
Abstract
The urinary tract is highly innervated by autonomic nerves which are essential in urinary tract development, the production of growth factors, and the control of homeostasis. These neural signals may become dysregulated in several genitourinary (GU) disease states, both benign and malignant. Accordingly, the autonomic nervous system is a therapeutic target for several genitourinary pathologies including cancer, voiding dysfunction, and obstructing nephrolithiasis. Adrenergic receptors (adrenoceptors) are G-Protein coupled-receptors that are distributed throughout the body. The major function of α1-adrenoceptors is signaling smooth muscle contractions through GPCR and intracellular calcium influx. Pharmacologic intervention of α-and β-adrenoceptors is routinely and successfully implemented in the treatment of benign urologic illnesses, through the use of α-adrenoceptor antagonists. Furthermore, cell-based evidence recently established the antitumor effect of α1-adrenoceptor antagonists in prostate, bladder and renal tumors by reducing neovascularity and impairing growth within the tumor microenvironment via regulation of the phenotypic epithelial-mesenchymal transition (EMT). There has been a significant focus on repurposing the routinely used, Food and Drug Administration-approved α1-adrenoceptor antagonists to inhibit GU tumor growth and angiogenesis in patients with advanced prostate, bladder, and renal cancer. In this review we discuss the current evidence on (a) the signaling events of the autonomic nervous system mediated by its cognate α- and β-adrenoceptors in regulating the phenotypic landscape (EMT) of genitourinary organs; and (b) the therapeutic significance of targeting this signaling pathway in benign and malignant urologic disease.
Video abstract
Similar content being viewed by others

Introduction
The urinary tract is highly innervated by autonomic nerves. These nerves are essential in the development of the urinary tract, the production of trophic factors, and the regulation of homeostatic function [1,2,3]. The prostate gland is higly innervated due to its unique anatomical positioning that allows for facile manipulation of sympathetic nervous system (SNS) and parasympathetic nervous system (PSNS) signals [1]. Emerging evidence suggests that these neural signals may become dysregulated in several genitourinary (GU) organs and disease states, both benign and malignant [3, 4]. The current understanding of the mechanisms underlying α1-adrenoreceptor signaling, led to the identification of therapeutic benefits of blockade of these receptors in patients with urological diseases. a1-adrenoceptor antagonists are widely established as the first treatment choice to treat men with lower urinary tract symptoms (LUTS), most commonly associated with benign prostatic hyperplasia (BPH). The mechanism of action of α-blockade is a relaxation of smooth muscles in the bladder, urethra, and prostate by α1-adrenoceptor antagonists, which results in enhanced urinary flow and decreases LUTS.
Compelling evidence from diverse investigative teams, has implicated both α-and β- adrenergic signaling exerts regulatory control on critical cellular processes contributing to human cancer including, sustained proliferative signaling, resisting anoikis, inducing cell polarity and epithelial-mesenchymal transition (EMT), promoting angiogenesis, immune system evasion, promotion of local invasion and metastasis, cancer cell energy metabolism, increased inflammation and neovascularity of the tumor microenvironment (TME).
Distribution of selective α- and β-adrenergic receptors (adrenoceptors)
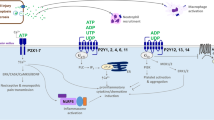
Adrenergic receptors (adrenoceptors) are G-Protein coupled-receptors that are distributed throughout the body. They serve as receptors for catecholamines (noradrenaline and epinephrine) secreted from the autonomic sympathetic nervous system and adrenal medulla and play an important role in the regulation of a wide range of diverse physiological processes in human biological systems [5]. Alpha (α) adrenoceptors mediate smooth muscle contraction and vasoconstriction, while beta (β) receptors mediate vasodilation, smooth muscle relaxation, bronchodilation, and excitatory cardiac function [6, 7]. The α-adrenoceptors are divided into α1 and α2 subtypes. The α1 adrenoceptors play an important signaling role in all vasculature (including vasculature of the urinary tract) and mediate vasoconstriction (Fig. 2). Exogenous and endogenous epinephrine and norepinephrine activate these receptors through the Gq family of G proteins, which stimulates the hydrolysis of membrane phospholipids and subsequent generation of inositol phosphate and diacylglycerol (DAG) [8,9,10]. The binding of inositol phosphate to its receptor then leads to mobilization of calcium from intracellular storage sites and subsequent smooth muscle contraction [8]. Contrary to α1-adrenoceptoors, α2 adrenoceptors respond to epinephrine and norepinephrine through the Gi family of G proteins, which inhibits adenyl cyclase and subsequently results in inhibition of norepinephrine release from presynaptic neurons. Thus α2-adrenoceptors provide negative feedback on α1-adrenoceptors. While potentially important for their functional interactions with α1 adrenoceptors, α2-adrenoceptors have not been studied for in urologic disease [11].
The α1-adrenoceptors are further sub-divided into α1A, α1B, and α1D, with the α1A subtype of therapeutic interest in urologic pathophysiology because of its location in the prostate, vas deferens, and urethra in humans [7, 12,13,14] (Fig. 2). Additional evidence demonstrated the expression of α1A in the cortex, pelvis, calyces, blood vessels and tubules of the kidney [15,16,17]. The α1A-subtype predominantly regulates smooth muscle tone in the urethra, prostate and bladder neck [18, 19]. About 70% of the prostatic adrenoceptor mRNA expression has been found to be of the α1A-subtype, though all three subtypes are found throughout the prostatic epithelial and stromal components [20]. Notably, the majority of α1D-adrenoceptors were found in the detrusor muscle of the bladder, the bladder neck, and the sacral region of the spinal cord [19, 21, 22]. Prazosin was found to have lower affinity to lower urinary tract α1A-adrenoceptors than α1-adrenoceptors in other systems of the body, and thus this particular sub-classification of α1A-adrenoceptors in the lower urinary tract have been named α1L-arenorecptors. While pharmacologically distinct, α1L-adrenoceptors are products of the α1-adrenoceptor gene, but its altered phenotype is still under investigation [23].
The α1-adrenoceptor antagonists, doxazosin, tamsulosin, terazosin and prazosin, can alleviate urinary tract obstructions (LUTS) and treat voiding dysfunction by relaxing ureter, prostatic and bladder smooth muscle [24]. Moreover, given that these α1-adrenoceptors can induce gene expression of pro-inflammatory cytokines, such as interleukin 6 (IL-6) (driven by the α1A subtype), signal transducer and activator of transcription 3 (STAT3) and glycoprotein 130 (driven by α1A and α1D), one recognizes the magnitude of potential anti-inflammatory effects by α1-adrenoceptor blockade [25]. α1-adrenoceptor antagonists may also lead to inhibition of receptor activation on urothelium, vasodilation of blood vessels, and suppression of excitatory afferent nerve signaling throughout the urinary tract [26, 27]. Specifically, prazosin and tamsulosin effectively reduce the frequency of prostatic arterial contractions in pig models [27]. Studies using in vivo pre-clinical models of rats, demonstrated that the α1D-receptor antagonist naftopidil suppressed the excitatory effects induced by bladder distension and prolonged the intercontraction interval of bladder cells [26]. Interestingly α1-adrenoceptor blockade can induce synthesis of new α2-adrenoceptors, which subsequently override the post-junctional domain and function of α1-adrenoceptor antagonists. This effect has mostly been investigated in the context of treatment of hypertension and renal vascular denervation, as α2-adrenoceptors have been found to overcome sympathetic stimulation and increase sodium excretion from renal tubules [28]. In addition to α1-adrenoceptors, β-adrenoceptors mediate many of the effects of norepinephrine released from the sympathetic nervous system and epinephrine released from the adrenal medulla through the Gs family of G proteins that activate adenylyl cyclase [5, 6, 29]. While it is established that the β1 and β2 subtypes mediate most signaling in the urinary tract, emerging evidence defines the role of the third subtype β3 in inducing smooth muscle relaxation, with potential therapeutic targeting value for cancer treatment [29]. The β2 and β3 adrenoceptors are found throughout the GU tract in the ureter, bladder, prostate, and urethra, while β1 adrenoceptors are mainly present in the ureter and bladder [29].
In the human ureter, \(\mathrm{\beta }\)-adrenoceptors are involved in ureteral relaxation, though norepinephrine appears to mainly act on α1-adrenoceptors with subsequent contraction as opposed to relaxation in vitro and in vivo [30, 31]. However, the net catecholaminergic effect may depend on the specific segment of the ureter, as the distal segments exhibit greater β-adrenoceptor effects [32]. In the bladder, β-adrenergic stimulation can reduce symptoms of overactive bladder contractions. Bladder relaxation occurs in the detrusor of the bladder, as opposed to the bladder neck, and is driven predominantly by β3-adrenoceptors [29]. While β-adrenergic stimulation may have little effect on basal prostate tone, β2-adrenoceptors may inhibit α1-adrenoceptor-independent prostate contraction (Fig. 2) [33, 34]. In vitro studies have shown that norepinephrine stimulates cell growth, and progression of autochthonous prostate cancer mouse models, which in turn can be inhibited through β-adrenoceptor antagonism [4, 35, 36].
Significance of targeting of adrenergic signaling mechanisms
α1-adrenoceptor antagonists are currently used for hypertension but have also been widely used to treat obstructing kidney stones by decreasing intra-ureteral pressure and increasing fluid passage [37]. The clinical efficacy of α1-adrenoreceptor antagonists in the treatment of benign prostatic hyperplasia (BPH) and prostate cancer has been attributed to the ability of these agents to not only reduce smooth tone and relieve overall obstruction, but to also induce apoptosis and overcome anoikis resistance [38,39,40,41,42]. The quinazoline-derived α1-antagonists induce apoptosis and overcome anoikis resistance in human castration-resistant prostate cancer (CRPC) cells and androgen-sensitive prostate cancer cells, suggesting a strong molecular rationale for the use of these compounds as anti-tumor agents [43,44,45]. Indeed rapidly expanding evidence supports the therapeutic value of these FDA-approved drugs for the treatment of prostate, bladder, colorectal, and renal cell carcinoma [38, 40, 43, 46, 47]. As schematically illustrated on Fig. 3, the mechanisms of apoptosis induction involve: 1) Smad activation of transforming growth factor (TGF)-β1 signaling, which controls cell proliferation, apoptosis and EMT in several cell types [47, 48], 2) transcription of the NF-κB inhibitor IκBα, 3) activation of the death receptor Fas-assosciated death domain (FADD)-mediated caspase-8 activation [47, 48]. The α1-adrenoceptor antagonists can impair VEGF-induced angiogenesis and target Akt survival mechanisms [47, 49]. At the cellular level, α1-adrenoceptor antagonists have the ability to:(a) block cellular adhesion and invasion by targeting cell–cell interaction and impairing cell tight junctions (and also between epithelial and endothelial cells with the extracellular matrix [ECM]); (b) functionally drive the phenotypic interconversions between epithelial-mesenchymal-transition (EMT) to mesenchymal-epithelial-transition (MET); and (c) increase cellular vulnerability to death via anoikis [49] (Fig. 3). Additional mechanistic targets have been identified involving integrin-linked kinase (ILK), a serine and threonine protein kinase, that plays a key role in anoikis resistance by interacting with the cytoplasmic domains of β1-integrin and β3-integrin, which are pivotal in regulating cell adhesion, fibronectin-ECM assembly, and anchorage-dependent cell growth [50,51,52,53,54]. Within the tumor microenvironment, ILK is activated in its phosphorylated form by focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase (PI3-kinase)/Akt pathways [50, 55, 56]. By inhibiting ILK, quinazoline-derived α1-adrenoceptor antagonists disrupt these cell-survival signals towards anoikis induction [57, 58]. As resistance to anoikis is one of the major hallmarks of metastatic cancer cells, the ability to prevent such resistance is a distinct therapeutic benefit afforded by α1 adrenoceptor antagonists.
