-xL. J Immunol. 2015;194:5388–96.
Barker H, Aaltonen M, Pan P, Vähätupa M, Kaipiainen P, May U, et al. Role of carbonic anhydrases in skin wound healing. Exp Mol Med. 2017;49: e334.
Genah S, Angeli A, Supuran CT, Morbidelli L. Effect of Carbonic Anhydrase IX inhibitors on human endothelial cell survival. Pharmacol Res. 2020;159: 104964.
Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017;284:4177–95.
O’Reilly LA, Kruse EA, Puthalakath H, Kelly PN, Kaufmann T, Huang DCS, et al. MEK/ERK-mediated phosphorylation of bim is required to ensure survival of T and B lymphocytes during mitogenic stimulation. J Immunol. 2009;183:261–9.
Yue J, López JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci. 2020;21:2346.
Xu F, Uh J, Brier MR, Hart J, Yezhuvath US, Gu H, et al. The influence of carbon dioxide on brain activity and metabolism in conscious humans. J Cereb Blood Flow Metab. 2011;31:58–67.
Bandopadhyay S, Prasad P, Ray U, Das Ghosh D, Roy SS. SIRT6 promotes mitochondrial fission and subsequent cellular invasion in ovarian cancer. FEBS Open Bio. 2022;12:1657–76.
Simula L, Corrado M, Accordi B, di Rita A, Nazio F, Antonucci Y, et al. JNK1 and ERK1/2 modulate lymphocyte homeostasis via BIM and DRP1 upon AICD induction. Cell Death Differ. 2020;27:2749–67.
Prieto J, León M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, et al. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. 2016;7:11124.
Gao M, Wang J, Lu N, Fang F, Liu J, Wong C-W. Mitogen-activated protein kinase kinases promote mitochondrial biogenesis in part through inducing peroxisome proliferator-activated receptor γ coactivator-1β expression. Biochim Biophys Acta. 2011;1813:1239–44.
Wu H, Zhao J, Chen M, Wang H, Yao Q, Fan J, et al. The anti-aging effect of erythropoietin via the ERK/Nrf2-ARE pathway in aging rats. J Mol Neurosci. 2017;61:449–58.
Lucero M, Suarez AE, Chambers JW. Phosphoregulation on mitochondria: integration of cell and organelle responses. CNS Neurosci Ther. 2019;25:837–58.
Blombach B, Takors R. CO2 - intrinsic product, essential substrate, and regulatory trigger of microbial and mammalian production processes. Front Bioeng Biotechnol. 2015;3:108.
Boron WF, Michenkova M, Taki S, Blosser MC, Hwang HJ, Kowatz T, et al. Carbon dioxide transport across membranes. Interface Focus. 2021;11:20200090.
Occhipinti R, Boron WF. Role of carbonic anhydrases and inhibitors in acid-base physiology: insights from mathematical modeling. Int J Mol Sci. 2019;20:3841.
Asghar MS, Haider Kazmi SJ, Khan NA, Akram M, Hassan M, Rasheed U, et al. Poor prognostic biochemical markers predicting fatalities caused by COVID-19: a retrospective observational study from a developing country. Cureus. 2020;12: e9575.
Sinha P, Calfee CS, Cherian S, Brealey D, Cutler S, King C, et al. Prevalence of phenotypes of acute respiratory distress syndrome in critically ill patients with COVID-19: a prospective observational study. Lancet Respir Med. 2020;8:1209–18.
Bezuidenhout MC, Wiese OJ, Moodley D, Maasdorp E, Davids MR, Koegelenberg CF, et al. Correlating arterial blood gas, acid-base and blood pressure abnormalities with outcomes in COVID-19 intensive care patients. Ann Clin Biochem. 2021;58:95–101.
Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368: m1091.
Ouyang S-M, Zhu H-Q, Xie Y-N, Zou Z-S, Zuo H-M, Rao Y-W, et al. Temporal changes in laboratory markers of survivors and non-survivors of adult inpatients with COVID-19. BMC Infect Dis. 2020;20:952.
Dheir H, Karacan A, Sipahi S, Yaylaci S, Tocoglu A, Demirci T, et al. Correlation between venous blood gas indices and radiological involvements of COVID-19 patients at first admission to emergency department. Rev Assoc Med Bras (1992). Brazil; 2021;67Suppl 1:51–6.
Kafan S, Tadbir Vajargah K, Sheikhvatan M, Tabrizi G, Salimzadeh A, Montazeri M, et al. Predicting risk score for mechanical ventilation in hospitalized adult patients suffering from COVID-19. Anesth Pain Med. 2021;11: e112424.
Yitao Z, Mu C, Ling Z, Shiyao C, Jiaojie X, Zhichong C, et al. Predictors of clinical deterioration in non-severe patients with COVID-19: a retrospective cohort study. Curr Med Res Opin. 2021;37:385–91.
O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–65.
Vassiliou AG, Jahaj E, Ilias I, Markaki V, Malachias S, Vrettou C, et al. Lactate kinetics reflect organ dysfunction and are associated with adverse outcomes in intensive care unit patients with COVID-19 pneumonia: preliminary results from a GREEK single-centre study. Metabolites. 2020;10:386.
