
Abstract
Background
Neutrophils depend heavily on glycolysis for energy production under normal conditions. In contrast, neutrophils require energy supplied by mitochondrial oxidative phosphorylation (OXPHOS) during chemotaxis. However, the mechanism by which the energy supply changes from glycolysis to OXPHOS remains unknown. Leucine-rich repeat kinase 2 (LRRK2) is partially present in the outer mitochondrial membrane fraction. Lrrk2-deficient cells show mitochondrial fragmentation and reduced OXPHOS activity. We have previously reported that mitofusin (MFN) 2 is involved in chemotaxis and OXPHOS activation upon chemoattractant N-formyl-Met-Leu-Phe (fMLP) stimulation in differentiated HL-60 (dHL-60) cells. It has been previously reported that LRRK2 binds to MFN2 and partially colocalizes with MFN2 at the mitochondrial membranes. This study investigated the involvement of LRRK2 in chemotaxis and MFN2 activation in neutrophils and dHL-60 cells.
Methods
Lrrk2 knockout neutrophils and Lrrk2 knockdown dHL-60 cells were used to examine the possible involvement of LRRK2 in chemotaxis. Lrrk2 knockdown dHL-60 cells were used a tetracycline-inducible small hairpin RNA (shRNA) system to minimize the effects of LRRK2 knockdown during cell culture. The relationship between LRRK2 and MFN2 was investigated by measuring the GTP-binding activity of MFN2 in Lrrk2 knockdown dHL-60 cells. The effects of LRRK2 kinase activity on chemotaxis were examined using the LRRK2 kinase inhibitor MLi-2.
Results
fMLP-induced chemotactic activity was reduced in Lrrk2 knockout neutrophils in vitro and in vivo. Lrrk2 knockdown in dHL-60 cells expressing Lrrk2 shRNA also reduced fMLP-induced chemotactic activity. Lrrk2 knockdown dHL-60 cells showed reduced OXPHOS activity and suppressed mitochondrial morphological change, similar to Mfn2 knockdown dHL-60 cells. The amount of LRRK2 in the mitochondrial fraction and the GTP-binding activity of MFN2 increased upon fMLP stimulation, and the MFN2 GTP-binding activity was suppressed in Lrrk2 knockdown dHL-60 cells. Furthermore, the kinase activity of LRRK2 and Ser935 phosphorylation of LRRK2 were reduced upon fMLP stimulation, and LRRK2 kinase inhibition by MLi-2 increased the migration to fMLP.
Conclusions
LRRK2 is involved in neutrophil chemotaxis and the GTP-binding activity of MFN2 upon fMLP stimulation. On the other hand, the kinase activity of LRRK2 shows a negative regulatory effect on fMLP-induced chemotactic activity in dHL-60 cells.
Video Abstract
Similar content being viewed by others


Background
Neutrophils are important innate immune cells that act as the first line of defense against pathogens. Most neutrophil chemoattractants, including the bacterial product N-formyl-Met-Leu-Phe (fMLP) and chemokines, bind to G protein-coupled receptors. Gßγ subunits, which released from heterotrimeric G protein, leads activation of various chemotaxis-related molecules including RAC1 and RAC2 through the activation of phosphoinositide 3‑kinase γ [1, 2]. During this process, the mitochondrial membrane potential of neutrophils increases after fMLP stimulation. Inhibition of mitochondrial oxidative phosphorylation (OXPHOS) suppresses the neutrophil chemotaxis [3, 4].
Mutations in Leucine-rich repeat kinase 2 (LRRK2) are a leading cause of familial Parkinson’s disease (PD) [5, 6]. LRRK2 is a large protein with two catalytic domains (GTPase and protein kinase) and multiple protein–protein interacting domains [7]. LRRK2 is present mainly in the cytoplasm, and approximately 10% is present in the mitochondrial fraction [8]. LRRK2 is associated with the outer mitochondrial membrane [9]. LRRK2 binds to mitofusin (MFN) 1, MFN2, optic atrophy 1 (OPA1), and dynamin-related protein 1 and partially colocalizes with MFN1, MFN2, and OPA1 at the mitochondrial membranes [10]. Lrrk2 knockout cells show mitochondrial fragmentation and reduce OXPHOS activity and endoplasmic reticulum (ER)-mitochondrial interactions compared to wild-type cells [11, 12]. The kinase activity of LRRK2 is critical for mitochondrial morphology and function. The G2019S mutation in LRRK2 (LRRK2[G2019S]) is the most prevalent mutation in PD and leads to increased kinase activity of LRRK2 [13, 14]. LRRK2(G2019S)-expressing mouse embryonic fibroblasts (MEFs) showed mitochondrial fragmentation and reduced OXPHOS activity and ER-mitochondrial interactions compared to wild-type LRRK2 expressing MEFs [11]. In contrast, kinase-dead LRRK2(D1994A)-expressing MEFs showed increased of citrate synthase and OXPHOS activities compared to wild-type LRRK2 expressing MEFs [11].
MFN2 is a member of the dynamin superfamily and is mainly localized in the mitochondrial outer membrane [15]. MFN2 mediates mitochondrial outer membrane fusion and ER-mitochondria tethering via its GTPase activity [16]. Studies have reported that suppressing Mfn2 expression using antisense RNA causes mitochondrial fragmentation and reduce OXPHOS activity [17].
We previously reported that Mfn2 knockdown by small hairpin RNA (shRNA) suppresses mitochondrial morphological changes, OXPHOS, and chemotactic activity upon fMLP stimulation in differentiated HL-60 (dHL-60) cells [18]. Furthermore, Mfn2 knockout neutrophils were reported to show reduced migration in zebrafish [19]. Here, we investigated whether LRRK2, which is reported to bind to MFN2, is involved in neutrophils chemotaxis. Lrrk2 knockout neutrophils and Lrrk2 knockdown dHL-60 cells were used to examine the relationship between LRRK2 and chemotaxis, and between LRRK2 and MFN2. These results suggest that LRRK2 is involved in neutrophil chemotaxis and is required for the GTP-binding activity of MFN2.