Due to the effects in relaxing smooth muscles in the ureter and prostate, β2 and β3 adrenoceptor agonists have been implicated in targeting ureteral stones [29, 59]. Compelling mechanistic evidence suggests that β adrenoceptor antagonists exert anti-neoplastic effects by suppressing the MAPK pathway, modulating inflammation and oxidative stress, and inhibiting of nitric oxide synthase (NOS) expression.[6] The use of video-microscopy and microarray analysis demonstrated that neurotransmitters like norepinephrine can induce tumor cell migration in human prostate cancer cells. Because norepinephrine acts mostly via β2 receptors in the prostate gland, β-adrenoceptor antagonists may serve as a potential preventative measure for metastasis [60]. Clinically, the long-term usage [3,4,5] of atenolol may be beneficial in reducing the risk for prostate cancer, as compared to those not on β-adrenoceptor antagonists or those taking other β-adrenoceptor antagonists, such as carvedilol and metoprolol [61]. A systematic literature review and meta-analysis of 16,825 patients found that this β-blocker use was associated with lower cancer-specific mortality in patients with prostate cancer. Furthermore, β-adrenoceptor antagonists were reported to increase anti-angiogenic effects of chemotherapeutic regimens as well as prevent metastases in breast and ovarian cancers [62].
Adrenoceptors serve as receptors for catecholamines (noradrenaline and epinephrine) secreted from the autonomic sympathetic nervous system and play an important role in the regulation of a wide range of physiological systems in the body (Fig. 1) [5]. Though both autonomically innervated, α- and β-adrenoceptors largely mediate alternative functions. α- adrenoceptors mediate smooth muscle contraction and vasoconstriction, while β-adrenoceptors mediate vasodilation, smooth muscle relaxation, bronchodilation, and excitatory cardiac function [6, 7].

RNA expression profile of α1- adrenoceptors from a data set in 43 human tissue types. In GU organs, high expression of α1A-adrenoceptors is detected in the prostate and seminal vesicles, while urinary bladder and kidney exhibit significantly lower levels of mRNA for α1A- adrenoceptor expression
Table 1 summarizes the ubiquitous nature of α- and β‐adrenoceptor mRNA expression in the human body and their distribution in specific tissue types [63, 64]. The consequential receptor-mediated signaling in a tissue-specific pattern regulating diverse physiological processes, presents a rational targeting platform by pharmacological intervention, thus the clinically available, the Food and Drug Administration (FDA) approved α-and β-adrenoceptor antagonists [63]. In this context, comparative RNA expression analysis of adrenergic receptors in 37 tissue types from human protein atlas (HPA) tissue gene expression profiles to put forth a combined mapping of the adrenoceptors in the human body [65], revealed that human liver, adipose tissue, prostate, seminal vesicles contain the most abundant α1A adrenoceptor RNA expression (Table 1) [65]. The adrenoceptor α1D, however, was highly enriched in cervix, uterine, and prostate. The β-adrenoceptors were primarily abundant in placenta, ovary, heart, prostate, and bladder (Table 1). This in-depth comparison provide a rational for why α-adrenoceptor antagonists have been beneficial for the treatment of BPH, erectile dysfunction, and heart failure [63], while β-adrenoceptor antagonists are specifically beneficial to cardiac arrhythmia and Angina pectoris [66]. Consequently, BPH, prostatitis, and obstructing ureteral stone are effectively treated with α1-adrenoceptor antagonists [67]. Significantly, β-adrenoceptor antagonists are used for the treatment of patients with aortic aneurysm, cardiovascular abnormalities, and BPH.
Thus one may argue that the relative distribution of adrenergic receptors is unique to tissue types (Fig. 1) and in-depth analyses of the target receptors followed by their subsequent mapping may provide a rationale of targeting both α- versus β- adrenoceptors for urological diseases. This goal is benefited with the advent of novel genomic and proteomic technologies being applied in prostate and renal cancer [68]. We are entering in to a new area of large scale data analyses, which may help find better adrenergic targets for the next-generation therapeutic interventions of urological diseases [68, 69].
α1-adrenoceptor antagonists are currently used for hypertension but have also been widely used to treat kidney stones by decreasing intra-ureteral pressure and increasing fluid passage [37], BPH and more recently to suppress prostate tumor growth and vascularity [38,39,40,41,42]. Quinazoline-derived compounds are not only effective in inducing apoptosis in benign prostate epithelial cells but also in both castration-resistant prostate cancer (CRPC) cells and androgen-sensitive prostate cancer cells [43,44,45] Moreover these α1-antagonists have been found effective in the treatment of other GU tumors including, bladder cancer, colorectal cancer, and renal cell carcinoma.[38, 40, 43, 46, 47].
Growing evidence suggests the pro-apoptotic effects of quinazoline-derived α-adrenoceptor antagonists against prostate cancer epithelial and endothelial cells, mechanistically via inducing apoptosis via activation of transforming growth factor (TGF)-β1 signaling, which controls EMT and differentiation, in diverse cell types,[47, 48], transcription of the NF-κB inhibitor IκBα, and engaging the death receptor Fas-associated death domain (FADD)-mediated caspase-8 activation,[47, 48]. The action of these antagonists against tissue vascularity proceeds via inhibition of the vascular endothelial growth factor (VEGF)-induced angiogenesis and Akt survival mechanisms [47, 49]. Furthermore, α1-adrenoceptor antagonists have the ability to block adhesion, migration, and invasion due to loss of appropriate interaction between epithelial and endothelial cells with the ECM, thus increasing anoikis vulnerability [49]. Considering that resistance to anoikis is hallmark feature of the metastatic journey of cancer cells, the ability to overcome such resistance provides a distinct therapeutic benefit by α1-adrenoceptor antagonists towards impairing metastasis.
Due to the effects in relaxing smooth muscles in the ureter and prostate, the β2 and β3 adrenoceptor agonists have been found to be therapeutic in the treatment of ureteral stones; β3 adrenoceptor agonists can specifically inhibit prostatic smooth muscle contraction and reduce secondary to BPH symptoms [29, 59]. Pharmacologic targeting of β-adrenoceptor signaling results in tumor suppression by inhibiting the MAPK signaling pathway, modulating inflammation and oxidative stress [6] (Fig. 3). The use of video-microscopy and microarray analysis demonstrated that neurotransmitters like norepinephrine induces tumor cell migration in prostate cancer cells, supporting the use of β-blockers as a preventative measure for prostate cancer metastasis [60]. Clinically, the long-term usage [3,4,5] of atenolol may be beneficial in reducing the risk for prostate cancer, compared to those not on β-adrenoceptor antagonists or those taking other β-adrenoceptor antagonists, such as carvedilol and metoprolol [61]. A systematic literature review and meta-analysis of 16,825 patients found that exposure to β-adrenoceptor antagonists was associated with lower cancer-specific mortality in patients with prostate cancer. Significantly, β-adrenoceptor antagonists can enhance the anti-angiogenic effects of chemotherapeutic regimens and prevent metastases in breast and ovarian cancers [62].
Pharmacologic targeting of adrenergic signaling in benign urologic disease
Pharmacologic targeting of α- and β-adrenoceptors is clinically applied in the treatment of benign urologic illnesses. Most commonly, α-adrenoceptor antagonists are used in the treatment of benign prostatic hyperplasia (BPH). As men age, the prostate undergoes benign growth which may obstruct the flow of urine or alter bladder dynamics leading to lower urinary tract symptoms (LUTS) [70, 71]. α1A adrenoceptors are the most common subtype within the prostate functioning to facilitate smooth muscle tone[72] (Fig. 2). The α1-adrenoceptor antagonists target this receptor signaling, resulting in prostatic smooth muscle relaxation, which alleviate prostatic obstruction and improve urinary flow [73]. Tamsulosin, a selective α1A adrenoceptor antagonist [74], is employed as a first line pharmacotherapy for LUTS in men and is one of the most commonly prescribed medications in the US [75].

α- and β- adrenoceptor Expression and Distribution in the Urinary Tract. Schematic representation of the location and distribution of α and β -adrenoceptors and their subtypes in the bladder, prostate gland and urethra (a), and the kidney and ureter (b). The α-adrenoceptors are divided into α1 and α2 subtypes. The α1 receptors are important receptors in the urinary tract vasculature via their ability to mediate vasoconstriction. Exogenous and endogenous epinephrine and norepinephrine activate these receptors through the Gq family of G proteins, which stimulates the hydrolysis of membrane phospholipids and subsequent generation of inositol phosphate and diacylglycerol (DAG). α and β-adrenoceptors respond to signals from catecholamines (noradrenaline and epinephrine) secreted from the autonomic sympathetic nervous system through the hypogastric and renal nerves to regulate physiological functions and cellular processes. (Insert, color-coding key illustrating the various adrenoceptor subtypes)
Αlpha (α)-adrenoceptors are also found in the bladder and similarly facilitate smooth muscle tone [73, 76]. Regarding voiding function, relaxation of the bladder’s detrusor muscle has two opposing functions depending on anatomic location. The most common α1adrenoceptor subtype in the bladder is the α1d adrenoceptor [76], making for a therapeutic target in patients with storage-type LUTS. Indeed, naftopidil, a selective α1d receptor antagonist, has shown efficacy in improving urinary storage symptoms [77]. The use of α-blockers for LUTS symptoms has been associated with ejaculatory dysfunction believed to be secondary to a reduction in seminal fluid emission due to central nervous system sympathetic inhibition [78].
In addition to the treatment of voiding dysfunction, α1-adrenoceptor antagonists are routinely used in the management of kidney stone disease. As discussed earlier α-adrenoceptors are expressed throughout the ureter and the lower urinary tract, functionally modulating smooth muscle tone [79, 80]. The use of α-adrenoceptor antagonists to relax smooth muscle within the ureter in patients with obstructing ureteral stones may increase luminal diameter and facilitate spontaneous stone passage (Fig. 2). The distal ureter has a greater muscular component and greater α-receptor expression than the proximal ureter [79, 80]. Accordingly, α1-blockade is most clinically beneficial for medical expulsive therapy in patients with obstructing distal ureteral stones [81]. Similarly important is the use of α-adrenoceptor antagonists for the treatment of ureteral stent colic. Ureteral stents are regularly used to alleviate upper urinary tract obstruction; this approach however is often associated with severe discomfort causing a condition known as “stent colic.” Αlpha-adrenoceptor antagonists have been shown to alleviate symptoms of stent colic, likely secondary to their regulatory (relaxation) effect on the ureteral smooth muscle [82]. The most common receptor subtype within the human ureter is the α1a adrenoceptor, making tamsulosin the α1-antagonist of choice for the treatment of stent colic [80].
Beta-3 sympathetic receptors are expressed in the detrusor muscle of the human bladder and are a pharmacologic target for the treatment of overactive bladder (OAB) [83]. Historically, the mainstay of pharmacotherapy for OAB was anticholinergic medications which indirectly increased relative sympathetic tone by blunting parasympathetic nervous system input via antagonism of muscarinic receptors [84]. However, anticholinergic medications have several side effects such as dry mouth, dry eyes, and constipation and have poor long-term adherence for many patients [84, 85]. Mirabegron, a medication approved a decade ago by the FDA, functions as a β3 selective adrenoceptor agonist that promotes detrusor relaxation and in turn alleviates symptoms of OAB [86]. β3 adrenoceptors are a common subtype within the human bladder [87] and the selective nature of mirabegron may explain the greater tolerability of mirabegron when compared to anticholinergics in patients with OAB [88]. As our understanding of sympathetic receptor expression within the urinary tract continues to evolve, our therapeutic armamentarium for the treatment of benign urologic disease will likely include β-blockade.
In the kidney, the expression and distribution of α1-adrenoreceptors are in the cortex, pelvis, calyces, blood vessels and renal tubules, suggesting potential effects of α1 adrenoceptor antagonists in renal pathophysiology [89,90,91]. The α1- and α2-adrenoceptors are present in the renal vasculature and mediate vasoconstriction of exogenous and endogenous noradrenaline [92].