Price-Haywood EG, Burton J, Fort D, Seoane L. Hospitalization and mortality among black patients and white patients with Covid-19. N Engl J Med. 2020;382:2534–43.
Sinha P, Furfaro D, Cummings MJ, Abrams D, Delucchi K, Maddali MV, et al. Latent class analysis reveals COVID-19-related acute respiratory distress syndrome subgroups with differential responses to corticosteroids. Am J Respir Crit Care Med. 2021;204:1274–85.
Ehresman J, Cottrill E, Caplan JM, McDougall CG, Theodore N, Nyquist PA. Neuroprotective role of acidosis in ischemia: review of the preclinical evidence. Mol Neurobiol. 2021;58:6684–96.
Fan Y-Y, Shen Z, He P, Jiang L, Hou W, Shen Y, et al. A novel neuroprotective strategy for ischemic stroke: transient mild acidosis treatment by CO2 inhalation at reperfusion. J Cereb Blood Flow Metab. 2014;34:275–83.
Inserte J, Ruiz-Meana M, Rodríguez-Sinovas A, Barba I, Garcia-Dorado D. Contribution of delayed intracellular pH recovery to ischemic postconditioning protection. Antioxid Redox Signal. 2011;14:923–39.
Kaku DA, Giffard RG, Choi DW. Neuroprotective effects of glutamate antagonists and extracellular acidity. Science. 1993;260:1516–8.
Baxter AL, Schwartz KR, Johnson RW, Kuchinski A-M, Swartout KM, Srinivasa Rao ASR, et al. Rapid initiation of nasal saline irrigation to reduce severity in high-risk COVID+ outpatients. Ear Nose Throat J. 2022;014556132211237.
Mody K. Effect of 8.4% soda-bicarbonate steam inhalation on the course of disease in mild to moderate cases of Covid-19. Acta Scientific Orthopaedics. 2021;4:35–43.
Wardeh A, Conklin J, Ko M. Case reports of observed significant improvement in patients with ARDS due to COVID-19 and maximum ventilatory support after inhalation of sodium bicarbonate. J Clin Intensive Care Med. 2020;5:15–9.
Shigemura M, Lecuona E, Angulo M, Homma T, Rodríguez DA, Gonzalez-Gonzalez FJ, et al. Hypercapnia increases airway smooth muscle contractility via caspase-7-mediated miR-133a-RhoA signaling. Sci Transl Med. 2018;10:eaat1662.
Viswanathan R, Lodi ST, Subramanian S, Radha TG. Pulmonary vascular response to ventilation hypercapnia in man. Respiration. 1976;33:165–78.
Cheng Y, Sun F, Wang L, Gao M, Xie Y, Sun Y, et al. Virus-induced p38 MAPK activation facilitates viral infection. Theranostics. 2020;10:12223–40.
Wang L, Xia Z, Tang W, Sun Y, Wu Y, Kwok HF, et al. p38 activation and viral infection. Expert Rev Mol Med. 2022;24: e4.
Jiao A, Sun C, Wang X, Lei L, Liu H, Li W, et al. DExD/H-box helicase 9 intrinsically controls CD8(+) T cell-mediated antiviral response through noncanonical mechanisms. Sci Adv. 2022;8:eabk2691.
Wehbe Z, Hammoud S, Soudani N, Zaraket H, El-Yazbi A, Eid AH. Molecular insights into SARS COV-2 interaction with cardiovascular disease: role of RAAS and MAPK signaling. Front Pharmacol. 2020;11:836.
Kumar R, Khandelwal N, Thachamvally R, Tripathi BN, Barua S, Kashyap SK, et al. Role of MAPK/MNK1 signaling in virus replication. Virus Res. 2018;253:48–61.
Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR, et al. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol. 2001;3:301–5.
Li CC, Wang XJ, Wang HCR. Repurposing host-based therapeutics to control coronavirus and influenza virus. Drug Discov Today. 2019;24:726–36.
Cai Y, Liu Y, Zhang X. Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J Virol. 2007;81:446–56.
Börgeling Y, Schmolke M, Viemann D, Nordhoff C, Roth J, Ludwig S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J Biol Chem. 2014;289:13–27.
Meng Y, Yu C-H, Li W, Li T, Luo W, Huang S, et al. Angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-κB pathway. Am J Respir Cell Mol Biol. 2014;50:723–36.
Hung Y-H, Hsieh W-Y, Hsieh J-S, Liu F-C, Tsai C-H, Lu L-C, et al. Alternative roles of STAT3 and MAPK signaling pathways in the MMPs activation and progression of lung injury induced by cigarette smoke exposure in ACE2 knockout mice. Int J Biol Sci. 2016;12:454–65.
Lin C-I, Tsai C-H, Sun Y-L, Hsieh W-Y, Lin Y-C, Chen C-Y, et al. Instillation of particulate matter 2.5 induced acute lung injury and attenuated the injury recovery in ACE2 knockout mice. Int J Biol Sci. 2018;14:253–65.
Chen I-Y, Chang SC, Wu H-Y, Yu T-C, Wei W-C, Lin S, et al. Upregulation of the chemokine (C-C motif) ligand 2 via a severe acute respiratory syndrome coronavirus spike-ACE2 signaling pathway. J Virol. 2010;84:7703–12.
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9.