Materials and methods
Cell lines
HL-60 cells were purchased from American Type Culture Collection (Manassas, VA, USA). HL-60 cells were cultured in RPMI 1640 medium supplemented with L-glutamine (Nacalai Tesque, Kyoto, Japan) and 10% fetal bovine serum (Gibco, Billings, MT, USA). Cells were passaged once every two days. For differentiation into neutrophil-like cells, the cells were cultured in a medium supplemented with 1.25% dimethyl sulfoxide (DMSO; Merck, Rahway, NJ, USA) for 4 days.
Animal models
C57BL/6-Lrrk2tm1.1Miff/J mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). All the mice were housed under standard laboratory conditions in a controlled pathogen-free room. Food and water were provided ad libitum. The protocols used for all animal experiments in this study were approved by Hokkaido University, Japan.
Antibodies and chemicals
The following antibodies were used in this study: mouse monoclonal antibodies against ß-actin (Merck) and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and rabbit monoclonal antibodies against MFN2 (Abcam, Tokyo, Japan), LRRK2 (Abcam), LRRK2 pS935 (Abcam), RAB10 (Abcam), RAB10 pT73 (Abcam), and TOMM20 (Cell Signaling Technology, Danvers, MA, USA). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit donkey IgG antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Alexa Fluor 488-conjugated anti-rabbit goat IgG and Alexa Fluor 555-conjugated anti-mouse goat IgG antibody were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Cis-2,6-dimethyl-4-(6-(5-(1-methylcyclopropoxy)-1H-indazol-3-yl)pyrimidin-4-yl)morpholine (MLi-2) was purchased from Cayman Chemical Co (Ann Arbor, MI, USA).
Bone marrow neutrophils
Bone marrow neutrophils were prepared by centrifugation using Percoll gradients, as previously described [20]. Purified neutrophils were suspended in ice-cold Hank’s balanced salt solution (HBSS) containing 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.2) and 1% bovine serum albumin (BSA, Rockland Immunochemicals Pottstown, PA, USA) and stored on ice until further use.
Gene silencing
LRRK2 was silenced by infecting HL-60 cells with lentiviruses constructed by inserting Lrrk2 shRNA sequence (sh1: TRCN0000021462; sh2: TRCN0000021460, Merck) into Tet-pLKO-puro (Addgene, Watertown, MA, USA). Tet-pLKO-puro was a gift from Dr. Dmitri Wiederschain [21]. The targeting sequence of the irrelevant (Irr) shRNA has been previously reported [22]. The infected HL-60 cells were selected with 0.8 µg/ml puromycin (Merck). For gene silencing in dHL-60 cells, HL-60 cells expressing tetracycline-inducible Lrrk2 shRNA or Irr shRNA were cultured in the presence of 1.25% DMSO for 1 day, followed by 3 days in a medium supplemented with 1.25% DMSO and 0.5 µg/ml doxycycline (Dox; FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan).
Chemotaxis assays
Transwell chemotaxis assays were performed as previously described [18, 23]. For neutrophils, transwell chemotaxis assays were performed using 24-well transwell chambers (pore size, 3.0 μm; Corning, Corning, NY, USA). For dHL-60 cells, transwell chemotaxis assays were performed using 24-well transwell chambers (pore size, 5.0 μm; Corning). Cells were placed on the upper side of the top chambers, and cells that migrated to the lower side of the chamber membrane were stained with Diff-Quick (Sysmex, Kobe, Japan) and counted. Five microscope fields of view were analyzed for each experiment.
For two-dimensional chemotaxis assays, a Dunn chamber (Hawksley, Sussex, UK) was used. After washing cells, the cells were attached to a coverslip and incubated at 37 °C for 10 min in HBSS containing 20 mM HEPES (pH 7.2) and 1% BSA. For neutrophils, coverslips were placed on a Dunn chamber, in which the outer well of the chamber was filled with the same solution supplemented with 10 µM fMLP (MP Biomedicals, Irvine, CA, USA) and a 1:10 dilution of 10% gelatin (Nacalai Tesque) in H2O. Cell migration was recorded by capturing images every 15 s for 20 min. For dHL-60 cells, the outer wells of the Dunn chambers were filled with the same solution, supplemented with 10 nM fMLP. Cell migration was recorded by capturing images every 15 s for 30 min. Image of each cell migration were recorded using an inverted microscope (Axiovert135, Carl Zeiss, Oberkochen, Germany) equipped with a digital camera (AxioCam and Axiovison software; Carl Zeiss). Stacks of images were then analyzed by ImageJ Fiji software (National Institutes of Health, Bethesda, MA, USA), with Chemotaxis and Migration Tool plug-in (Ibidi) for analysis of the cell tracking, velocity, and directional index of each moving cell. Cells that migrated more than 20 µm in their tracks during a 20 min incubation period were used for analysis.
Air pouch analysis
Air pouch analysis of local inflammation was performed as described previously [24]. Mice were anesthetized by intraperitoneal injection of pentobarbital sodium and injected with 5 ml of air to create a subcutaneous dorsal pouch. After three days, the pouches were reinjected with 3 ml of air. Six days after the first injection, the pouches were injected with 1 ml of 0.5 μg/ml human interleukin-8 (IL-8) (Shenandoah Biotechnology, Warwick, PA, USA) and 0.5% carboxymethylcellulose (FUJIFILM Wako) in PBS. After 4 h, the mice were anesthetized, and the pouches were washed with 2 ml of PBS. The lavage fluid was immediately chilled on ice, the volume was recorded, and the neutrophils were counted.