Adrenoceptors are involved in both acute kidney injury (AKI) and chronic kidney disease (CKD) pathogenesis. Modulation of either α- or β-adrenoceptors protects renal ischemia/reperfusion injury (IRI) and nephrotoxic AKIs [93, 94], suggesting fine-tuning of its activity as a promising target to develop effective treatment for these clinical entities. α2-adrenoceptor blockers, yohimbine and atipamezole, and JP-1302, an α2C-adrenoceptor specific inhibitor, attenuate renal tubular damage in rat models of renal mass reduction and in mice subjected to unilateral IRI [95, 96]. In sharp contrast, activation of α2A-adrenoceptor using α2A-adrenoceptor agonists, clonidine and moxonidine inhibit activation of sympathetic nerves, resulting in protection against IRI, in unilateral IRI rabbits and rats that removed the contralateral kidney [97, 98], suggesting injury-dependent mechanism of α2-adrenoceptor antagonism. In the cisplatin AKI, yohimbine and JP-1302, α2-adrenoceptor antagonists, or nebivolol, β1-adrenoceptor antagonist, suppress renal tubular damage and dysfunction through reduction of plasma norepinephrine (NE) levels and inhibition of pro-inflammatory cytokine expression [99, 100].
A number of clinical and experimental studies suggest that maladaptive recovery from AKI predisposes to CKD progression [101,102,103]. A recent meta-analysis revealed that hospitalized patients experiencing AKI have a higher risk for CKD and cardiovascular complication [101]. Experimental studies from our group and other investigators, demonstrated that blockade of either β-adrenoceptor by propranolol, or α2-adrenoceptor by atipamezole, or α2C-adrenoceptor subtype by JP-1302 prevents long-term sequelae of ischemic AKI, including chronic inflammation and fibrosis [95, 104, 105]. Pharmacological inhibition of α2-adrenoceptor subtype(s) markedly suppressed unilateral ureteral obstruction (UUO)- or IRI-induced kidney fibrosis, along with reduced tubular injury and inflammation [95, 106]. In a CKD model of subtotal nephrectomy, inhibition of either α1-adrenoceptor or β-adrenoceptor mitigates the renal injury, and a combinational inhibition of α1- and β-adrenoceptors shows additive beneficial effects in renal injury [107].
Blockade using doxazosin, an α1-adrenoceptor antagonist, and atenolol, a β1-adrenoceptor antagonist blunts obesity-associated hypertension and increase of heart rate in both clinical and experimental studies [89,90,91, 108]. Diabetic kidney disease is the main cause of CKD and end-stage kidney disease, commonly accompanied by hypertension and additional cardiovascular complications [109, 110]. Beta (β)-blockers attenuate albuminuria, hypertension, and cardiovascular complications in patients with diabetic nephropathy [92]. β1‐cardioselective adrenoceptor antagonists, metoprolol and atenolol, recommended as antihypertensive agents in CKD or ESKD patients with hypertension, effectively lower blood pressure (BP) without significant decline of glomerular filtration rate (GFR) and renal blood flow (RBF) [92, 111,112,113]. Peripherally acting α-blockade against vascularity of vessel walls can be used either as a part of combination therapy or first-line therapy for the management of CKD and hypertension [114,115,116].
In kidney diseases, the beneficial effect of adrenoceptor antagonists is most likely associated with suppression of renal inflammation, tubular cell cycle arrest, cell death, and fibrosis. Upon kidney injury, inflammatory cells including monocytes/macrophages are recruited into the site of injury and secrete proinflammatory/fibrogenic cytokines, resulting in persistent tubular injury and fibrosis [117]. Monocytes, macrophages as well as renal tubular cells expresses most adrenoceptor subtypes, including α2-adrenoceptor. Activation of α2-adenoceptor in macrophage upregulates pro-inflammatory cytokines like tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), while that of β2-adrenoceptor confers anti-inflammatory response [118]. Inhibition of α2-adrenoceptors prevented interstitial fibrosis in IRI and UUO models, along with reduction of profibrotic factors, TGF-β1 and Smad3, and thus suppressing myofibroblast activation and collagen deposition [95, 106]. In line with our results, α2-AR agonist upregulates TNF-α in macrophages [119], accelerates mesenchymal cell apoptosis [120] and triggers cell cycle arrest in oligodendrocyte progenitors [121]. Contrary to the results from kidney, blockade of α2A-adrenoceptor and α2C-adrenoceptor subtypes increases the susceptibility to develop heart failure after chronic pressure overload in mice [122, 123], suggesting tissue-dependent role of α2-adrenoceptors.
Another potential mechanism for α-adrenoceptor antagonism in the AKI and CKD model is associated with nitric oxide (NO) availability [124]. In the kidney, NO has numerous physiological roles including the modulation of renal sympathetic nerve activity [125, 126]. Renal NO synthase activity is reduced in both AKI and CKD [127,128,129]. Downregulation of NO synthesis are prevented by prior administration of an α2-adrenoceptor antagonist [130, 131] Administration of an α2-adrenoceptor agonist, B-TH933 to the denervated kidney restores both glomerular and tubular injury due to inhibition of NO [132]. Decline of NOS activity by L-NAME, a non-selective inhibitor of NOS, was significantly restored by quinazoline-based α1-adrenoceptors antagonists, prazosin, or doxazosin, and mitigated the renal injury [133,134,135]. Furthermore, it was also shown that β-adrenoceptor antagonists induce relaxation of renal vascular and glomerular endothelial cells [136, 137], while selective β-adrenoceptor blockers, such as carvedilol and nebivolol, protect gentamycin-induced nephrotoxicity, nephrectomy-induced CKD, or hypertensive CKD through upregulation of NO release [138,139,140,141].
The renin-angiotensin system (RAS) interacts with renal sympathetic nerve-derived signaling [142]. Renin that catalyzes cleavage of angiotensin (Ang) to Ang II release from the juxtaglomerular apparatus of the kidney and acts as a determinant of renal damage [89, 91]. Conversely, Ang II can enhance NE level by acting on sympathetic nerve terminals [143, 144]. Moreover, genetic inhibition of α2A-adrenoceptor or pharmacological inhibition of α2-adrenoceptor attenuates Ang II-mediated norepinephrine release in isolated kidneys with nephrectomy-induced CKD [145]. However, exploitation of the α-adrenoceptor signaling in the regulation of intrarenal RAS directly in benign kidney disease is ongoing.
Significance of adrenergic signaling in phenotypic landscaping of the microenvironment
Pharmacologic targeting of α1-adrenoceptors in GU malignancies may proceed through induction of apoptosis. Quinazoline-based α1-adrenoceptor antagonists have been found to suppress growth of androgen-dependent and castration resistant prostate cancer [146] as well as renal cancer cells through induction of the extrinsic mechanism of apoptosis [45, 147]. As illustrated on Fig. 3, α-adrenoceptor antagonists can dictate the canonical TGF-β receptor signaling mechanism to induce apoptosis and EMT [43,44,45, 148]. Other α-adrenoceptor (quinazoline-based) antagonists including prazosin promote apoptotic activity by inducing cell cycle arrest as a result of DNA strand breaks, phosphorylation of cyclin-dependent kinase (CDK) causing CDK1 inactivation and G2 checkpoint arrest [149]. Novel quinazoline-based α-antagonists, (DZ-50) have been developed by our group, impair primary growth and progression of renal and prostate cancer to metastasis by inhibiting focal adhesion kinase (FAK) phosphorylation and protein kinase B (AKT) signaling pathways by inducing anoikis. This is consequential to the disruution of adhesion to the ECM via targeting the integrin β1 focal adhesion complexes (Fig. 3) [47, 150]. Further mechanistic evidence suggests that piperazine based α1-adrenoceptor antagonists may have an anti-tumor effect on cancer cells in vitro and in vivo. Naftopidil induced apoptosis in bladder cancer cell lines and xenograft tumors by activation of the caspase-3 cascade [151]. In contrast, sulfonamide-based α-adrenoceptor antagonists (tamsulosin) failed to induce apoptosis in prostate cancer cells suggesting that the chemical structure of α-adrenoceptor antagonists is functionally an important factor driving the apoptotic action of the drugs [45].

Molecular regulation of the phenotypic landscape by targeting α- and β-adrenoceptor signaling pathways. Top panel, Mechanistic signaling of α- adrenoceptor signaling and β-adrenoceptor eliciting effects on phenotypic EMT, apoptosis and cell proliferation. The α1-adrenoceptor signaling, activates the survival pathways NFκB and MAPK inducing transcriptional activation of growth genes (fos, myc). In response to α1-adrenoceptor pharmacologic targeting, in addition to blocking the α-adrenoceptor pathway, there is also inhibition of the TGF-β/ TGF-β receptor RI/RII/Smad signaling that induces apoptosis as well as phenotypic EMT. In response to β-adrenoceptor signaling there is activation of protein kinase A mechanism (though cAMP), promoting cell proliferation and survival via CREB activation and Bcl-2 overexpression, respectively. Lower panel, Impact of pharmacologic targeting of α1-adrenoeptor antagonists of the phenotypic interconversions (mesenchymal-EMT to epithelial-MET) within the tumor microenvironment landscape via regulation E-cadherin, IGFBP-3 and vimentin. Upon detachment from the ECM, cells unable to enter the phenotypic interconversion cycle mediated by TGF-β undergo anoikis. This provides the molecular targeting platform for α-adrenoceptor antagonists in tumor cells in the various GU organs, including the prostate and kidney
In human androgen-sensitive prostate cancer cells, that continuous exposure to catecholamine epinephrine is protective against apoptosis [152]. Through binding of agonists to β-adrenoceptor, there is upregulation of intracellular cyclic adenosine monophosphate (cAMP) to bind to PKA, leading to the phosphorylation of the cAMP responsive element binding protein (CREB) [153]. Stimulation of CREB activity induces expression of survival protein B-cell lymphoma 2 (Bcl-2) [153, 154]. The cAMP/PKA signaling further increases resistance to apoptosis by inhibition of the Bcl-2-associated death promotor (BAD) in in vitro and in vivo models of prostate cancer [153,154,155]. While the apoptotic effect of β-adrenoceptor antagonists has not been investigated in urologic tumors, there is evidence suggesting the ability of β-adrenoceptor antagonists, such as propranolol, to induce apoptosis in other cancers. Studies utilizing liver cancer and melanoma cell lines demonstrated that treatment with propranolol induced cell cycle arrest and apoptosis [156, 157]. In late stage breast cancer patients, neoadjuvant treatment with propranolol decreased the expression of survival protein Bcl-2 and increased tumor suppressor P53 in tumor specimens [158].
The adrenergic connection to angiogenesis and tumor vascularity has been defined at both the α- and β-adrenoceptor level. Angiogenesis under normal physiological conditions is primarily involved in the ovarian cycle and in wound healing and repair, existing only transiently in adults as their complete and mature vasculature is derived from the network of blood vessels formed during embryonic development [153, 159]. Angiogenesis supports tumor growth by establishing a network of blood vessels that provide nutrients and oxygen as well as waste removal to cells within the tumor microenvironment [160]. Adrenergic nerves are situated closely to the arterioles and capillaries within the stromal component of tissues, thus are ideally positioned to contribute to vasculature stimulation [1]. Angiogenesis occurs when the balance between pro- and anti-angiogenic factors is tipped in favor of supporting growth, an event known as the “angiogenic switch” [4, 161]. A hypoxic environment is created that stimulates transcriptional activity of hypoxia-inducible factor 1 (HIF1), driving transcription of pro-angiogenic genes [160]. Prominent pro-angiogenic factors include members of the VEGF family, transforming growth factors (TGF)-α and TGF-β, TNF-α, platelet-derived endothelial growth factor and fibroblast growth factor (FGF) [161, 162]. Inhibitors of angiogenesis include angiotensin, plasminogen activator-inhibitor-1, and several cytokines and metalloproteases [161]. β-Adrenoceptor signaling contributes significantly to enhanced tissue vascularity via activating the above pathways and promoters of anhiogenesis in human solid cancers, including GU tumors [162].