Jia H. Pulmonary angiotensin-converting Enzyme 2 (ACE2) and inflammatory lung disease. Shock. 2016;46:239–48.
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3.
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–6.
Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. 2005;79:14614–21.
Bi Z, Hong W, Que H, He C, Ren W, Yang J, et al. Inactivated SARS-CoV-2 induces acute respiratory distress syndrome in human ACE2-transgenic mice. Signal Transduct Target Ther. 2021;6:439.
Colunga Biancatelli RML, Solopov PA, Sharlow ER, Lazo JS, Marik PE, Catravas JD. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Κ18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2021;321:L477–84.
Barreda D, Santiago C, Rodríguez JR, Rodríguez JF, Casasnovas JM, Mérida I, et al. SARS-CoV-2 spike protein and its receptor binding domain promote a proinflammatory activation profile on human dendritic cells. Cells. 2021;10:3279.
Shirato K, Takanari J, Kizaki T. Standardized extract of asparagus officinalis stem attenuates SARS-CoV-2 Spike Protein-Induced IL-6 and IL-1β production by suppressing p44/42 MAPK and Akt phosphorylation in murine primary macrophages. Molecules. 2021;26:6189.
Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, et al. The Global Phosphorylation landscape of SARS-CoV-2 infection. Cell. 2020;182:685-712.e19.
Olajide OA, Iwuanyanwu VU, Lepiarz-Raba I, Al-Hindawi AA. Induction of exaggerated cytokine production in human peripheral blood mononuclear cells by a recombinant SARS-CoV-2 Spike Glycoprotein S1 and its inhibition by dexamethasone. Inflammation. 2021;44:1865–77.
Del Re A, Corpetti C, Pesce M, Seguella L, Steardo L, Palenca I, et al. Ultramicronized palmitoylethanolamide inhibits NLRP3 inflammasome expression and pro-inflammatory response activated by SARS-CoV-2 spike protein in cultured murine alveolar macrophages. Metabolites. 2021;11:592.
Olajide OA, Iwuanyanwu VU, Adegbola OD, Al-Hindawi AA. SARS-CoV-2 Spike Glycoprotein S1 induces neuroinflammation in BV-2 microglia. Mol Neurobiol. 2022;59:445–58.
Geng J, Chen L, Yuan Y, Wang K, Wang Y, Qin C, et al. CD147 antibody specifically and effectively inhibits infection and cytokine storm of SARS-CoV-2 and its variants delta, alpha, beta, and gamma. Signal Transduct Target Ther. 2021;6:347.
Avolio E, Carrabba M, Milligan R, Kavanagh Williamson M, Beltrami AP, Gupta K, et al. The SARS-CoV-2 Spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: a potential non-infective mechanism of COVID-19 microvascular disease. Clin Sci (Lond). 2021;135:2667–89.
Qian Y, Lei T, Patel PS, Lee CH, Monaghan-Nichols P, Xin H-B, et al. Direct activation of endothelial cells by SARS-CoV-2 nucleocapsid protein is blocked by simvastatin. J Virol. 2021;95: e0139621.
Klann K, Bojkova D, Tascher G, Ciesek S, Münch C, Cinatl J. Growth factor receptor signaling inhibition prevents SARS-CoV-2 replication. Mol Cell. 2020;80:164-174.e4.
Ahsan N, Rao RSP, Wilson RS, Punyamurtula U, Salvato F, Petersen M, et al. Mass spectrometry-based proteomic platforms for better understanding of SARS-CoV-2 induced pathogenesis and potential diagnostic approaches. Proteomics. 2021;21: e2000279.
Weckbach LT, Schweizer L, Kraechan A, Bieber S, Ishikawa-Ankerhold H, Hausleiter J, et al. Association of complement and MAPK Activation With SARS-CoV-2-associated myocardial inflammation. JAMA Cardiol. 2022;7:286–97.
Iwanski J, Kazmouz SG, Li S, Stansfield B, Salem TT, Perez-Miller S, et al. Antihypertensive drug treatment and susceptibility to SARS-CoV-2 infection in human PSC-derived cardiomyocytes and primary endothelial cells. Stem Cell Reports. 2021;16:2459–72.
An S, Li Y, Lin Y, Chu J, Su J, Chen Q, et al. Genome-wide profiling reveals alternative polyadenylation of innate immune-related mRNA in patients with COVID-19. Front Immunol. 2021;12: 756288.
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med. Massachusetts Medical Society; 2020;383:120–8.
Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7:e575–82.
Sardu C, Gambardella J, Morelli MB, Wang X, Marfella R, Santulli G. Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? a comprehensive evaluation of clinical and basic evidence. J Clin Med. 2020;9:1417.
Pons S, Fodil S, Azoulay E, Zafrani L. The vascular endothelium: the cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit Care. 2020;24:353.
Booz GW, Altara R, Eid AH, Wehbe Z, Fares S, Zaraket H, et al. Macrophage responses associated with COVID-19: A pharmacological perspective. Eur J Pharmacol. 2020;887: 173547.
Yang L, Xie X, Tu Z, Fu J, Xu D, Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct Target Ther. 2021;6:255.
Janovec V, Aouar B, Font-Haro A, Hofman T, Trejbalova K, Weber J, et al. The MEK1/2-ERK pathway inhibits type I IFN production in plasmacytoid dendritic cells. Front Immunol. 2018;9:364.