Immunofluorescence microscopy
Briefly, dHL-60 cells were attached to coverslips with HBSS containing 20 mM HEPES (pH 7.2) and 1% BSA for 10 min at 37 °C. After treatment with or without 10 nM fMLP for 5 min, the cells were fixed with 2% paraformaldehyde at 37 °C for 10 min, washed with PBS, and fixed with methanol at -20 °C for 5 min. The cells were then permeabilized with 0.1% Triton X-100 in PBS for 5 min, washed with PBS, and then incubated with 1% BSA in PBS for 30 min. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Nacalai Tesque). High-resolution structured illumination microscopy (SIM) images were acquired using an N-SIM microscope (Nikon, Tokyo, Japan) and NIS-elements software (Nikon), as previously described [18]. The percentage of cells with tubular mitochondria was calculated by counting cells with tubular mitochondria (length > 3 µm) and dividing this number by the total number of cells observed within each microscopic field, as described previously [18].
Mitochondrial OXPHOS activity
The mitochondrial respiratory capacity was measured at 37 °C by using a high-resolution respirometer (Oxygraph-2 k, Oroboros Instruments, Innsbruck, Austria) as described previously [18]. Briefly, 2 × 106 dHL-60 cells were added to the respirometer chamber filled with 2 ml of MiR05 medium (110 mM sucrose, 60 mM potassium lactobionate, 0.5 mM ethylene glycol tetraacetic acid, 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES [pH 7.1], and 1% BSA). The cells were then stimulated with 1 µM fMLP for 5 min and permeabilized with 5 µM digitonin (Merck). Substrates of OXPHOS and ADP were added to the respirometer chamber in the following order: (1) 2 mM malate, 10 mM glutamate, and 5 mM pyruvate (complex I-linked substrates); (2) 5 mM ADP + 3 mM MgCl2; and (3) 10 mM succinate (complex II-linked substrates). The O2 consumption rates (OCR) were expressed as O2 flux normalized to 1 × 106 cells. Data acquisition and analysis of data were performed using DatLab software (Oroboros Instruments), as described previously [18].
Mitochondrial fractionation
Mitochondrial fractionation was performed as previously described with some modifications [25]. dHL-60 cells were pre-incubated in HBSS containing 20 mM HEPES (pH 7.2) for 5 min at 37 °C and then stimulated with or without 1 µM fMLP at 37 °C for 5 min. The cells were lysed in hypotonic buffer (10 mM Tris-MOPS [pH7.4], 1 mM EDTA-Tris [pH7.4]) with rotation at 4 °C for 20 min. The lysed cells were homogenized by 15 repeated passes through a 30-gauge needle attached to a 1 ml syringe. The homogenate was centrifuged at 700 × g and 4 °C for 10 min, and the supernatant was centrifuged again at 700 × g and 4 °C for 10 min. The supernatant was then centrifuged at 7,000 × g and 4 °C for 10 min. The pellet was suspended with isotonic buffer (10 mM Tris-MOPS [pH7.4], 1 mM EDTA-Tris [pH7.4], 200 mM sucrose) and was centrifuged at 7,000 × g and 4 °C for 10 min. The pellets were again suspended with isotonic buffer and was centrifuged at 7,000 × g and 4 °C for 10 min. The pellets were resuspended in RIPA buffer (150 mM NaCl, 20 mM Tris–HCl [pH 7.4], 5 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1 mM Na3VO4, 20 mM NaF, 10 mM sodium diphosphate, and a protease inhibitor cocktail for Use with Mammalian Cell and Tissue Extracts [Nacalai Tesque]) and incubated on ice for 10 min. The lysates were sonicated and centrifuged at 21,900 × g and 4 °C for 30 min. This supernatant was used as the mitochondrial fraction.
GTP-binding assay
The GTP-binding assay for MFN2 was performed as previously described with some modifications [26]. dHL-60 cells were pre-incubated in HBSS containing 20 mM HEPES (pH 7.2) for 5 min at 37 °C, and then stimulated with or without 1 µM fMLP at 37 °C for 5 min. After stopping the reaction by adding ice-cold HBSS containing 20 mM HEPES (pH 7.2), the cells were centrifuged at 200 × g and 4 °C for 3 min. The cells were lysed in lysis buffer (150 mM NaCl, 50 mM Tris–HCl [pH 7.4], 1% Triton X-100, 5 mM MgOAc, 1 mM DTT, 1 mM Na3VO4, 20 mM NaF, 10 mM sodium diphosphate, and a protease inhibitor cocktail) on ice for 10 min. Cell lysates were sonicated and centrifuged at 21,900 × g and 4 °C for 10 min. After preclear, 250 µg cell lysates were incubated with GTP-agarose beads (Abcam) for 2 h at room temperature. The beads were then washed four times with lysis buffer, and proteins were eluted from the beads using the SDS sample buffer. The GTP-bound form of MFN2 was detected by western blotting with an anti-MFN2 antibody.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version.8 (GraphPad Software, Inc., Boston, MA, USA). Differences between multiple groups were calculated using one-way ANOVA followed by the Tukey–Kramer test. Differences between the two groups were calculated using a two-tailed Student’s t-test.
Results
Lrrk2 knockout neutrophils show reduced fMLP-induced chemotactic activity
Previously, it was reported that Lrrk2 knockout causes mitochondrial fragmentation and reduces OXPHOS activity [11, 12]. Since the inhibition of OXPHOS activity is reported to reduce neutrophil chemotactic activity, we first examined whether Lrrk2 is involved in neutrophil chemotaxis using transwell chambers. The fMLP-induced chemotactic activity of Lrrk2 knockout neutrophils was lower than that of wild-type neutrophils in transwell chamber assays (Fig. 1A). Next, we examined two-dimensional chemotaxis using Dunn chambers to assess the velocity and directionality. Lrrk2 knockout neutrophils showed decreased velocity compared to wild-type neutrophils, whereas no change was observed in the directionality index of Lrrk2 knockout neutrophils compared to that of wild-type neutrophils (Fig. 1B, C, S1, Additional files 1, 2). Furthermore, we examined the chemotaxis of Lrrk2 knockout neutrophils in vivo using an air pouch model. The accumulation of neutrophils in an air-pouches during the 4 h interval after the injection of IL-8 into the air pouches was lower in Lrrk2 knockout mice than in wild-type mice (Fig. 1D). These results suggest that LRRK2 is involved in neutrophil chemotaxis in vitro and in vivo.