The human prostate gland is highly innervated [154], and is ideal as a model to study the coontribution of innervation to cancer development, as it is positioned fully enabling for facile manipulation of SNS and PSNS signaling [1]. Furthering this notion is the fact that the majority of prostate tumors emerge from the peripheral zone, a region that contains the highest abundance of nerves for the organ [154]. Within the TME, β-adrenoceptors are stimulated via adrenergic signals delivered by sympathetic nerves (Fig. 1.) [4]. Seminal work by Zahalka et al. First established that the expression of β2-adrenoceptors within prostate epithelial cells is necessary and critical to maintaining them at the proper metabolic state (i.e. performing aerobic glycolysis) essential for angiogenesis [4]. Orthotopic implantation of human prostate cancer xenografts (derived from PC-3 cells) revealed that xenografts grown in mice expressing WT β2-adrenoceptor grew exponentially post-day 18 compared to xenografts implanted in β2-adrenoceptor and β3-adrenoceptor-knockout (KO) mice [4]. Furthermore when sympathectomy or conditional cell-specific deletion of β2-adrenoceptor in endothelial cells was performed, angiogenesis was inhibited via upregulation of cytochrome C oxidase assembly factor 6 (Coa6), a protein involved in the electron transport chain and oxidative phosphorylation [1, 4]. Recent insights into the functional role the β2-adrenoceptor plays in angiogenesis in prostate cancer revealed a dose-dependent increase in VEGF expression in LNCaP human prostate cancer cells, upon stimulation of β2-adrenoceptor via epinephrine [154]. In an independent study, Park et al. observed similar upregulation in VEGF expression levels, as well as expression of hypoxia-inducible factor (HIF)-1α in androgen-independent human prostate cancer cells PC-3, treated with norepinephrine and isoproterenol, an effect that was mechanistically mediated through increased activity of the cAMP/PKA pathway [154, 163]. Conditioned media from prostate cancer cells exposed to norepinephrine lead to capillary tube formation [164], while in vivo treatment of rats with propranolol significantly reduced the ventral prostate volume [154, 164].
Adrenergic signaling controls epithelial mesenchymal transition (EMT) and cell polarity
A rapidly expanding body of work has shed the light on the molecular and cellular mechanisms involved in the cross-talk between α- and β-adrenergic signaling and EMT [41, 165]. Phenotypic EMT is a tightly regulated cellular process directed by TGF-β signaling and where cancer cells adopt migratory and invasive properties including loss of intercellular junctions, anchorage-independent survival, and metalloprotease-dependent matrix degradation [166]. EMT has been recognized as an important functional contributor the increased number of myofibroblasts in cancer and fibrotic diseases. Renal sympathetic nerve activity is elevated in patients and pre-clinical CKD models and contributes to renal interstitial fibrosis in obstructive nephropathy. The underlying mechanisms of sympathetic overactivation in renal fibrosis remain revealed that norepinephrine (NE), the main sympathetic neurotransmitter, promotes TGF-β1-induced EMT. Of direct translational significance is recent evidence on the impact of pharmacological targeting of α1-adrenoceptors on reducing the kidney injury and renal fibrosis in a unilateral ureteral obstruction pre-clinical model by suppressing EMT in the kidneys [167]. Thus, sympathetic overactivation facilitates EMT of renal epithelial cells and fibrosis via the α1-adrenoceptor driven TGF-β signaling mechanism, inhibition of which by an α1-adrenoceptor antagonist provides a promising approach for the treatment of renal fibrosis (Fig. 3).
EMT has been shown to play a role in airway remodeling induced by eosinophils. Procaterol, a selective and β2-adrenoceptor agonist, clinically for asthmatic attacks as a controller, was recently shown to functionally suppress EMT of airway epithelial cells induced by eosinophils [168]. Mechanistic studies revealed that Butoxamine, a specific β2-adrenoceptor antagonist, significantly abrogated changes induced by procaterol. In addition, procaterol inhibited expression of adhesion molecules induced during the interaction between eosinophils and bronchial epithelial cells, supporting the involvement of adhesion molecules in EMT. Procaterol significantly inhibited in vitro associated morphological changes of bronchial epithelial cells, decreased vimentin, and increased E-cadherin expression [168].
Released by sympathetic nerves or through leakage from vascular neuromuscular junctions, catecholamines are found in high concentrations within the TME. Through binding of β-adrenoceptors on tumor cells, catecholamines have been shown to promote integrin-dependent cell adhesion, calcium-dependent cytoskeleton remodeling, formation of invasive protrusions (invadopodia) and release of ECM-degrading proteases [169,170,171,172,173,174,175,176]. While the functional role of β-adrenoceptor signaling has been investigated in cancer, less is known about the pro-tumorigenic role of α-adrenoceptors. Recently, preclinical and clinical data have supported the antitumor action of α-adrenoceptor antagonists on bladder cancer growth and migration and extended progression-free survival in bladder cancer patients [177]. Pharmacologic blockade of α-adrenoceptor signaling is capable of reversing EMT to MET by targeting insulin-like growth factor binding protein-3 (IGFBP3) and the TGF-β induced of transcriptional activation of EMT-associated regulators [178]. (Fig. 3).
A growing number of studies indicated that the β2-adrenoceptor mediated signaling pathway contributes to tumor initiation and progression in human solid cancers, including in breast cancer [179], melanoma, prostate cancer [154], gastric cancer [180], and tongue squamous cell carcinoma (TSCC) [181]. The β2-adrenoceptor is a transmembrane, G protein-coupled receptor required for the response to adrenaline and noradrenaline. As shown on Fig. 3, activation of β2-adrenoceptor signaling can be functionally involved in EMT, contributing to invasion and metastasis in colon and gastric cancer and its deregulation in TSCC confirmed a role in EMT and TSCC tumor progression [181, 182].
More recently, our team provided new evidence suggesting the anti-tumor effects of the α-adrenoceptor blockade through modifications of the EMT phenotypic landscape within the tumor microenvironment. Molecular profiling identified the EMT effector, IGFBP-3 as the primary target for DZ-50, a novel quinazoline derivative of the α-adrenoceptor antagonist doxazosin [178]. Treatment of prostate and renal cancer cells with this new compound downregulates nuclear IGFBP-3, through TGF-β, consequently leading to the phenotypic re-differentiation of prostate cancer cells from EMT to MET and ultimately overcoming therapeutic resistance through lifting anoikis resistance [178, 183]. IGFBP-3 holds major clinical significance, as its expression is predictive of prostate cancer progression, and these studies provide the first evidence for IGFBP-3 as a target to treat therapeutically resistant prostate cancer [183]. (Fig. 3). Further studies aim to explore the role of the α-blockade and EMT reversal through IGFBP-3 in optimizing treatment strategies in both urologic tumors and benign disease.
Catecholamines can also contribute to the metastatic journey within the genitourinary tumor microenvironment of the primary tumor localization and or distant site. In prostate cancer, β-adrenoceptor signaling in osteoblasts promotes the secretion of CXCL12 and binding to CXCR4 on surface of prostate cancer cells resulting in increases bone metastasis formation [184]. Tumor-associated endothelial cells are also sensitive to catecholamines [4, 49, 172], consequently implicating targeting of tumor angiogenesis and metastatic spread by β-adrenoceptor antagonists. Recent evidence found that β-adrenoceptor signaling promotes growth of tumor-associated endothelial cells via increasing tumor cell access to oxygen and nutrients [4, 172]. Exposure of endothelial cells to the α1-adrenoceptor antagonist doxazosin led to significant inhibition of angiogenic properties (based on endothelial tube formation) [49]. Taken together these lines of evidence support the concept that the antiangiogenic activity of α- and β-adrenoceptor antagonists may prevent tumor progression and dissemination via reduced tumor perfusion and neovascularization.
While the pro-metastatic role of α- and β-adrenoceptors is still under intense investigation, it is not universal but rather disease- or cell-type specific [185,186,187,188,189]. As shown on Fig. 3, in the presence of agonist or hormone binding, β-adrenoceptor upregulates PKA activity, which in return, abolishes ERK signaling cascade and ultimately cancer cell proliferation and migration [185, 186]. A more recent study underscored the antitumor activity of α2A-aadrenoceptor in cervical cancer and suppression of cancer cell migration and invasion through inhibition of the PI3K/Akt/mTOR survival pathway [189]. The β-adrenoceptor antagonists are commonly used to treat hypertension and recent findings on the role of adrenergic signaling in tumor progression have stimulated drug repurposing of β-adrenoceptor antagonists as anticancer therapies. Several retrospective studies analyzed the outcome of breast cancer patients treated with β-adrenoceptor antagonists for hypertension [190,191,192,193,194,195]. Overall, these clinical studies highlight that use of β-adrenoceptor antagonists are associated with prolonged recurrence-free survival and overall survival in breast cancer patients. Recently, a phase II randomized trial is being conducted to evaluate the anti-metastatic properties of β-adrenoceptor antagonists (propranolol) in patients undergoing surgical resection of primary breast cancer [196]. Transcriptomic profiling of resected tumors treated with propranolol showed reduced expression of genes functionally involved in TGF-β-induced EMT including Snail, Slug and Smad (Fig. 3) [196, 197].
Clinical landscape of targeting of adrenoceptor signaling in GU cancers
Prostate cancer
Experimental studies in cell-based and preclinical models have reported that (with a quinazoline chemical structure) α-adrenoceptor antagonists induce apoptosis among the tumor epithelial cells, leading to significant suppression of prostate tumor growth, indicating that these medications may have therapeutic value in targeting prostate tumor dynamics for the treatment of prostate cancer patients with advanced disease [45].
The current clinical evidence on prostate cancer incidence in men treated with α-adrenoceptor antagonists for LUTS is conflicting. The lack of randomized trials in the area raises concerns in interpreting evidence, given the risk for detection bias caused by increased symptom-related care-contacts. Several large non-randomized studies have evaluated the incidence of prostate cancer in men treated with α-blockers due to LUTS. In a nationwide population-based case–control study in men diagnosed with prostate cancer from Sweden, evaluating 26 735 cases and 133 671 matched controls, results showed that men using α-adrenoceptor antagonists had an overall increased risk of prostate cancer (OR = 1.33, 95% CI = 1.27 to 1.39). [198]. However, the increased risk was only seen in men with Gleason Score 6–7, not in men with Gleason Score 8–10. The same pattern was observed in a nationwide cohort study of 3,009,258 men from Denmark [199]. Additionally, a Finnish prostate cancer screening trial showed no significant difference in the overall risk of prostate cancer incidence among those patients exposed to α-adrenoceptor antagonists was detected [200]. There was however a lower incidence of men with high-grade tumors in α-adrenoceptor users compared to non-users.
Epidemiological evidence derived from a larger retrospective cohort study of 249 986 men, of whom 7 764 were α-adrenoceptor antagonists, also showed a higher risk of diagnosis with Gleason Score 8–10 cancer in men exposed to α-blockade compared with those who were not; but an 11% lower risk of getting diagnosed with prostate cancer. However, the increased risk of high-grade prostate cancer in α-blocker users compared with non-users was not translated into a higher incidence of prostate cancer mortality [201]. Similarly, Sarkar et al. reported no effect on prostate cancer-specific mortality in men treated with α-adrenoceptor antagonists in their population-based cohort study of 80 875 men with stage I-IV prostate cancer within the Veterans Affairs health care system. Their data also contained information on α-blocker use, and their results show that 12-year cumulative incidence of prostate cancer -specific mortality and all-cause mortality was higher in 5α-reductase inhibitors (5-ARI) users, but no significant difference was seen in men treated with α-adrenoceptor antagonists. Further analyses showed that pre-diagnostic use of α-adrenoceptor antagonists was associated with a delayed diagnosis, which might explain the difference seen in mortality, again emphasizing the risk for detection bias [202].