Adu-Amankwaah J, Adzika GK, Adekunle AO, Ndzie Noah ML, Mprah R, Bushi A, et al. ADAM17, A key player of cardiac inflammation and fibrosis in heart failure development during chronic catecholamine stress. Front Cell Dev Biol. 2021;9: 732952.
Roy RK, Sharma U, Wasson MK, Jain A, Hassan MI, Prakash H. Macrophage activation syndrome and COVID 19: impact of MAPK driven immune-epigenetic programming by SARS-Cov-2. Front Immunol. 2021;12: 763313.
Esposito G, Perrino C, Schiattarella GG, Belardo L, di Pietro E, Franzone A, et al. Induction of mitogen-activated protein kinases is proportional to the amount of pressure overload. Hypertension. 2010;55:137–43.
Sampaio WO, Henrique de Castro C, Santos RAS, Schiffrin EL, Touyz RM. Angiotensin-(1–7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension. 2007;50:1093–8.
Hoffmann BR, Stodola TJ, Wagner JR, Didier DN, Exner EC, Lombard JH, et al. Mechanisms of Mas1 receptor-mediated signaling in the vascular endothelium. Arterioscler Thromb Vasc Biol. 2017;37:433–45.
Sousa-Lopes A, de Freitas RA, Carneiro FS, Nunes KP, Allahdadi KJ, Webb RC, et al. Angiotensin (1–7) Inhibits Ang II-mediated ERK1/2 activation by stimulating MKP-1 activation in vascular smooth muscle cells. Int J Mol Cell Med. 2020;9:50–61.
Beltrán AE, Briones AM, García-Redondo AB, Rodríguez C, Miguel M, Alvarez Y, et al. p38 MAPK contributes to angiotensin II-induced COX-2 expression in aortic fibroblasts from normotensive and hypertensive rats. J Hypertens. 2009;27:142–54.
Ju H, Behm DJ, Nerurkar S, Eybye ME, Haimbach RE, Olzinski AR, et al. p38 MAPK inhibitors ameliorate target organ damage in hypertension: Part 1. p38 MAPK-dependent endothelial dysfunction and hypertension. J Pharmacol Exp Ther. 2003;307:932–8.
Lu Y, Sun X, Peng L, Jiang W, Li W, Yuan H, et al. Angiotensin II-Induced vascular remodeling and hypertension involves cathepsin L/V- MEK/ERK mediated mechanism. Int J Cardiol. 2020;298:98–106.
Sartori M, Ceolotto G, Papparella I, Baritono E, Ciccariello L, Calò L, et al. Effects of angiotensin II and insulin on ERK1/2 activation in fibroblasts from hypertensive patients. Am J Hypertens. 2004;17:604–10.
Zhang L, Zhang Y, Wu Y, Yu J, Zhang Y, Zeng F, et al. Role of the balance of akt and MAPK pathways in the exercise-regulated phenotype switching in spontaneously hypertensive rats. Int J Mol Sci. 2019;20:5690.
Ricard N, Scott RP, Booth CJ, Velazquez H, Cilfone NA, Baylon JL, et al. Endothelial ERK1/2 signaling maintains integrity of the quiescent endothelium. J Exp Med. 2019;216:1874–90.
Novack P, Shenkin HA, Bortin L, Goluboff B, Soffe AM. The effects of carbon dioxide inhalation upon the cerebral blood flow and cerebral oxygen consumption in vascular disease. J Clin Invest. 1953;32:696–702.
Németh B, Kiss I, Ajtay B, Péter I, Kreska Z, Cziráki A, et al. Transcutaneous carbon dioxide treatment is capable of reducing peripheral vascular resistance in hypertensive patients. In Vivo. 2018;32:1555–9.
Santamarina MG, Beddings I, Lomakin FM, Boisier Riscal D, Gutiérrez Claveria M, Vidal Marambio J, et al. Sildenafil for treating patients with COVID-19 and perfusion mismatch: a pilot randomized trial. Crit Care. 2022;26:1.
Kvietys PR, Fakhoury HMA, Kadan S, Yaqinuddin A, Al-Mutairy E, Al-Kattan K. COVID-19: lung-centric immunothrombosis. Front Cell Infect Microbiol. 2021;11: 679878.
Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. 2020;217(12):e20201129.
Caillon A, Trimaille A, Favre J, Jesel L, Morel O, Kauffenstein G. Role of neutrophils, platelets, and extracellular vesicles and their interactions in COVID-19-associated thrombopathy. J Thromb Haemost. 2022;20:17–31.
Kotani M, Kotani T, Li Z, Silbajoris R, Piantadosi CA, Huang Y-CT. Reduced inspiratory flow attenuates IL-8 release and MAPK activation of lung overstretc. Eur Respir J. 2004;24:238–46.
Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13:120.
Govender N, Khaliq OP, Moodley J, Naicker T. Insulin resistance in COVID-19 and diabetes. Prim Care Diabetes. 2021;15:629–34.
Codo AC, Davanzo GG, Monteiro L de B, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020;32:437–446.e5.