Inactivation of LRRK2 results in decreased neutrophil chemotactic activities. A Neutrophil chemotaxis assays in the transwell. Neutrophil migration in response to 10 µM fMLP was measured for 90 min in transwell chambers (n = 6 wells). The activity of wild-type neutrophils was taken as 100%. B, C Two-dimensional chemotaxis assays. Neutrophil migration in response to 10 µM fMLP was measured for 20 min in Dunn chambers (n > 45 cells). B Velocity. C Directionality index. D Air pouch analysis. Neutrophil numbers in exudates isolated 4 h after injection of IL-8 into subcutaneous dorsal air pouches (n = 10 mice). wt, wild-type mice. Lrrk2 KO, Lrrk2 knockout mice. Error bars, SEM. ** p < 0.01 compared with wild-type mice. NS, not significant
Lrrk2 knockdown dHL-60 cells show reduced fMLP-induced chemotactic activity
HL-60 cells are differentiated into a granulocytic lineage by DMSO and are used as a model for neutrophils [19, 27, 28]. To thoroughly investigate the role of LRRK2 in chemotaxis, we analyzed dHL-60 cells. We used a tetracycline-inducible system to minimize the effects of LRRK2 knockdown on cell growth during cell culture. Tetracycline-induced Lrrk2 shRNA suppressed LRRK2 expression without altering MFN2 expression in dHL-60 cells (Fig. 2A). We then used transwell chambers to verify whether Lrrk2 knockdown dHL-60 cells exhibited reduced chemotactic activity. Lrrk2 knockdown dHL-60 cells showed reduced chemotactic activity compared to Irr shRNA-expressing dHL-60 cells (Fig. 2B). We found that Lrrk2 knockdown in dHL-60 cells, like neutrophils, slowed without changing the directional index in a two-dimensional chemotaxis assay (Fig. 2C, D, Supplementary Figure S2, Additional files 3, 4, 5). In contrast, Irr shRNA-expressing and Lrrk2 knockdown dHL-60 cells cultured without doxycycline showed no significant difference in chemotactic activity in the transwell chambers and two-dimensional chemotaxis assays (Supplementary Fig. S3). These results suggest that LRRK2 is involved in the chemotaxis of dHL-60 cells.

Lrrk2 knockdown results in a decrease in the chemotactic activities of dHL-60 cells. A Expression pattern of proteins in Lrrk2 knockdown dHL-60 cells. dHL-60 cells expressing tetracycline-inducible Lrrk2 shRNA or irrelevant (Irr) shRNA were cultured in the presence of 1.25% DMSO with or without 0.5 µg/ml doxycycline (Dox) for 3 days, and analyzed for expression of the indicated proteins. B Transwell chemotaxis assays of dHL-60 cells. dHL-60 cells expressing tetracycline-inducible Lrrk2 shRNA or Irr shRNA were cultured with 0.5 µg/ml Dox for 3 days. dHL-60 cells migration in response to 10 nM fMLP was measured for 120 min in transwell chambers (n = 6 wells). The activity of Irr shRNA expressing dHL-60 cells was taken as 100%. C, D Two-dimensional chemotaxis assays. dHL-60 cells expressing tetracycline-inducible Lrrk2 shRNA or Irr shRNA were cultured with 0.5 µg/ml Dox for 3 days. dHL-60 cells migration in response to 10 nM fMLP was measured for 30 min in Dunn chambers (n > 25 cells). C Velocity. D Directionality index. ** p < 0.01 compared with Irr shRNA expressing dHL-60 cells. NS, not significant
Knockdown of Lrrk2 reduces OXPHOS activity and suppresses mitochondrial morphological changes
Lrrk2 knockdown dHL-60 cells showed reduced fMLP-induced chemotactic activity, similar to Mfn2 knockdown dHL-60 cells. Next, we examined whether Lrrk2 knockdown dHL-60 cells showed a reduction in OXPHOS activity, similar to Mfn2 knockdown dHL-60 cells. We found that OXPHOS activity at complex I and complex I + complex II was reduced in Lrrk2 knockdown dHL-60 cells (Fig. 3A, B).

Lrrk2 knockdown results in a decrease in OXPHOS activities of dHL-60 cells. A, B dHL-60 cells expressing tetracycline-inducible Lrrk2 shRNA or irrelevant (Irr) shRNA were cultured in 1.25% DMSO with 0.5 µg/ml doxycycline (Dox) for 3 days. The OXPHOS activities of these cells were measured after treatment with 1 µM fMLP for 5 min (n = 4). Error bars, SEM. A complex I. B complex I + II. C Representative immunofluorescence microscope images of mitochondria. dHL-60 cells expressing tetracycline-inducible Lrrk2 shRNA or irrelevant (Irr) shRNA were cultured in 1.25% DMSO with 0.5 µg/ml Dox for 3 days. dHL-60 cells attached to coverslips were then incubated with or without 10 nM fMLP for 5 min. Green; anti-TOMM20, Red; anti-ß-actin, Blue; DAPI. Bar, 10 µm. D Percentages of cells with mitochondria of tubular form after fMLP stimulation. > 50 cells were analyzed in four independent experiments. Error bars, SEM. ** p < 0.01 compared with Irr shRNA expressing dHL-60 cells
Immunofluorescence microscopy using an anti-TOMM20 antibody showed that the mitochondrial morphology changed from spherical to tubular upon fMLP stimulation in dHL-60 cells [18]. We examined whether Lrrk2 knockdown dHL-60 cells suppressed mitochondrial morphological changes similar to Mfn2 knockdown dHL-60 cells. In Irr shRNA-expressing dHL-60 cells, 87.8% of the transformed into cells with tubular mitochondria upon fMLP stimulation (Fig. 3C, D). In contrast, Lrrk2 knockdown dHL-60 cells transformed into cells with tubular mitochondria at approximately 31% cells, suggesting that Lrrk2 knockdown suppresses mitochondrial morphology changes upon fMLP stimulation in dHL-60 cells (Fig. 3C, D).