We recognize that we must proceed with caution in the interpretation of the current evidence on prostate cancer incidence in men treated with α-adrenoceptor antagonists especially concerning causality due to the considerable risk for detection bias. It is also essential to recognize that prostate cancer rarely presents with obstructive voiding symptoms, decreasing the risk of missing the diagnosis due to the drug masking the symptoms [203]. Additionally, unlike 5-ARIs that suppress prostate specific antigen (PSA) by 50% after 3 months of use, there is no clinical evidence that α-blockers decrease PSA, which would reduce the risk of a delayed diagnosis. Moreover, Orsted et al. and Sarkar et al. performed studies to evaluate the effect of 5-ARI use and not α-adrenoceptor antagonists, which may introduce bias in the methodology [199, 202]. The current non-randomized evidence shows an overall non-significant effect of α-adrenoceptor antagonist use on both incidence and mortality, but the results in different risk-groups are conflicting. Ongoing retrospective studies and randomized trials are designed to evaluate the impact of α-adrenoceptor antagonists on prostate cancer incidence and mortality.
Bladder cancer
On the basis of recent studies, manipulation of the adrenergic system for the purposes of therapeutic targeting has shown promise for the systemic treatment for bladder cancer. Current evidence has focused mainly on pharmacologic blockade of either α- or β-adrenergic receptors with in vivo and in vitro studies in pre-clinical models.
The quinazoline based α-adrenoreceptor antagonist group of drugs have been shown to have potential efficacy in both prevention and treatment of bladder cancer in patients. A retrospective observational study of 27,183 male patients in Kentucky linked to the SEER program of the NCI, was able to compare bladder cancer incidence in those exposed to quinazolone α1 blockade vs those not receiving this treatment [204]. Analysis of cumulative bladder cancer incidence was 0.24% in the α-blocker exposed vs 0.42% in the α-blocker unexposed group. This indicates that the number of men diagnosed with bladder cancer was 1.8 less per 1000 men in the exposed group, with a relative risk reduction of 43% for those exposed to alpha blockers [204]. Examination of bladder tumor tissue specimens post radical cystectomy using immunohistochemical methods to detect microvessel density and apoptosis, revealed that previous long-term exposure of patients to quinazolone α1 adrenoceptor, terazosin, had reduced tumor vascularity and increased apoptosis. Despite the obvious limitation of having only a small number of tumor specimens examined [9 cystectomy samples exposed vs 13 cystectomy samples unexposed to terazosin], this is consistent with the established apoptotic effect of terazosin on prostate cancer clinical specimens [205]. Similarly, in vitro studies assessing cell viability assays in human bladder cancer cell lines, found prazosin at concentrations of greater than 30microml/l was able to reduce cell viability and induce apoptosis [40]. In the same experiment, naftopidil, a piperazine based α1-adrenoceptor antagonist which is highly selective for α-1A and α-1D receptor subtypes (Fig. 2), was able to have the stronger effect in reducing bladder cancer cell viability at concentrations of 10-100microml/l [40]. Other studies have also showed orally administered naftopidil was able to reduce bladder tumor volume in mouse xenografts models via the activation of apoptosis [151]. In an in vitro study, doxazosin, a quinazoline based α-adrenoceptor antagonist, was able to inhibit the growth of HT137 bladder cancer cells, an effect that was reduced by pre-treatment with 5-hydroxytryptamine (5-HT, serotonin). This was thought to be due to the similar structure of adrenergic and 5-HT receptors [38, 206]. Interestingly, the expression of ELK1, a marker for tumor progression post radical cystectomy for bladder cancer, is inhibited by Silodosin, which also inhibits ELK1 bladder cancer cell survival and migration [207]. Based on this cell-derived evidence, Silodosin was proposed to potentiate the efficacy of cisplatin chemotherapy for patients with ELK1 bladder tumors, who have developed chemoresistance [38, 207].
Our current understanding of the neurobiology of cancer supports that β- adrenergic signaling influences tumorigenesis not only by landscaping the tissue phenotype within TME, but also impairing DNA repair mechanisms facilitating genomic instability [162]. Neural progenitor cells and sensory neurons can differentiate into adrenergic cells in the TME as a source of catecholamines [208, 209]. Further in vitro studies using murine cells pretreated with catecholamines identified a significantly increased DNA damage, an effect that was prevented by propranolol [210]. Growing evidence from rigorous molecular work has shown β-adrenergic signaling exerts a significant regulatory control on several cancer-promoting cellular processes including sustained proliferative signaling, resisting cell death, inducing cell migration, angiogenesis, immune evasion, inflammation, metabolic energy and TME landscape dynamics, all critical contributors to metastasis [162, 211]. Molecular profiling of bladder cancer pathobiology, has implicated the repurposing of propranolol as an anti-cancer agent with high therapeutic value in advanced stage tumors [212]. Moreover, there are trials due to start recruiting directly assessing propranolol as an adjuvant agent in combination with BCG for high risk non muscle invasive bladder cancer, assessing long term disease recurrence and progression and survival as outcomes (https://clinicaltrials.gov/ct2/show/NCT04493489).
Renal cancer
The most common genetic abnormality for clear cell RCC (ccRCC) is the chromosome 3p deletion and inactivation of the von Hippel Lindau (VHL) tumor suppressor gene, present in almost all familial and up to 60% of sporadic RCCs [47, 213]. Loss of the VHL gene leads to the upregulation of hypoxia-inducible factor (HIF) and activation of vascular endothelial growth factor receptors (VEGFR), leading to tumorigenesis with an aggressive angiogenic phenotype [47, 213]. For patients with advanced localized renal cancer treatment with adjuvant VEGFR TKI sunitinib is approved by the FDA, although the modest clinical benefit and concern for the potential side effects has largely limited its clinical application to date [214]. For metastatic ccRCC, a number of approaches combining immune checkpoint inhibitors or with VEGFR-TKIs have now become the standard of care after demonstrating a definitive survival benefit in the first-line setting compared to sunitinib alone [215]. The current systemic therapeutic approaches include: targeting pathways of angiogenesis, immune checkpoint blockade, and mTOR inhibition. By inducing smooth muscle relaxation and vasodilation, these drugs are used for the treatment of hypertension (HTN) and obstructive symptoms associated with BPH (as discussed above) [216, 217]. This mechanism of action is also utilized in the treatment of renal and ureteric stones, as α1 blockers reduce intra-ureteral pressure and increase fluid passage [216].
As we discussed earlier adrenoceptor signaling has a well-established role in renal physiology. Immunohistochemical profiling shows the presence of both α- and β-adrenoceptors in the majority of kidney structures and cell types [215, 218]. Similar results are confirmed through in situ hybridization measurement of α- and β-adrenoceptor mRNA expression levels [219] (summarized on Fig. 1). These receptors play a role in many vital renal functions, rationalizing the presence of aberrant adrenoceptor signaling in a host of kidney diseases such as diabetic nephropathy and acute kidney injury [220, 221]. A molecular and immunohistochemical expression profile of β-adrenoceptors across a panel of different cancer types, revealed increased expression in clear cell RCC relative to normal kidney tissue [222]. At the translational setting, abnormal α- and β-adrenoceptor signaling is functionally linked to the pathophysiology of clinical hypertension, while epidemiological evidence has established a clear association between hypertension and RCC [223]. This provides a strong rationale supporting that modulation of adrenoceptor signaling using an α- or β-adrenoceptor antagonist may have therapeutic impact in RCC (Fig. 2).
Despite the widespread use of α-adrenoceptor antagonists among patients with RCC for comorbid conditions, direct analyses of the effect of these medications in RCC are limited to preclinical and observational studies. Cell-based studies support the anti-cancer effect of these drugs. Doxazosin and naftodipil, selective α1-adrenoceptor antagonists, inhibit the proliferation of RCC cells both in vitro and in vivo human tumor xenografts in mice [47, 213]. Propranolol induces loss of cell viability in RCC cells in vitro by increasing gene expression involved in apoptosis, decreasing HIF-α expression, and inhibiting the NFκB survival signaling [216, 217]. α-1 blockade has been linked to an increased RCC incidence, although the study is limited by confounding factors such as concurrent BPH or HTN [224]. On the other hand, the clinical evidence indicates that β-blockade is not associated with RCC risk in patients with HTN [225], and patients with resected localized RCC who use β-blockade did not have better outcomes [226, 227]. In a retrospective analysis of patients with metastatic RCC, concomitant use of β-blockers with immunotherapy is associated with improved clinical outcomes. However, this effect may be attributed to the blockade of β-adrenoceptor signaling on tumor immunosuppression [228, 229].
Given the recent evidence implicating both α- and β-adrenoceptors as functional contributors to RCC pathogenesis and progression, further translational studies are warranted to better understand the targeting value of the adrenergic signaling in advanced RCC and overcoming resistance to current therapeutic modalities.
Impact of pharmacologic α- and β-adrenergic blockade on quality of life
Studies that examined the impact of the pharmacologic α- and β-adrenoceptor antagonists on patient quality of life and functional status focused primarily on patient populations with cardiovascular diseases (e.g., HTN, cardiovascular diseases) and BPH. Although shown to be beneficial in primary and secondary prevention of cardiovascular diseases, and the reduction of reduced symptom bother in BPH patients, concerns about the side effects of β-adrenoceptor antagonists were reported by patients and physicians because of their impact on patient sexual function [230]. Further clinical evidence suggests that β-adrenoceptor antagonists disrupt the balance between α- and β-adrenergic nerve fibers thus leading to increased deterioration of sexual function/ erectile dysfunction in men [231]; the molecular mechanisms underlying this effect remain unclear. Similarly, among the side effects commonly reported with the use of β-adrenoceptor antagonists in these patient populations, are symptoms related to the central nervous system including fatigue, reduced cognitive function, and lethargy leading to poor treatment compliance [232]. Evidence from clinical studies also demonstrated the effects of β-adrenoceptor antagonists on mood state and depression [233,234,235,236], as well as with increased sleep disturbances [237, 238], sleep quality [rapid eye movement (REM) and non-REM sleep], and increased risk of sleep apnea, thus further contributing to fatigue, reduced cognitive function and quality of life [239].
As discussed above, the increasing use of pharmacologic α- and β-adrenoceptor inhibition is well documented in patients with urologic disease including kidney stones, BPH, and bladder, prostate, and renal cancer with side effects varying by the disease population [38, 40, 43, 46, 47]. The impact of pharmacologic α- and β-adrenoceptor antagonists on patient quality of life and functioning status among kidney stones, BPH, and bladder, prostate, and renal cell cancer patients, has revealed a significant effect on increasing heart palpitation, fatigue, dizziness, and increased risk for erectile dysfunction and renal cell carcinoma independent of hypertension [224].
Conclusions
In summary, this review discusses the promise of repurposing of the α- and β-adrenoceptor antagonists for the treatment of benign and malignant urologic diseases. α1-adrenoceptor antagonists may be of value in the treatment of GU malignancies as anti-tumor-modalities as well as in the therapeutic management of benign urologic conditions such as obstructing kidney stones and voiding function. Retrospective epidemiological studies are underway to assess the impact of α1 adrenoceptor antagonists as chemopreventive agents, and prospective clinical trials designed to investigate their efficacy in pre-surgical, post-surgical, and in-patient settings of metastatic disease. Ongoing research efforts exploit the action of α- and β-adrenoceptor signaling mechanisms at: (a) the cellular level, by interrogating the functional exchanges between apoptosis/anoikis signaling and phenotypic configurations within the prostate, bladder and kidney tumor microenvironment; and (b) the clinical setting by undertaking retrospective epidemiological studies and prospective clinical trials to determine the therapeutic value of α1- and β-adrenoceptor antagonists in patients with metastatic disease.