Formoso G, Chen H, Kim J, Montagnani M, Consoli A, Quon MJ. Dehydroepiandrosterone mimics acute actions of insulin to stimulate production of both nitric oxide and endothelin 1 via distinct phosphatidylinositol 3-kinase- and mitogen-activated protein kinase-dependent pathways in vascular endothelium. Mol Endocrinol. 2006;20:1153–63.
Wolff D, Nee S, Hickey NS, Marschollek M. Risk factors for Covid-19 severity and fatality: a structured literature review. Infection. 2021;49:15–28.
Lim S, Bae JH, Kwon H-S, Nauck MA. COVID-19 and diabetes mellitus: from pathophysiology to clinical management. Nat Rev Endocrinol. 2021;17:11–30.
Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18:1094–9.
Vernia S, Morel C, Madara JC, Cavanagh-Kyros J, Barrett T, Chase K, et al. Excitatory transmission onto AgRP neurons is regulated by cJun NH2-terminal kinase 3 in response to metabolic stress. Elife. 2016;5: e10031.
Prusty D, Park B-H, Davis KE, Farmer SR. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARgamma ) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J Biol Chem. 2002;277:46226–32.
Macura M, Ban Frangez H, Cankar K, Finžgar M, Frangez I. The effect of transcutaneous application of gaseous CO(2) on diabetic chronic wound healing-A double-blind randomized clinical trial. Int Wound J. 2020;17:1607–14.
Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, Bruttocao A, et al. NETosis delays diabetic wound healing in mice and humans. Diabetes. 2016;65:1061–71.
Salinet ASM, Minhas JS, Panerai RB, Bor-Seng-Shu E, Robinson TG. Do acute stroke patients develop hypocapnia? a systematic review and meta-analysis. J Neurol Sci. 2019;402:30–9.
Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, et al. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–9.
Athiraman U, Sultan-Qurraie A, Nair B, Tirschwell DL, Ghodke B, Havenon AD, et al. Endovascular treatment of acute ischemic stroke under general anesthesia: predictors of good outcome. J Neurosurg Anesthesiol. 2018;30:223–30.
Yang WC, Wang Q, Chi LT, Wang YZ, Cao HL, Li WZ. Therapeutic hypercapnia reduces blood-brain barrier damage possibly via protein kinase Cϵ in rats with lateral fluid percussion injury. J Neuroinflammation. 2019;16(1):36.
Siemkowicz E, Hansen AJ. Brain extracellular ion composition and EEG activity following 10 minutes ischemia in normo- and hyperglycemic rats. Stroke. 1981;12:236–40.
Devaux JBL, Hedges CP, Birch N, Herbert N, Renshaw GMC, Hickey AJR. Acidosis maintains the function of brain mitochondria in hypoxia-tolerant triplefin fish: a strategy to survive acute hypoxic exposure? Front Physiol. 2018;9:1941.
Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, et al. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun. 2014;5:3550.
Ippolito JE, Brandenburg MW, Ge X, Crowley JR, Kirmess KM, Som A, et al. Extracellular pH modulates neuroendocrine prostate cancer cell metabolism and susceptibility to the mitochondrial inhibitor niclosamide. PLoS ONE. 2016;11: e0159675.
Tsonas AM, Botta M, Horn J, Morales-Quinteros L, Artigas A, Schultz MJ, et al. Clinical characteristics, physiological features, and outcomes associated with hypercapnia in patients with acute hypoxemic respiratory failure due to COVID-19–-insights from the PRoVENT-COVID study. J Crit Care. 2022;69: 154022.
Morales-Quinteros L, Camprubí-Rimblas M, Bringué J, Bos LD, Schultz MJ, Artigas A. The role of hypercapnia in acute respiratory failure. Intensive Care Med Exp. 2019;7:39.
Tiruvoipati R, Pilcher D, Buscher H, Botha J, Bailey M. Effects of hypercapnia and hypercapnic acidosis on hospital mortality in mechanically ventilated patients. Crit Care Med. 2017;45:e649–56.
Nin N, Muriel A, Peñuelas O, Brochard L, Lorente JA, Ferguson ND, et al. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med. 2017;43:200–8.
Gendreau S, Geri G, Pham T, Vieillard-Baron A, Mekontso DA. The role of acute hypercapnia on mortality and short-term physiology in patients mechanically ventilated for ARDS: a systematic review and meta-analysis. Intensive Care Med. 2022;48:517–34.
Helmerhorst HJF, Schouten LRA, Wagenaar GTM, Juffermans NP, Roelofs JJTH, Schultz MJ, et al. Hyperoxia provokes a time- and dose-dependent inflammatory response in mechanically ventilated mice, irrespective of tidal volumes. Intensive Care Med Exp. 2017;5:27.
Brueckl C, Kaestle S, Kerem A, Habazettl H, Krombach F, Kuppe H, et al. Hyperoxia-induced reactive oxygen species formation in pulmonary capillary endothelial cells in situ. Am J Respir Cell Mol Biol. 2006;34:453–63.
Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI. Hypoxia-induced endocytosis of Na, K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest. 2003;111:1057–64.
Grant GJ, Mimche PN, Paine R 3rd, Liou TG, Qian W-J, Helms MN. Enhanced epithelial sodium channel activity in neonatal Scnn1b mouse lung attenuates high oxygen-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2021;321:L29-41.