LRRK2 affects the GTP-binding activity of MFN2
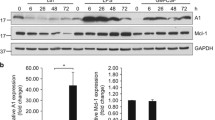
Because Lrrk2 knockdown in dHL-60 cells reduced OXPHOS activity and suppressed mitochondrial morphology changes upon fMLP stimulation, we examined the effects of LRRK2 on the amount of MFN2 in the mitochondrial fraction. We found that the mitochondrial fraction of LRRK2 increased upon fMLP stimulation, whereas no notable changes were detected in the mitochondrial fraction of MFN2 upon fMLP stimulation (Fig. 4A-C). The effect of Lrrk2 knockdown on the amount of MFN2 in the mitochondrial fraction was negligible (Fig. 4A, C).

Mitochondrial fraction of LRRK2 and GTP binding activity of MFN2 were increased upon fMLP stimulation. A-C The mitochondrial fraction of LRRK2 was increased upon fMLP stimulation. dHL-60 cells expressing tetracycline-inducible Lrrk2 shRNA or irrelevant (Irr) shRNA were cultured in 1.25% DMSO with 0.5 µg/ml doxycycline (Dox) for 3 days. The mitochondrial fraction of LRRK2 and MFN2 were measured with or without 1 µM fMLP for 5 min. D, E Lrrk2 knockdown results in a decrease in MFN2 GTP-binding activity of dHL-60 cells. dHL-60 cells expressing tetracycline-inducible Lrrk2 shRNA or irrelevant (Irr) shRNA were cultured in 1.25% DMSO with 0.5 µg/ml Dox for 3 days. The GTP-binding activity of MFN2 was measured with or without 1 µM fMLP for 5 min. A, D Representative western blotting images. B The rate of LRRK2/TOMM20 in the mitochondrial fraction of Irr shRNA expressing dHL-60 cells without fMLP stimulation was taken as 1.0 (n = 3). ** p < 0.01 compared with Irr shRNA, Lrrk2 sh1, or sh2 expressing dHL-60 cells without fMLP. C The rate of MFN2/TOMM20 in the mitochondrial fraction of Irr shRNA expressing dHL-60 cells without fMLP stimulation was taken as 1.0 (n = 3). NS, not significant. E GTP-binding activity of Irr shRNA expressing dHL-60 cells without fMLP was taken as 1.0 (n = 4). Error bars, SEM. ** p < 0.01 and * p < 0.05 compared with Irr shRNA expressing dHL-60 cells with or without 1 µM fMLP. NS, not significant
We examined the GTP-binding activity of MFN2 following fMLP stimulation. The GTP-binding activity of MFN2 increased upon fMLP stimulation (Fig. 4D, E). In contrast, no increase in the GTP-binding activity of MFN2 was detected in Lrrk2 knockdown dHL-60 cells upon fMLP stimulation (Fig. 4D, E). These results suggest that LRRK2 affects the GTP-binding activity of MFN2, rather than the amount of MFN2 in the mitochondrial fraction, upon fMLP stimulation.
Inhibition of LRRK2 Kinase activity causes increased fMLP-induced chemotactic activity of dHL-60 cells
LRRK2 phosphorylates various proteins, including RAB10, RAB8 [29], and WAVE2 [30], and their phosphorylation affects membrane trafficking [31] and cytoskeleton remodeling [30], which are important for chemotaxis. The kinase activity of LRRK2 can be assessed based on the Thr73 phosphorylation level of RAB10 [32]. Therefore, we analyzed the Thr73 phosphorylation level of RAB10 to assess the kinase activity of LRRK2. The Thr73 phosphorylation level of RAB10 decreased upon fMLP stimulation, suggesting decreased kinase activity of LRRK2 (Fig. 5A-C). We also found that Ser935 phosphorylation of LRRK2 decreased upon fMLP stimulation (Fig. S4). Furthermore, we examined the effect of the LRRK2 kinase inhibitor MLi-2 on the chemotaxis of dHL-60 cells. We found that fMLP-induced chemotactic activity in dHL-60 cells was increased by MLi-2 treatment compared to that in untreated cells (Fig. 5D-F, S5, Additional files 6, 7). These results show that the kinase activity of LRRK2 negatively regulates chemotactic activity upon fMLP stimulation in dHL-60 cells.

LRRK2 kinase inhibitor, MLi-2 activates chemotaxis of dHL-60 cells. A-C Effects of MLi-2 on kinase activity of LRRK2. dHL-60 cells were cultured in the presence of indicated concentrations of MLi-2 or 0.1% DMSO (vehicle) for 30 min. The cells were preincubated in HBSS containing 20 mM HEPES (pH 7.2) admixed with MLi-2 at indicated concentrations for 5 min at 37 °C and stimulated with or without 1 µM fMLP at 37 °C for 5 min. (A) Representative western blotting images. B The rate of Thr73 phosphorylation of RAB10 without fMLP/RAB10 without fMLP was taken as 1.0 (n = 3). (C) The rate of Thr73 phosphorylation of RAB10 without fMLP/GAPDH without fMLP was taken as 1.0 (n = 3). Error bars, SEM. ** p < 0.01 and * p < 0.05 compared to condition of 0.1% DMSO without fMLP. D Transwell chemotaxis assays of MLi-2 treated dHL-60 cells. dHL-60 cells were cultured with 30 nM MLi-2 or 0.1% DMSO (vehicle) for 30 min. The cell migration in response to 10 nM fMLP was measured with 30 nM MLi-2 or 0.1% DMSO for 120 min in transwell chambers (n = 6 wells). The activity of dHL-60 cells treated with 0.1% DMSO was taken as 100%. E, F Two-dimensional chemotaxis assays of MLi-2 treated dHL-60 cells. dHL-60 cells were cultured with 30 nM MLi-2 or 0.1% DMSO (vehicle) for 30 min. The cell migration in response to 10 nM fMLP was measured with 30 nM MLi-2 or 0.1% DMSO for 30 min in Dunn chambers (n > 16 cells). E Velocity. F Directionality index. ** p < 0.01 compared to the cells treated with 0.1% DMSO. NS, not significant
Discussion
In the present study, we found that Lrrk2 knockout neutrophils and Lrrk2 knockdown dHL-60 cells showed reduced chemotactic activity upon fMLP stimulation, and knockdown of Lrrk2 reduced OXPHOS activity in dHL-60 cells. Furthermore, the amount of LRRK2 in the mitochondrial fraction increased upon fMLP stimulation, and LRRK2 affected the GTP-binding activity of MFN2. These results suggest that fMLP stimulation might translocate LRRK2 to the mitochondrial fraction and that LRRK2 might activate OXPHOS through MFN2 and cause chemotactic migration.