The present study not only describes the evidence on the beneficial effects of α- and β- adrenoceptor antagonists in long-term sequelae of AKI and progression of CKD, but also recognizes the adverse outcomes in kidney function like GFR and RBF, despite BP- and/or blood glucose-lowering effects. Furthermore the α- and β adrenoreceptor antagonists have distinct effects depending on tissue or injury, that would be exploited for tissue-targeted approaches using conditional genetic ablation and cell targeted drug delivery systems. The analytical insights presented here are of high relevance in understanding the molecular and phenotypic effects of pharmacologic action of clinically available adrenoceptor antagonists and their targeting of the α- and β- adrenoceptor subtypes in kidney, bladder and prostatic disease.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- 5-ARI:
-
5α-Reductase inhibitor
- 5-HT:
-
5-Hydroxytryptamine
- ADRA1D:
-
Adrenoceptor α1D
- AKI:
-
Acute kidney injury
- Akt:
-
Protein kinase B
- Ang:
-
Angiotensin
- Ang II:
-
Angiotensin II
- BAD:
-
Bcl-2-associated death promotor
- BCG:
-
Bacillus Calmette–Guerin
- Bcl-2:
-
B-cell lymphoma 2
- BPH:
-
Benign prostatic hyperplasia
- cAMP:
-
Cyclic adenosine monophosphate
- ccRCC:
-
Clear cell renal cell carcinoma
- CDK:
-
Cyclin-dependent kinase
- CDK1:
-
Cyclin-dependent kinase 1
- CI:
-
Confidence interval
- CKD:
-
Chronic kidney disease
- Coa6:
-
Cytochrome c oxidase assembly factor 6
- CREB:
-
CAMP responsive element binding protein
- CRPC:
-
Castration-resistant prostate cancer
- DAG:
-
Diacylglycerol
- DNA:
-
Deoxyribonucleic acid
- dUTP:
-
Deoxyuridine triphosphate
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-mesenchymal transition
- ESKD:
-
End stage kidney disease
- FADD:
-
Fas-associated death domain
- FAK:
-
Focal adhesion kinase
- FDA:
-
Food and Drug Administration
- FGF:
-
Fibroblast growth factor
- GFR:
-
Glomerular filtration rate
- GU:
-
Genitourinary
- HIF:
-
Hypoxia-inducible factor
- HIF1:
-
Hypoxia-inducible factor 1
- HIF-1α:
-
Hypoxia-inducible factor α
- HPA:
-
Human Protein Atlas
- HTN:
-
Hypertension
- IGFBP-3:
-
Insulin-like growth factor-binding protein-3
- IL-6:
-
Interleukin 6
- ILK:
-
Integrin-linked kinase
- IRI:
-
Ischemia/reperfusion injury
- LUTS:
-
Lower urinary tract symptoms
- MAPK:
-
Mitogen-activated protein kinase
- MET:
-
Mesenchymal-epithelial transition
- mRNA:
-
Messenger RNA
- mTOR:
-
Mammalian target of rapamycin
- NCI:
-
National Cancer Institute
- NE:
-
Norepinephrine
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- OAB:
-
Overactive bladder
- OR:
-
Overall risk
- PCa:
-
Prostate cancer
- PI3-kinase:
-
Phosphatidylinositol 3-kinase
- PKA:
-
Protein kinase A
- PSA:
-
Prostate specific antigen
- RAS:
-
Renin-angiotensin system
- RBF:
-
Renal blood flow
- RCC:
-
Renal cell carcinoma
- REM:
-
Rapid eye movement
- RNA:
-
Ribonucleic acid
- STAT3:
-
Signal transducer and activator of transcription 3
- TGF-α:
-
Transforming growth factor α
- TGF-β:
-
Transforming growth factor-β
- TGF-β1:
-
Transforming growth factor-β1
- TSCC:
-
Tongue squamous cell carcinoma
- TKI:
-
Tyrosine kinase inhibitor
- TME:
-
Tumor microenvironment
- TNF-α:
-
Tumor necrosis factor α
- UUO:
-
Ureteral obstruction
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- VEGF:
-
Vascular endothelial growth factor
- VEGF:
-
Vascular endothelial growth factor receptor
- VHL:
-
Von Hippel Lindau
References
Zahalka AH, Frenette PS. Nerves in cancer. Nat Rev Cancer. 2020;20(3):143–57.
de Groat WC, Yoshimura N. Anatomy and physiology of the lower urinary tract. Handb Clin Neurol. 2015;130:61–108.
Nguyen MJ, Higashi R, Ohta K, Nakamura KI, Hashitani H, Lang RJ. Autonomic and sensory nerve modulation of peristalsis in the upper urinary tract. Auton Neurosci Basic Clin. 2016;200:1–10.
Zahalka AH, Arnal-Estapé A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science (New York, NY). 2017;358(6361):321–6.
Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–55.
Quốc Lu’o’ng KV, Nguyễn LT. The roles of beta-adrenergic receptors in tumorigenesis and the possible use of beta-adrenergic blockers for cancer treatment: possible genetic and cell-signaling mechanisms. Cancer Manag Res. 2012;4:431–45.
Civantos Calzada B, Aleixandre de Artiñano A. Alpha-adrenoceptor subtypes. Pharmacol Res. 2001;44(3):195–208.
Hawrylyshyn KA, Michelotti GA, Cogé F, Guénin SP, Schwinn DA. Update on human alpha1-adrenoceptor subtype signaling and genomic organization. Trends Pharmacol Sci. 2004;25(9):449–55.
Hesse IF, Johns EJ. An in vivo study of the alpha-adrenoreceptor subtypes on the renal vasculature of the anaesthetized rabbit. J Auton Pharmacol. 1984;4(3):145–52.
Michelotti GA, Price DT, Schwinn DA. Alpha 1-adrenergic receptor regulation: basic science and clinical implications. Pharmacol Ther. 2000;88(3):281–309.
Giovannitti JA Jr, Thoms SM, Crawford JJ. Alpha-2 adrenergic receptor agonists: a review of current clinical applications. Anesth Prog. 2015;62(1):31–9.
Kobayashi S, Tang R, Shapiro E, Lepor H. Characterization and localization of prostatic alpha 1 adrenoceptors using radioligand receptor binding on slide-mounted tissue section. J Urol. 1993;150(6):2002–6.
Marshall I, Burt RP, Chapple CR. Noradrenaline contractions of human prostate mediated by alpha 1A-(alpha 1c-) adrenoceptor subtype. Br J Pharmacol. 1995;115(5):781–6.
Taniguchi N, Ukai Y, Tanaka T, Yano J, Kimura K, Moriyama N, et al. Identification of alpha 1-adrenoceptor subtypes in the human prostatic urethra. Naunyn Schmiedebergs Arch Pharmacol. 1997;355(3):412–6.
Karabacak OR, Sener NC, Yilmazer D, Karabacak Y, Goktug HN, Yigitbasi O, et al. Alpha adrenergic receptors in renal pelvis and calyces: can rat models be used? Int Braz J Urol. 2014;40(5):683–9.
Minamisawa K, Umemura S, Hirawa N, Hayashi S, Toya Y, Ishikawa Y, et al. Characteristic localization of alpha 1- and alpha 2-adrenoceptors in the human kidney. Clin Exp Pharmacol Physiol. 1993;20(7–8):523–6.
Uhlén S, Lindblom J, Kindlundh A, Mugisha P, Nyberg F. Nandrolone treatment decreases the level of rat kidney alpha(1B)-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol. 2003;368(2):91–8.
Guimarães S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev. 2001;53(2):319–56.
Yamada S, Ito Y. α(1)-Adrenoceptors in the urinary tract. Handb Exp Pharmacol. 2011;202:283–306.
Price DT, Schwinn DA, Lomasney JW, Allen LF, Caron MG, Lefkowitz RJ. Identification, quantification, and localization of mRNA for three distinct alpha 1 adrenergic receptor subtypes in human prostate. J Urol. 1993;150(2 Pt 1):546–51.
Kawabe K. Current status of research on prostate-selective alpha 1-antagonists. Br J Urol. 1998;81(Suppl 1):48–50.
Smith MS, Schambra UB, Wilson KH, Page SO, Schwinn DA. Alpha1-adrenergic receptors in human spinal cord: specific localized expression of mRNA encoding alpha1-adrenergic receptor subtypes at four distinct levels. Brain Res Mol Brain Res. 1999;63(2):254–61.
White CW, da Silva Junior ED, Lim L, Ventura S. What makes the α(1A) -adrenoceptor gene product assume an α(1L) -adrenoceptor phenotype? Br J Pharmacol. 2019;176(14):2358–65.
Michel MC. The forefront for novel therapeutic agents based on the pathophysiology of lower urinary tract dysfunction: alpha-blockers in the treatment of male voiding dysfunction—how do they work and why do they differ in tolerability? J Pharmacol Sci. 2010;112(2):151–7.
Gonzalez-Cabrera PJ, Gaivin RJ, Yun J, Ross SA, Papay RS, McCune DF, et al. Genetic profiling of alpha 1-adrenergic receptor subtypes by oligonucleotide microarrays: coupling to interleukin-6 secretion but differences in STAT3 phosphorylation and gp-130. Mol Pharmacol. 2003;63(5):1104–16.
Ishihama H, Momota Y, Yanase H, Wang X, de Groat WC, Kawatani M. Activation of alpha1D adrenergic receptors in the rat urothelium facilitates the micturition reflex. J Urol. 2006;175(1):358–64.
Recio P, Orensanz LM, Martínez MP, Navarro-Dorado J, Bustamante S, García-Sacristán A, et al. Noradrenergic vasoconstriction of pig prostatic small arteries. Naunyn Schmiedebergs Arch Pharmacol. 2008;376(6):397–406.
Pettinger WA, Jackson EK. α (2)-Adrenoceptors: Challenges and Opportunities-Enlightenment from the Kidney. Cardiovasc Ther. 2020;2020:2478781.
Michel MC. β-Adrenergic Receptor Subtypes in the Urinary Tract. Handb Exp Pharmacol. 2011;202:307–18.
Hernández M, Prieto D, Simonsen U, Rivera L, Barahona MV, García-Sacristán A. Noradrenaline modulates smooth muscle activity of the isolated intravesical ureter of the pig through different types of adrenoceptors. Br J Pharmacol. 1992;107(4):924–31.
Morita T, Ando M, Kihara K, Oshima H. Function and distribution of autonomic receptors in canine ureteral smooth muscle. Neurourol Urodyn. 1994;13(3):315–21.
Tindall AR. Preliminary observations on the mechanical and electrical activity of the rat ureter. J Physiol. 1972;223(3):633–47.
Tsujii T, Azuma H, Yamaguchi T, Oshima H. A possible role of decreased relaxation mediated by beta-adrenoceptors in bladder outlet obstruction by benign prostatic hyperplasia. Br J Pharmacol. 1992;107(3):803–7.
Drescher P, Eckert RE, Madsen PO. Smooth muscle contractility in prostatic hyperplasia: role of cyclic adenosine monophosphate. Prostate. 1994;25(2):76–80.
Palm D, Lang K, Niggemann B, Drell TLT, Masur K, Zaenker KS, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer. 2006;118(11):2744–9.
Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science (New York, NY). 2013;341(6142):1236361.
Campschroer T, Zhu Y, Duijvesz D, Grobbee DE, Lock M. Alpha‐blockers as medical expulsive therapy for ureteral stones. Cochrane Database of Systematic Reviews. 2014(4).
Batty M, Pugh R, Rathinam I, Simmonds J, Walker E, Forbes A, et al. The role of α1-adrenoceptor antagonists in the treatment of prostate and other cancers. Int J Mol Sci. 2016;17(8).
Harris AM, Warner BW, Wilson JM, Becker A, Rowland RG, Conner W, et al. Effect of alpha1-adrenoceptor antagonist exposure on prostate cancer incidence: an observational cohort study. J Urol. 2007;178(5):2176–80.
Gotoh A, Nagaya H, Kanno T, Nishizaki T. Antitumor action of α(1)-adrenoceptor blockers on human bladder, prostate and renal cancer cells. Pharmacology. 2012;90(5–6):242–6.