Tung Y-T, Wei C-H, Yen C-C, Lee P-Y, Ware LB, Huang H-E, et al. Aspirin attenuates hyperoxia-induced acute respiratory distress syndrome (ARDS) by suppressing pulmonary inflammation via the NF-κB signaling pathway. Front Pharmacol. 2021;12: 793107.
Huppert LA, Matthay MA. Alveolar fluid clearance in pathologically relevant conditions: in vitro and in vivo models of acute respiratory distress syndrome. Front Immunol. 2017;8:371.
Fischer H, Widdicombe JH. Mechanisms of acid and base secretion by the airway epithelium. J Membr Biol. 2006;211:139–50.
Rehman T, Thornell IM, Pezzulo AA, Thurman AL, Romano Ibarra GS, Karp PH, et al. TNFα and IL-17 alkalinize airway surface liquid through CFTR and pendrin. Am J Physiol Cell Physiol. 2020;319:C331–44.
Thornell IM, Rehman T, Pezzulo AA, Welsh MJ. Paracellular bicarbonate flux across human cystic fibrosis airway epithelia tempers changes in airway surface liquid pH. J Physiol. 2020;598:4307–20.
Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in Hospitalized Patients with Covid-19. N Engl J Med. 2021;384:693–704.
Li L, Goleva E, Hall CF, Ou L-S, Leung DYM. Superantigen-induced corticosteroid resistance of human T cells occurs through activation of the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK-ERK) pathway. J Allergy Clin Immunol. 2004;114:1059–69.
Tsitoura DC, Rothman PB. Enhancement of MEK/ERK signaling promotes glucocorticoid resistance in CD4+ T cells. J Clin Investig. 2004;113:619–27.
Goel S, Saheb Sharif-Askari F, Saheb Sharif Askari N, Madkhana B, Alwaa AM, Mahboub B, et al. SARS-CoV-2 Switches “on” MAPK and NFκB Signaling via the Reduction of Nuclear DUSP1 and DUSP5 Expression. Front Pharmacol. 2021;12:631879.
Saheb Sharif-Askari F, Saheb Sharif-Askari N, Goel S, Hafezi S, Assiri R, Al-Muhsen S, et al. SARS-CoV-2 attenuates corticosteroid sensitivity by suppressing DUSP1 expression and activating p38 MAPK pathway. Eur J Pharmacol. 2021;908: 174374.
Theoharides TC. COVID-19, pulmonary mast cells, cytokine storms, and beneficial actions of luteolin. BioFactors. 2020;46:306–8.
Qu C, Fuhler G, Pan Y. Could histamine H1 receptor antagonists be used for treating COVID-19? Int J Mol Sci. 2021;22:5672.
Janowitz T, Gablenz E, Pattinson D, Wang TC, Conigliaro J, Tracey K, et al. Famotidine use and quantitative symptom tracking for COVID-19 in non-hospitalised patients: a case series. Gut. 2020;69:1592–7.
Freedberg DE, Conigliaro J, Wang TC, Tracey KJ, Callahan MV, Abrams JA. Famotidine use is associated with improved clinical outcomes in hospitalized COVID-19 patients: a propensity score matched retrospective cohort study. Gastroenterology. 2020;159:1129-1131.e3.
Changeux JP, Amoura Z, Rey FA, Miyara M. A nicotinic hypothesis for Covid-19 with preventive and therapeutic implications. C R Biol Academie des sciences. 2020;343:33–9.
Farsalinos K, Barbouni A, Niaura R. Smoking, vaping and hospitalization for COVID-19. Qeios. 2020; https://www.qeios.com/read/Z69O8A.2
Miyara M, Tubach F, Pourcher V, Morélot-Panzini C, Pernet J, Haroche J, et al. Lower rate of daily smokers with symptomatic COVID-19: a monocentric self-report of smoking habit study. Front Med (Lausanne). 2021;8: 668995.
Farsalinos K, Barbouni A, Niaura R. Systematic review of the prevalence of current smoking among hospitalized COVID-19 patients in China: could nicotine be a therapeutic option? Intern Emerg Med. 2020;15:845–52.
Lippi G, Henry BM. Active smoking is not associated with severity of coronavirus disease 2019 (COVID-19). Eur J Intern Med. 2020;75:107–8.
Emami A, Javanmardi F, Pirbonyeh N, Akbari A. Prevalence of underlying diseases in hospitalized patients with COVID-19: a systematic review and meta-analysis. Arch Acad Emerg Med. 2020;8: e35.
Guan W-J, Liang W-H, Zhao Y, Liang H-R, Chen Z-S, Li Y-M, et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: a nationwide analysis. Eur Respir J. 2020;55(5):2000547.
Rossato M, Russo L, Mazzocut S, di Vincenzo A, Fioretto P, Vettor R. Current smoking is not associated with COVID-19. Eur Respir J. 2020;55:2001290.
Guan W-J, Ni Z-Y, Hu Y, Liang W-H, Ou C-Q, He J-X, et al. Clinical characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;382:1708–20.
Gupta I, Sohail MU, Elzawawi KE, Amarah AH, Vranic S, Al-Asmakh M, et al. SARS-CoV-2 infection and smoking: What is the association? a brief review. Comput Struct Biotechnol J. 2021;19:1654–60.