Chemotaxis requires a complex series of intracellular events such as cytoskeleton remodeling, membrane trafficking, and OXPHOS activation [3, 4, 33, 34]. LRRK2 is involved in membrane trafficking [31] and cytoskeleton remodeling [30]. We also observed reduced OXPHOS activity (Fig. 3A, B) and reduced GTP-binding activity of MFN2 (Fig. 4D, E) in Lrrk2 knockdown dHL-60 cells. MFN2 is involved in OXPHOS activity [18] and actin cytoskeleton regulation [19] in dHL-60 cells. Therefore, the reduced chemotactic activity in Lrrk2 knockout neutrophils and Lrrk2 knockdown dHL-60 cells could be attributed to LRRK2, and MFN2-mediated effects.
We found that inhibition of LRRK2 kinase activity by MLi-2 in dHL-60 cells increased in fMLP-induced chemotactic activity (Fig. 5D-F). Kinase-dead LRRK2 (D1994A) in MEFs increased citrate synthase activity, OXPHOS activity, mitochondrial Ca2+ concentration, and the physical interaction between the ER and mitochondria compared to wild-type cells, although the facilitation of ATP production was undetectable [11]. It has been reported that a reduction in mitochondrial Ca2+ concentration due to the inhibition of the mitochondrial Ca2+ uniporter suppresses chemotactic activity in neutrophils [35]. Therefore, an increased mitochondrial Ca2+ concentration possibly increase the fMLP-induced chemotactic activity of dHL-60 cells. On the other hand, it was recently reported that inhibition of LRRK2 kinase activity by MLi-2 enhanced lysosomal degradative activity and increased the expression of multiple lysosomal hydrolases in induced pluripotent stem cell-derived macrophages [36]. Thus, the inhibition of LRRK2 kinase activity by MLi-2 may induce the expression of OXPHOS-associated and chemotaxis-associated proteins in dHL-60 cells. As described above, there are multiple possible mechanisms by which the inhibition of LRRK2 kinase activity by MLi-2 increases fMLP-induced chemotactic activity. Further studies are required to clarify how inhibition of LRRK2 kinase activity by MLi-2 facilitates fMLP-induced chemotactic activity.
In the present study, we found that the Ser935 phosphorylation of LRRK2 was reduced upon fMLP stimulation (Fig. S4). In general, protein phosphorylation is regulated by kinases and phosphatases. Ser935 of LRRK2 is phosphorylated by protein kinase A [37], an inhibitor of nuclear factor κB kinases [38], and casein kinase 1 α [39]. In contrast, Ser935 of LRRK2 is dephosphorylated by protein phosphatase 1 (PP1). Furthermore, it is reported that PP1α, which is expressed in neutrophils, preferentially interacts with LRRK2 [40]. Ser935 of LRRK2 is basally phosphorylated in cells, and LRRK2 has a long half-life [41]. Therefore, the short-term decrease in Ser935 phosphorylation of LRRK2 by fMLP stimulation may result from increased dephosphorylation of LRRK2 by phosphatases rather than decreased phosphorylation of LRRK2 by kinases. Phosphorylated Ser935 of LRRK2 is a 14–3-3 binding site [42], and the binding of 14–3-3 to LRRK2 affects its subcellular localization of LRRK2 [42]. We found that the mitochondrial fraction of LRRK2 increased upon fMLP stimulation (Fig. 4A, B). Reduced Ser935 phosphorylation of LRRK2 upon fMLP stimulation may affect the translocation of LRRK2 to the mitochondrial fraction by reducing 14–3-3 binding to LRRK2. On the other hand, Ser935 of LRRK2 was partially dephosphorylate, and the kinase activity of LRRK2 was partly decreased upon fMLP stimulation (Fig. 5A-C and Fig. S4). LRRK2 phosphorylate RAB proteins, including RAB5 and RAB10 [43]. RAB5 is present on early phagosomes, and regulate their fusion with early endosomes [44, 45]. LRRK2 forms a complex with the Wiskott–Aldrich syndrome protein-family verproline 2, and colocalizes with RAB5A during the fusion of phagosomes and early endosomes. Bone marrow-derived macrophages expressing LRRK2(G2019S) show increased phagocytic activity compared to wild-type macrophages [30]. Therefore, the kinase activity of LRRK2 is required, and may be partially required, for neutrophil and dHL-60 cell functions after fMLP stimulation.
We showed that the amount of LRRK2 in the mitochondrial fraction was increased upon fMLP stimulation, although the alteration of MFN2 level in the mitochondrial fraction was undetectable (Fig. 4A-C). The GTP-binding activity of MFN2 increased upon fMLP stimulation in Irr shRNA-expressing dHL-60 cells, whereas no notable changes were observed in Lrrk2 knockdown dHL-60 cells (Fig. 4D, E). Therefore, it appears that the GTP-binding activity of MFN2 is related to the mitochondrial fraction of LRRK2. LRRK2 increases the GTP-binding activity of MFN1 in LRRK2 and MFN1 overexpressing cells [10]. Because MFN1 and MFN2 share approximately 80% sequence similarity, increased LRRK2 level in the mitochondrial fraction might increase the GTP-binding activity of MFN2 upon fMLP stimulation.