Wade CA, Goodwin J, Preston D, Kyprianou N. Impact of α-adrenoceptor antagonists on prostate cancer development, progression and prevention. Am J Clin Exp Urol. 2019;7(1):46–60.
Bilbro J, Mart M, Kyprianou N. Therapeutic value of quinazoline-based compounds in prostate cancer. Anticancer Res. 2013;33(11):4695–700.
Kyprianou N, Benning CM. Suppression of human prostate cancer cell growth by alpha1-adrenoceptor antagonists doxazosin and terazosin via induction of apoptosis. Can Res. 2000;60(16):4550–5.
Anglin IE, Glassman DT, Kyprianou N. Induction of prostate apoptosis by alpha1-adrenoceptor antagonists: mechanistic significance of the quinazoline component. Prostate Cancer Prostatic Dis. 2002;5(2):88–95.
Benning CM, Kyprianou N. Quinazoline-derived alpha1-adrenoceptor antagonists induce prostate cancer cell apoptosis via an alpha1-adrenoceptor-independent action. Can Res. 2002;62(2):597–602.
Suzuki N, Niikura R, Ihara S, Hikiba Y, Kinoshita H, Higashishima N, et al. Alpha-blockers as colorectal cancer chemopreventive: findings from a case-control study, human cell cultures, and in vivo preclinical testing. Cancer Prev Res (Phila). 2019;12(3):185–94.
Sakamoto S, Schwarze S, Kyprianou N. Anoikis disruption of focal adhesion-Akt signaling impairs renal cell carcinoma. Eur Urol. 2011;59(5):734–44.
Garrison JB, Kyprianou N. Novel targeting of apoptosis pathways for prostate cancer therapy. Curr Cancer Drug Targets. 2004;4(1):85–95.
Keledjian K, Garrison JB, Kyprianou N. Doxazosin inhibits human vascular endothelial cell adhesion, migration, and invasion. J Cell Biochem. 2005;94(2):374–88.
Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. 1996;379(6560):91–6.
Li F, Liu J, Mayne R, Wu C. Identification and characterization of a mouse protein kinase that is highly homologous to human integrin-linked kinase. Biochem Biophys Acta. 1997;1358(3):215–20.
Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155(4):505–10.
Radeva G, Petrocelli T, Behrend E, Leung-Hagesteijn C, Filmus J, Slingerland J, et al. Overexpression of the integrin-linked kinase promotes anchorage-independent cell cycle progression. J Biol Chem. 1997;272(21):13937–44.
Cieslik K, Zembowicz A, Tang JL, Wu KK. Transcriptional regulation of endothelial nitric-oxide synthase by lysophosphatidylcholine. J Biol Chem. 1998;273(24):14885–90.
Attwell S, Roskelley C, Dedhar S. The integrin-linked kinase (ILK) suppresses anoikis. Oncogene. 2000;19(33):3811–5.
Fukuda T, Chen K, Shi X, Wu C. PINCH-1 is an obligate partner of integrin-linked kinase (ILK) functioning in cell shape modulation, motility, and survival. J Biol Chem. 2003;278(51):51324–33.
Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124(4):619–26.
Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13(5):555–62.
Calmasini FB, Candido TZ, Alexandre EC, D’Ancona CA, Silva D, de Oliveira MA, et al. The beta-3 adrenoceptor agonist, mirabegron relaxes isolated prostate from human and rabbit: new therapeutic indication? Prostate. 2015;75(4):440–7.
Lang K, Drell TLT, Lindecke A, Niggemann B, Kaltschmidt C, Zaenker KS, et al. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int J Cancer. 2004;112(2):231–8.
Zahalka AH, Fram E, Lin W, Mohn L, Frenette PS, Agalliu I, et al. Use of beta-blocker types and risk of incident prostate cancer in a multiethnic population. Urol Oncol. 2020;38(10):794.e11-e16.
Lu H, Liu X, Guo F, Tan S, Wang G, Liu H, et al. Impact of beta-blockers on prostate cancer mortality: a meta-analysis of 16,825 patients. Onco Targets Ther. 2015;8:985–90.
Schwinn DA. Adrenergic receptors: unique localization in human tissues. Adv Pharmacol (San Diego, Calif). 1994;31:333–41.
Price DT, Lefkowitz RJ, Caron MG, Berkowitz D, Schwinn DA. Localization of mRNA for three distinct alpha 1-adrenergic receptor subtypes in human tissues: implications for human alpha-adrenergic physiology. Mol Pharmacol. 1994;45(2):171–5.
Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science (New York, NY). 2015;348(6235):648–60.
Gilarevskii SR, Lantsova EV, Akimov AA. Efficacy and safety of combined treament with ivabradine and metoprolol in patients with stable angina pectoris—a systematic review. Kardiologiia. 2020;60(11):1357.
Romics I. The role of alpha-adrenoreceptors in the treatment of urological diseases. Neurochem Int. 2007;51(5):328–31.
Mihalopoulos M, Dogra N, Mohamed N, Badani K, Kyprianou N. COVID-19 and kidney disease: molecular determinants and clinical implications in renal cancer. Eur Urol Focus. 2020;6(5):1086–96.
Murillo OD, Thistlethwaite W, Rozowsky J, Subramanian SL, Lucero R, Shah N, et al. exRNA atlas analysis reveals distinct extracellular RNA cargo types and their carriers present across human biofluids. Cell. 2019;177(2):463-77.e15.
Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol. 1984;132(3):474–9.
Caine M. The present role of alpha-adrenergic blockers in the treatment of benign prostatic hypertrophy. J Urol. 1986;136(1):1–4.
Lepor H, Tang R, Meretyk S, Shapiro E. Alpha 1 adrenoceptor subtypes in the human prostate. J Urol. 1993;149(3):640–2.
Schwinn DA, Roehrborn CG. Alpha1-adrenoceptor subtypes and lower urinary tract symptoms. Int J Urol. 2008;15(3):193–9.
Kenny BA, Miller AM, Williamson IJ, O’Connell J, Chalmers DH, Naylor AM. Evaluation of the pharmacological selectivity profile of alpha 1 adrenoceptor antagonists at prostatic alpha 1 adrenoceptors: binding, functional and in vivo studies. Br J Pharmacol. 1996;118(4):871–8.
Fuentes AV, Pineda MD, Venkata KCN. Comprehension of top 200 prescribed drugs in the US as a resource for pharmacy teaching, training and practice. Pharmacy (Basel, Switzerland). 2018;6(2).
Malloy BJ, Price DT, Price RR, Bienstock AM, Dole MK, Funk BL, et al. Alpha1-adrenergic receptor subtypes in human detrusor. J Urol. 1998;160(3 Pt 1):937–43.
Nishino Y, Masue T, Miwa K, Takahashi Y, Ishihara S, Deguchi T. Comparison of two alpha1-adrenoceptor antagonists, naftopidil and tamsulosin hydrochloride, in the treatment of lower urinary tract symptoms with benign prostatic hyperplasia: a randomized crossover study. BJU international. 2006;97(4):747–51, discussion 51.
Kaplan SA. Side effects of alpha-blocker use: retrograde ejaculation. Rev Urol. 2009;11(Suppl 1):S14–8.
Malin JM Jr, Deane RF, Boyarsky S. Characterisation of adrenergic receptors in human ureter. Br J Urol. 1970;42(2):171–4.
Sigala S, Dellabella M, Milanese G, Fornari S, Faccoli S, Palazzolo F, et al. Evidence for the presence of alpha1 adrenoceptor subtypes in the human ureter. Neurourol Urodyn. 2005;24(2):142–8.
Lipkin M, Shah O. The use of alpha-blockers for the treatment of nephrolithiasis. Rev Urol. 2006;8(Suppl 4):35–42.
Dellis AE, Keeley FX Jr, Manolas V, Skolarikos AA. Role of α-blockers in the treatment of stent-related symptoms: a prospective randomized control study. Urology. 2014;83(1):56–61.
Michel MC, Vrydag W. Alpha1-, alpha2- and beta-adrenoceptors in the urinary bladder, urethra and prostate. Br J Pharmacol. 2006;147(Suppl 2):S88–119.
Hay-Smith J, Herbison P, Ellis G, Moore K. Anticholinergic drugs versus placebo for overactive bladder syndrome in adults. The Cochrane database of systematic reviews. 2002(3):Cd003781.
Veenboer PW, Bosch JL. Long-term adherence to antimuscarinic therapy in everyday practice: a systematic review. J Urol. 2014;191(4):1003–8.
Bragg R, Hebel D, Vouri SM, Pitlick JM. Mirabegron: a Beta-3 agonist for overactive bladder. Consult Pharm. 2014;29(12):823–37.
Igawa Y, Aizawa N, Homma Y. Beta3-adrenoceptor agonists: possible role in the treatment of overactive bladder. Korean J Urol. 2010;51(12):811–8.
Kelleher C, Hakimi Z, Zur R, Siddiqui E, Maman K, Aballéa S, et al. Efficacy and tolerability of mirabegron compared with antimuscarinic monotherapy or combination therapies for overactive bladder: a systematic review and network meta-analysis. Eur Urol. 2018;74(3):324–33.
Richards RJ, Thakur V, Reisin E. Obesity-related hypertension: its physiological basis and pharmacological approaches to its treatment. J Hum Hypertens. 1996;10(Suppl 3):S59-64.
Schiffl H, Lang SM. Obesity, acute kidney injury and outcome of critical illness. Int Urol Nephrol. 2017;49(3):461–6.
Wofford MR, Anderson DC Jr, Brown CA, Jones DW, Miller ME, Hall JE. Antihypertensive effect of alpha- and beta-adrenergic blockade in obese and lean hypertensive subjects. Am J Hypertens. 2001;14(7 Pt 1):694–8.
Bakris GL, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006;70(11):1905–13.
Noh MR, Jang HS, Kim J, Padanilam BJ. Renal sympathetic nerve-derived signaling in acute and chronic kidney diseases. Int J Mol Sci. 2020;21(5).
Jang HS, Kim J, Padanilam BJ. Renal sympathetic nerve activation via α(2)-adrenergic receptors in chronic kidney disease progression. Kidney Res Clin Pract. 2019;38(1):6–14.
Kim J, Padanilam BJ. Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int. 2015;87(2):350–8.
Shimokawa T, Tsutsui H, Miura T, Nishinaka T, Terada T, Takama M, et al. Renoprotective effect of yohimbine on ischaemia/reperfusion-induced acute kidney injury through α2C-adrenoceptors in rats. Eur J Pharmacol. 2016;781:36–44.
Solez K, Ideura T, Silvia CB, Hamilton B, Saito H. Clonidine after renal ischemia to lessen acute renal failure and microvascular damage. Kidney Int. 1980;18(3):309–22.
Tsutsui H, Sugiura T, Hayashi K, Ohkita M, Takaoka M, Yukimura T, et al. Moxonidine prevents ischemia/reperfusion-induced renal injury in rats. Eur J Pharmacol. 2009;603(1–3):73–8.
Tsutsui H, Shimokawa T, Miura T, Takama M, Nishinaka T, Terada T, et al. Inhibition of α2C-adrenoceptors ameliorates cisplatin-induced acute renal failure in rats. Eur J Pharmacol. 2018;838:113–9.
Morsy MA, Heeba GH. Nebivolol Ameliorates Cisplatin-Induced Nephrotoxicity in Rats. Basic Clin Pharmacol Toxicol. 2016;118(6):449–55.
Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81(5):442–8.
Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11(5):264–76.
Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI insight. 2017;2(18).
Shimokawa T, Tsutsui H, Miura T, Takama M, Hayashi K, Nishinaka T, et al. Post-treatment with JP-1302 protects against renal ischemia/reperfusion-induced acute kidney injury in rats. J Pharmacol Sci. 2019;139(3):137–42.
Chevalier RL, Finn WF. Effects of propranolol on post-ischemic acute renal failure. Nephron. 1980;25(2):77–81.