Li J, Long X, Zhang Q, Fang X, Li N, Fedorova B, et al. Tobacco smoking confers risk for severe COVID-19 unexplainable by pulmonary imaging. J Intern Med. 2021;289:574–83.
Huckstepp RTR, id Bihi R, Eason R, Spyer KM, Dicke N, Willecke K, et al. Connexin hemichannel-mediated CO2-dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J Physiol. 2010;588:3901–20.
Huckstepp RTR, Llaudet E, Gourine AV. CO2-Induced ATP-dependent release of acetylcholine on the ventral surface of the medulla oblongata. PLoS One. 2016;11:e0167861.
Shrestha G, Githumbi R, Oslanski B, Lachman N, Venkova D, Montgomery B, et al. A Phase IIa open label study to evaluate the safety, tolerability and efficacy of S-1226 administered by nebulization in subjects with cystic fibrosis lung disease. medRxiv. 2021;2021.12.10.21266937.
Swystun V, Green FHY, Dennis JH, Rampakakis E, Lalli G, Fadayomi M, et al. A phase IIa proof-of-concept, placebo-controlled, randomized, double-blind, crossover, single-dose clinical trial of a new class of bronchodilator for acute asthma. Trials. 2018;19:321.
Sakai Y, Miwa M, Oe K, Ueha T, Koh A, Niikura T, et al. A novel system for transcutaneous application of carbon dioxide causing an “artificial Bohr effect” in the human body. PLoS ONE. 2011;6: e24137.
Baddeley H, Brodrick PM, Taylor NJ, Abdelatti MO, Jordan LC, Vasudevan AS, et al. Gas exchange parameters in radiotherapy patients during breathing of 2%, 3.5% and 5% carbogen gas mixtures. Br J Radiol. 2000;73:1100–4.
Bradley SM, Simsic JM, Atz AM. Hemodynamic effects of inspired carbon dioxide after the Norwood procedure. Ann Thorac Surg. 2001;72:2084–8.
Ambrogio C, Lowman X, Kuo M, Malo J, Prasad AR, Parthasarathy S. Sleep and non-invasive ventilation in patients with chronic respiratory insufficiency. Intensive Care Med. 2009;35:306–13.
Hardin KA, Seyal M, Stewart T, Bonekat HW. Sleep in critically ill chemically paralyzed patients requiring mechanical ventilation. Chest. 2006;129:1468–77.
Friese RS, Diaz-Arrastia R, McBride D, Frankel H, Gentilello LM. Quantity and quality of sleep in the surgical intensive care unit: are our patients sleeping? J Trauma. 2007;63:1210–4.
Baird B, Castelnovo A, Riedner BA, Lutz A, Ferrarelli F, Boly M, et al. Human rapid eye movement sleep shows local increases in low-frequency oscillations and global decreases in high-frequency oscillations compared to resting wakefulness. eNeuro. 2018;5:ENEURO.0293–18.2018.
Datta S, Oliver MD. Cellular and molecular mechanisms of REM sleep homeostatic drive: a plausible component for behavioral plasticity. Front Neural Circuits. 2017;11:63.
Jouwena J, Eerlings SA, de Wolf AM, van Hoovels L, Neyrinck A, van de Velde M, et al. Arterial to end-tidal CO2 gradients during isocapnic hyperventilation. J Clin Monit Comput. 2023;37:311–7.
Tong J, Zhou X-D, Kolosov VP, Perelman JM. Role of the JNK pathway on the expression of inflammatory factors in alveolar macrophages under mechanical ventilation. Int Immunopharmacol. 2013;17:821–7.
Wood J, Tabacof L, Tosto-Mancuso J, McCarthy D, Kontorovich A, Putrino D. Levels of end-tidal carbon dioxide are low despite normal respiratory rate in individuals with long COVID. J Breath Res. 2022;16: 017101.
Szakmar E, Kovacs K, Meder U, Bokodi G, Andorka C, Lakatos A, et al. Neonatal encephalopathy therapy optimization for better neuroprotection with inhalation of CO(2): the HENRIC feasibility and safety trial. Pediatr Res. 2020;87:1025–32.
Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, et al. A cannabinoid link between mitochondria and memory. Nature. 2016;539:555–9.
Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156:825–35.
Liu J, Qi J, Chen X, Li Z, Hong B, Ma H, et al. Fear memory-associated synaptic and mitochondrial changes revealed by deep learning-based processing of electron microscopy data. Cell Rep. 2022;40: 111151.
Kataoka K, Bilkei-Gorzo A, Nozaki C, Togo A, Nakamura K, Ohta K, et al. Age-dependent alteration in mitochondrial dynamics and autophagy in hippocampal neuron of cannabinoid CB1 receptor-deficient mice. Brain Res Bull. 2020;160:40–9.
Helfenberger KE, Villalba NM, Buchholz B, Boveris A, Poderoso JJ, Gelpi RJ, et al. Subcellular distribution of ERK phosphorylation in tyrosine and threonine depends on redox status in murine lung cells. PLoS ONE. 2018;13: e0193022.
Alonso M, Melani M, Converso D, Jaitovich A, Paz C, Carreras MC, et al. Mitochondrial extracellular signal-regulated kinases 1/2 (ERK1/2) are modulated during brain development. J Neurochem. 2004;89:248–56.