In addition to PD, LRRK2 has been reported to be associated with inflammatory bowel diseases (IBD) which is composed of two major subtypes, Crohn’s disease and ulcerative colitis, based on genome-wide association studies [46,47,48,49]. IBD is characterized by chronic proinflammatory responses by immune cells to viruses or bacteria in the gastrointestinal tract [50]. LRRK2 is involved in inflammatory signaling pathways, such as type I interferon signaling, and phagocytosis [12, 36, 45] and is expressed at higher levels in neutrophils than in other peripheral blood immune cells [32]. Therefore, LRRK2 mutations may exacerbate IBD by disruption of neutrophils function.
Conclusions
This study showed that LRRK is involved in neutrophil chemotaxis and GTP binding activity of MFN2. Furthermore, it was suggested that the kinase activity of LRRK2 exerts a negative regulatory effect on chemotactic activity in dHL-60 cells upon fMLP stimulation. These results provide a better understanding of neutrophil chemotaxis and diseases involving LRRK2.
Availability of data and materials
All data used in this study are available from the corresponding author on reasonable requests.
Abbreviations
- ER:
-
Endoplasmic reticulum
- fMLP:
-
N-Formyl-Met-Leu-Phe peptide
- LRRK2:
-
Leucine-rich repeat kinase 2
- MEFs:
-
Mouse embryonic fibroblasts
- MFN:
-
Mitofusin
- OCR:
-
O2 consumption rates
- OPA1:
-
Optic atrophy 1
- OXPHOS:
-
Oxidative phosphorylation
References
de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16:378–91.
Gambardella L, Vermeren S. Molecular players in neutrophil chemotaxis–focus on PI3K and small GTPases. J Leukoc Biol. 2013;94:603–12.
Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, Edwards SW. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol. 2003;170:1964–72.
Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, Zhang J, Junger WG. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol. 2015;210:1153–64.
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004; 44:595–600.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7.
Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci. 2010;11:791–7.
West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–7.
Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–69.
Stafa K, Tsika E, Moser R, Musso A, Glauser L, Jones A, Biskup S, Xiong Y, Bandopadhyay R, Dawson VL, et al. Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum Mol Genet. 2014;23:2055–77.
Toyofuku T, Okamoto Y, Ishikawa T, Sasawatari S, Kumanogoh A. LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 2020;39: e100875.
Weindel CG, Bell SL, Vail KJ, West KO, Patrick KL, Watson RO. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife. 2020;9:e51071.
Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76:672–80.
Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A. French Parkinson’s Disease Genetics Study G: LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006;354:422–3.
Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–47.
Dorn GW 2nd. Mitofusin 2 Dysfunction and Disease in Mice and Men. Front Physiol. 2020;11:782.
Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003, 278:17190–17197.
Mazaki Y, Takada S, Nio-Kobayashi J, Maekawa S, Higashi T, Onodera Y, Sabe H. Mitofusin 2 is involved in chemotaxis of neutrophil-like differentiated HL-60cells. Biochem Biophys Res Commun. 2019;513:708–13.
Zhou W, Hsu AY, Wang Y, Syahirah R, Wang T, Jeffries J, et al. Mitofusin 2 regulates neutrophil adhesive migration and the actin cytoskeleton. J Cell Sci. 2020;133:jcs248880.
Mocsai A, Ligeti E, Lowell CA, Berton G. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J Immunol. 1999;162:1120–6.
Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C, et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8:498–504.
Mazaki Y, Onodera Y, Higashi T, Horinouchi T, Oikawa T, Sabe H. ARF1 recruits RAC1 to leading edge in neutrophil chemotaxis. Cell Commun Signal. 2017;15:36.
Mazaki Y, Hashimoto S, Tsujimura T, Morishige M, Hashimoto A, Aritake K, Yamada A, Nam JM, Kiyonari H, Nakao K, Sabe H. Neutrophil direction sensing and superoxide production linked by the GTPase-activating protein GIT2. Nat Immunol. 2006;7:724–31.
Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van Hinsbergh V, Sozzani S, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–25.
Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–95.
Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27:422–30.
Matzner Y, Gavison R, Rachmilewitz EA, Fibach E. Expression of granulocytic functions by leukemic promyelocytic HL-60 cells: differential induction by dimethylsulfoxide and retinoic acid. Cell Differ. 1987;21:261–9.
Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, Takuwa Y, Sugimoto N, Mitchison T, Bourne HR. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 2003;114:201–14.
Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, et al. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016;5:e12813.
Kim KS, Marcogliese PC, Yang J, Callaghan SM, Resende V, Abdel-Messih E, Marras C, Visanji NP, Huang J, Schlossmacher MG, et al. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson’s disease. Proc Natl Acad Sci U S A. 2018;115:E5164–73.
Schapansky J, Khasnavis S, DeAndrade MP, Nardozzi JD, Falkson SR, Boyd JD, Sanderson JB, Bartels T, Melrose HL, LaVoie MJ. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble alpha-synuclein in neurons. Neurobiol Dis. 2018;111:26–35.
Fan Y, Howden AJM, Sarhan AR, Lis P, Ito G, Martinez TN, Brockmann K, Gasser T, Alessi DR, Sammler EM. Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. Biochem J. 2018;475:23–44.
Sabe H, Onodera Y, Mazaki Y, Hashimoto S. ArfGAP family proteins in cell adhesion, migration and tumor invasion. Curr Opin Cell Biol. 2006;18:558–64.
Gamara J, Chouinard F, Davis L, Aoudjit F, Bourgoin SG. Regulators and effectors of Arf GTPases in neutrophils. J Immunol Res. 2015;2015: 235170.
Zheng X, Chen M, Meng X, Chu X, Cai C, Zou F. Phosphorylation of dynamin-related protein 1 at Ser616 regulates mitochondrial fission and is involved in mitochondrial calcium uniporter-mediated neutrophil polarization and chemotaxis. Mol Immunol. 2017;87:23–32.