Griffiths J, Grant SGN. Synapse pathology in Alzheimer’s disease. Semin Cell Dev Biol. 2023;139:13–23.
Torres AK, Jara C, Park-Kang HS, Polanco CM, Tapia D, Alarcón F, et al. Synaptic mitochondria: an early target of Amyloid-β and Tau in Alzheimer’s Disease. J Alzheimers Dis. 2021;84:1391–414.
Torres AK, Rivera BI, Polanco CM, Jara C, Tapia-Rojas C. Phosphorylated tau as a toxic agent in synaptic mitochondria: implications in aging and Alzheimer’s disease. Neural Regen Res. 2022;17:1645–51.
Reiss AB, Ahmed S, Dayaramani C, Glass AD, Gomolin IH, Pinkhasov A, et al. The role of mitochondrial dysfunction in Alzheimer’s disease: a potential pathway to treatment. Exp Gerontol. 2022;164: 111828.
Johri A. Disentangling mitochondria in Alzheimer’s Disease. Int J Mol Sci. 2021;22:11520.
Ganji R, Reddy PH. Impact of COVID-19 on mitochondrial-based immunity in aging and age-related diseases. Front Aging Neurosci. 2021;12: 614650.
Singh KK, Chaubey G, Chen JY, Suravajhala P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am J Physiol Cell Physiol. 2020;319:C258–67.
Saleh J, Peyssonnaux C, Singh KK, Edeas M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion. 2020;54:1–7.
André S, Picard M, Cezar R, Roux-Dalvai F, Alleaume-Butaux A, Soundaramourty C, et al. T cell apoptosis characterizes severe Covid-19 disease. Cell Death Differ. 2022;29:1486–99.
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–31.
Martínez D, Vermeulen M, Trevani A, Ceballos A, Sabatté J, Gamberale R, et al. Extracellular acidosis induces neutrophil activation by a mechanism dependent on activation of phosphatidylinositol 3-kinase/Akt and ERK pathways. J Immunol. 2006;176:1163–71.
Deng Y, Hu F, Ren L, Gao X, Wang Y. Effects of anoxia on survival and gene expression in Bactrocera dorsalis. J Insect Physiol. 2018;107:186–96.
Fagernes CE, Stensløkken K-O, Røhr ÅK, Berenbrink M, Ellefsen S, Nilsson GE. Extreme anoxia tolerance in crucian carp and goldfish through neofunctionalization of duplicated genes creating a new ethanol-producing pyruvate decarboxylase pathway. Sci Rep. 2017;7:7884.
Fago A. New insights into survival strategies to oxygen deprivation in anoxia-tolerant vertebrates. Acta Physiol (Oxf). 2022;235: e13841.
Nayak G, Prentice HM, Milton SL. Lessons from nature: signalling cascades associated with vertebrate brain anoxic survival. Exp Physiol. 2016;101:1185–90.
Nayak GH, Prentice HM, Milton SL. Neuroprotective signaling pathways are modulated by adenosine in the anoxia tolerant turtle. J Cereb Blood Flow Metab. 2011;31:467–75.
Tan L-R, Liu J-J, Deewan A, Lee JW, Xia P-F, Rao CV, et al. Genome-wide transcriptional regulation in Saccharomyces cerevisiae in response to carbon dioxide. FEMS Yeast Res. 2022;22:foac032.
Cao X, An T, Fu W, Zhang J, Zhao H, Li D, et al. Genome-wide identification of cellular pathways and key genes that respond to sodium bicarbonate stress in saccharomyces cerevisiae. Front Microbiol. 2022;13:831973.
Green O, Finkelstein P, Rivero-Crespo MA, Lutz MDR, Bogdos MK, Burger M, et al. Activity-based approach for selective molecular CO 2 sensing. J Am Chem Soc. 2022;144:8717–24.
King DT, Zhu S, Hardie DB, Serrano-Negrón JE, Madden Z, Kolappan S, et al. Chemoproteomic identification of CO(2)-dependent lysine carboxylation in proteins. Nat Chem Biol. 2022;18:782–91.
Tubita A, Tusa I, Rovida E. Playing the whack-a-mole game: ERK5 activation emerges among the resistance mechanisms to RAF-MEK1/2-ERK1/2- targeted therapy. Front Cell Dev Biol. 2021;9: 647311.
Schmetterer L, Lexer F, Findl O, Graselli U, Eichler HG, Wolzt M. The effect of inhalation of different mixtures of O2 and CO2 on ocular fundus pulsations. Exp Eye Res. 1996;63:351–5.
Acknowledgements
The authors thank Mirosława Dabert for her indispensable support.
Funding
This work was supported by the National Science Center, Poland [grant numbers 2015/19/D/NZ3/00479 to H.G., 2020/37/B/NZ1/01188 to W.J. and 2011/01/D/NZ3/02068 to Ł.G.].
Author information
Authors and Affiliations
Contributions
H.G., W.J., Ł.G.: funding acquisition, writing—original draft preparation, review and editing, and visualization. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gałgańska, H., Jarmuszkiewicz, W. & Gałgański, Ł. Carbon dioxide and MAPK signalling: towards therapy for inflammation. Cell Commun Signal 21, 280 (2023). https://doi.org/10.1186/s12964-023-01306-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01306-x