Yadavalli N, Ferguson SM. LRRK2 suppresses lysosome degradative activity in macrophages and microglia through MiT-TFE transcription factor inhibition. Proc Natl Acad Sci U S A. 2023;120: e2303789120.
Li X, Wang QJ, Pan N, Lee S, Zhao Y, Chait BT, Yue Z. Phosphorylation-dependent 14-3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS One. 2011;6.
Dzamko N, Inesta-Vaquera F, Zhang J, Xie C, Cai H, Arthur S, Tan L, Choi H, Gray N, Cohen P, et al. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS One. 2012;7: e39132.
Chia R, Haddock S, Beilina A, Rudenko IN, Mamais A, Kaganovich A, Li Y, Kumaran R, Nalls MA, Cookson MR. Phosphorylation of LRRK2 by casein kinase 1alpha regulates trans-Golgi clustering via differential interaction with ARHGEF7. Nat Commun. 2014;5:5827.
Lobbestael E, Zhao J, Rudenko IN, Beylina A, Gao F, Wetter J, Beullens M, Bollen M, Cookson MR, Baekelandt V, et al. Identification of protein phosphatase 1 as a regulator of the LRRK2 phosphorylation cycle. Biochem J. 2013;456:119–28.
Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, Chandran J, Lin X, Lai C, Yang WJ, et al. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci. 2008;28:3384–91.
Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR, Alessi DR. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J. 2010;430:393–404.
Lis P, Burel S, Steger M, Mann M, Brown F, Diez F, Tonelli F, Holton JL, Ho PW, Ho SL, et al. Development of phospho-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson’s disease kinase. Biochem J. 2018;475:1–22.
Gutierrez MG. Functional role(s) of phagosomal Rab GTPases. Small GTPases. 2013;4:148–58.
Wallings RL, Tansey MG. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem Soc Trans. 2019;47:1581–95.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62.
Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25.
Witoelar A, Jansen IE, Wang Y, Desikan RS, Gibbs JR, Blauwendraat C, Thompson WK, Hernandez DG, Djurovic S, Schork AJ, et al. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017;74:780–92.
Villumsen M, Aznar S, Pakkenberg B, Jess T, Brudek T. Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut. 2019;68:18–24.
Sun M, He C, Cong Y, Liu Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol. 2015;8:969–78.
Acknowledgements
We would like to thank Platforms for Advanced Technologies and Research Resources “Advanced Bioimaging Support” for their support (JSPS KAKENHI Grant Number JP16H06280).
Funding
This work was supported by JSPS KAKENHI Grant Number JP21K08487 to YM and JP16H06280.
Author information
Authors and Affiliations
Contributions
YM conceived the idea of the study and contributed to the design of the study. TH contributed to the design of the animal study. YM, HH, YF, and YO performed the experiments and analyzed the data. YM wrote the manuscript draft. All authors revised the manuscript draft. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were approved by the Institutional Animal Care and Use Committee of Hokkaido University and were conducted in accordance with the regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure S1. Representative images with individual trajectories of neutrophil chemotaxis to fMLP. (A) wild-type neutrophils. (B) Lrrk2 knockout neutrophils. Bar, 50 μm.
Additional file 2: Supplementary Figure S2. Representative images with individual trajectories of dHL-60 cells chemotaxis to fMLP. (A) irrelevant (Irr) shRNA expressing dHL-60 cells. (B) Lrrk2 sh1 expressing dHL-60 cells. (C) Lrrk2 sh2 expressing dHL-60 cells. Bar, 50 μm.
Additional file 3: Supplementary Figure S3. Transwell chemotaxis assays and two-dimensional chemotaxis assays of dHL-60 cells cultured without doxycycline. (A) dHL-60 cells expressing tetracycline-inducibleLrrk2 shRNA or irrelevant (Irr) shRNA were cultured without doxycycline. dHL-60 cells migration in response to 10 nM fMLP measured for 120 min in transwell chambers (n = 6 wells). The activity of Irr shRNA expressing dHL-60 cells was taken as 100%. (B, C) dHL-60 cells migration in response to 10 nM fMLP measured for 30 min in Dunn chambers (n > 20 cells). (B) Velocity (C) Directionality index. Error bars, SEM. NS, not significant.
Additional file 4: Supplementary Figure S4. Ser935 phosphorylation status of LRRK2 upon fMLP stimulation. dHL-60 cells were preincubated in HBSS containing 20 mM HEPES (pH 7.2) for 5 min at 37°C, and then stimulated with or without 1 mM fMLP at 37 °C for 5 min. (A) Representative western blotting images. (B) The rate of Ser935 phosphorylation of LRRK2 without fMLP/total LRRK2 without fMLP was taken as 1.0 (n = 4). Error bars, SEM. ** p < 0.01 compared to the no fMLP condition.
Additional file 5: Supplementary Figure S5. Representative images with individual trajectories of dHL-60 cells chemotaxis to fMLP. (A) Vehicle-treated dHL-60 cells. (B) 30 nM MLi-2 treated dHL-60 cells. Bar, 50 μm.
Additional file 6. Representative movie of chemotactic migration of dHL-60 cells treated without MLi-2. Migration of dHL-60 cells treated without MLi-2 in response to 10 nM fMLP, and individual trajectories.
Additional file 7. Representative movie of chemotactic migration of dHL-60 cells treated with 30 nM MLi-2. Migration of dHL-60 cells treated with 30 nM MLi-2 in response to 10 nM fMLP, and individual trajectories.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mazaki, Y., Handa, H., Fumoto, Y. et al. LRRK2 is involved in the chemotaxis of neutrophils and differentiated HL-60 cells, and the inhibition of LRRK2 kinase activity increases fMLP-induced chemotactic activity. Cell Commun Signal 21, 300 (2023). https://doi.org/10.1186/s12964-023-01305-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01305-y