Kim J, Padanilam BJ. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J Am Soc Nephrol. 2013;24(2):229–42.
Amann K, Koch A, Hofstetter J, Gross ML, Haas C, Orth SR, et al. Glomerulosclerosis and progression: effect of subantihypertensive doses of alpha and beta blockers. Kidney Int. 2001;60(4):1309–23.
Hall ME, DoCarmo JM, DaSilva AA, Juncos LA, Wang Z, Hall JE. Obesity, hypertension, and chronic kidney disease. Int J Nephrol Renovascular Dis. 2014;7:75–88.
Van Buren PN, Toto R. Hypertension in diabetic nephropathy: epidemiology, mechanisms, and management. Adv Chronic Kidney Dis. 2011;18(1):28–41.
Patney V, Whaley-Connell A, Bakris G. Hypertension management in diabetic kidney disease. Diabetes Spectr. 2015;28(3):175–80.
Tindall H, Urquhart S, Stickland M, Davies JA. Treatment with atenolol prevents progression of microalbuminuria in type I diabetic patients. Curr Med Res Opin. 1991;12(8):516–20.
Hjalmarson A. Cardioprotection with beta-adrenoceptor blockers. Does lipophilicity matter? Basic Res Cardiol. 2000;95(Suppl 1):41–5.
Jacob S, Rett K, Wicklmayr M, Agrawal B, Augustin HJ, Dietze GJ. Differential effect of chronic treatment with two beta-blocking agents on insulin sensitivity: the carvedilol-metoprolol study. J Hypertens. 1996;14(4):489–94.
Yildiz A, Hursit M, Celik AV, Kayacan SM, Yazici H, Akkaya V, et al. Doxazosin, but not amlodipine decreases insulin resistance in patients with chronic renal failure: a prospective, randomized-controlled study. Clin Nephrol. 2002;58(6):405–10.
Mori Y, Matsubara H, Nose A, Shibasaki Y, Masaki H, Kosaki A, et al. Safety and availability of doxazosin in treating hypertensive patients with chronic renal failure. Hypertens Res. 2001;24(4):359–63.
Erley CM, Haefele U, Heyne N, Braun N, Risler T. Microalbuminuria in essential hypertension. Reduction by different antihypertensive drugs. Hypertension (Dallas, Tex : 1979). 1993;21(6 Pt 1):810–5.
Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7(12):684–96.
Padro CJ, Sanders VM. Neuroendocrine regulation of inflammation. Semin Immunol. 2014;26(5):357–68.
Spengler RN, Allen RM, Remick DG, Strieter RM, Kunkel SL. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol (Baltimore, Md: 1950). 1990;145(5):1430–4.
Wang RX, Limbird LE. Distribution of mRNA encoding three alpha 2-adrenergic receptor subtypes in the developing mouse embryo suggests a role for the alpha 2A subtype in apoptosis. Mol Pharmacol. 1997;52(6):1071–80.
Ghiani CA, Eisen AM, Yuan X, DePinho RA, McBain CJ, Gallo V. Neurotransmitter receptor activation triggers p27(Kip1)and p21(CIP1) accumulation and G1 cell cycle arrest in oligodendrocyte progenitors. Development (Cambridge, England). 1999;126(5):1077–90.
Brede M, Philipp M, Knaus A, Muthig V, Hein L. alpha2-adrenergic receptor subtypes—novel functions uncovered in gene-targeted mouse models. Biol Cell. 2004;96(5):343–8.
Brede M, Wiesmann F, Jahns R, Hadamek K, Arnolt C, Neubauer S, et al. Feedback inhibition of catecholamine release by two different alpha2-adrenoceptor subtypes prevents progression of heart failure. Circulation. 2002;106(19):2491–6.
Schlaich MP, Socratous F, Hennebry S, Eikelis N, Lambert EA, Straznicky N, et al. Sympathetic activation in chronic renal failure. J Am Soc Nephrol. 2009;20(5):933–9.
Mount PF, Power DA. Nitric oxide in the kidney: functions and regulation of synthesis. Acta Physiol (Oxf). 2006;187(4):433–46.
Eppel GA, Denton KM, Malpas SC, Evans RG. Nitric oxide in responses of regional kidney perfusion to renal nerve stimulation and renal ischaemia. Pflugers Arch. 2003;447(2):205–13.
Aiello S, Noris M, Todeschini M, Zappella S, Foglieni C, Benigni A, et al. Renal and systemic nitric oxide synthesis in rats with renal mass reduction. Kidney Int. 1997;52(1):171–81.
Lee J. Nitric oxide in the kidney: its physiological role and pathophysiological implications. Electrolyte Blood Press E & BP. 2008;6(1):27–34.
Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72(2):151–6.
Bruck H, Gössl M, Spitthöver R, Schäfers RF, Kohnle M, Philipp T, et al. The nitric oxide synthase inhibitor L-NMMA potentiates noradrenaline-induced vasoconstriction: effects of the alpha2-receptor antagonist yohimbine. J Hypertens. 2001;19(5):907–11.
Kaur J, Young BE, Fadel PJ. Sympathetic Overactivity in Chronic Kidney Disease: Consequences and Mechanisms. Int J Mol Sci. 2017;18(8).
Thomson SC, Vallon V. Alpha 2-adrenoceptors determine the response to nitric oxide inhibition in the rat glomerulus and proximal tubule. J Am Soc Nephrol. 1995;6(5):1482–90.
Tojo A, Kobayashi N, Kimura K, Hirata Y, Matsuoka H, Yagi S, et al. Effects of antihypertensive drugs on nitric oxide synthase activity in rat kidney. Kidney Int Suppl. 1996;55:S138–40.
Wangensteen R, O'Valle F, Del Moral R, Vargas F, Osuna A. Chronic alpha1-adrenergic blockade improves hypertension and renal injury in L-NAME and low-renin L-NAME-DOCA hypertensive rats. Med Sci Monit. 2002;8(9):Br378–84.
Erley CM, Rebmann S, Strobel U, Schmidt T, Wehrmann M, Osswald H, et al. Effects of antihypertensive therapy on blood pressure and renal function in rats with hypertension due to chronic blockade of nitric oxide synthesis. Exp Nephrol. 1995;3(5):293–9.
Feng MG, Prieto MC, Navar LG. Nebivolol-induced vasodilation of renal afferent arterioles involves β3-adrenergic receptor and nitric oxide synthase activation. Am J Physiol Renal Physiol. 2012;303(5):F775–82.
Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, Jankowski M, Martyniec L, Angielski S, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation. 2003;107(21):2747–52.
Van den Branden C, Gabriels M, Vamecq J, Vanden Houte K, Verbeelen D. Carvedilol protects against glomerulosclerosis in rat remnant kidney without general changes in antioxidant enzyme status A comparative study of two beta-blocking drugs, carvedilol and propanolol. Nephron. 1997;77(3):319–24.
Kumar KV, Shifow AA, Naidu MU, Ratnakar KS. Carvedilol: a beta blocker with antioxidant property protects against gentamicin-induced nephrotoxicity in rats. Life Sci. 2000;66(26):2603–11.
Barone FC, Nelson AH, Ohlstein EH, Willette RN, Sealey JE, Laragh JH, et al. Chronic carvedilol reduces mortality and renal damage in hypertensive stroke-prone rats. J Pharmacol Exp Ther. 1996;279(2):948–55.
Whaley-Connell A, Habibi J, Johnson M, Tilmon R, Rehmer N, Rehmer J, et al. Nebivolol reduces proteinuria and renal NADPH oxidase-generated reactive oxygen species in the transgenic Ren2 rat. Am J Nephrol. 2009;30(4):354–60.
DiBona GF. Nervous kidney. Interaction between renal sympathetic nerves and the renin-angiotensin system in the control of renal function. Hypertension (Dallas, Tex : 1979). 2000;36(6):1083–8.
Schlaich MP, Kaye DM, Lambert E, Hastings J, Campbell DJ, Lambert G, et al. Angiotensin II and norepinephrine release: interaction and effects on the heart. J Hypertens. 2005;23(5):1077–82.
Hu Z, Wang Z, Wu H, Yang Z, Jiang W, Li L, et al. Ang II enhances noradrenaline release from sympathetic nerve endings thus contributing to the up-regulation of metalloprotease-2 in aortic dissection patients’ aorta wall. PLoS ONE. 2013;8(10):e76922.
Hoch H, Stegbauer J, Potthoff SA, Hein L, Quack I, Rump LC, et al. Regulation of renal sympathetic neurotransmission by renal α(2A)-adrenoceptors is impaired in chronic renal failure. Br J Pharmacol. 2011;163(2):438–46.
Forbes A, Anoopkumar-Dukie S, Chess-Williams R, McDermott C. Relative cytotoxic potencies and cell death mechanisms of α1 -adrenoceptor antagonists in prostate cancer cell lines. Prostate. 2016;76(8):757–66.
Garrison JB, Kyprianou N. Doxazosin induces apoptosis of benign and malignant prostate cells via a death receptor-mediated pathway. Can Res. 2006;66(1):464–72.
Partin JV, Anglin IE, Kyprianou N. Quinazoline-based alpha 1-adrenoceptor antagonists induce prostate cancer cell apoptosis via TGF-beta signalling and I kappa B alpha induction. Br J Cancer. 2003;88(10):1615–21.
Lin SC, Chueh SC, Hsiao CJ, Li TK, Chen TH, Liao CH, et al. Prazosin displays anticancer activity against human prostate cancers: targeting DNA and cell cycle. Neoplasia (New York, NY). 2007;9(10):830–9.
Garrison JB, Shaw YJ, Chen CS, Kyprianou N. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Can Res. 2007;67(23):11344–52.
Nakagawa YU, Nagaya H, Miyata T, Wada Y, Oyama T, Gotoh A. Piperazine-based alpha-1 AR blocker, naftopidil, selectively suppresses malignant human bladder cells via induction of apoptosis. Anticancer Res. 2016;36(4):1563–70.
Sastry KS, Karpova Y, Prokopovich S, Smith AJ, Essau B, Gersappe A, et al. Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J Biol Chem. 2007;282(19):14094–100.
Zhao Y, Li W. Beta-adrenergic signaling on neuroendocrine differentiation, angiogenesis, and metastasis in prostate cancer progression. Asian J Androl. 2019;21(3):253–9.
Braadland PR, Ramberg H, Grytli HH, Taskén KA. β-adrenergic receptor signaling in prostate cancer. Front Oncol. 2014;4:375.
Hassan S, Karpova Y, Baiz D, Yancey D, Pullikuth A, Flores A, et al. Behavioral stress accelerates prostate cancer development in mice. J Clin Investig. 2013;123(2):874–86.
Wang F, Liu H, Wang F, Xu R, Wang P, Tang F, et al. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol Med Rep. 2018;17(4):5213–21.
Zhou C, Chen X, Zeng W, Peng C, Huang G, Li X, et al. Propranolol induced G0/G1/S phase arrest and apoptosis in melanoma cells via AKT/MAPK pathway. Oncotarget. 2016;7(42):68314–27.
Montoya A, Varela-Ramirez A, Dickerson E, Pasquier E, Torabi A, Aguilera R, et al. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomedical journal. 2019;42(3):155–65.
Papetti M, Herman IM. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol. 2002;282(5):C947–70.
De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17(8):457–74.
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health risk Manag. 2006;2(3):213–9.
Mravec B, Horvathova L, Hunakova L. Neurobiology of cancer: the role of β-adrenergic receptor signaling in various tumor environments. Int J Mol Sci. 2020;21(21).
Park SY, Kang JH, Jeong KJ, Lee J, Han JW, Choi WS, et al. Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1α protein-dependent mechanism. Int J Cancer. 2011;128(10):2306–16.
Plećas B, Glavaski A, Solarović T. Propranolol treatment affects ventral prostate blood vessels and serum testosterone concentrations in adult rats. Andrologia. 1997;29(2):109–14.
Conceição F, Sousa DM, Paredes J, Lamghari M. Sympathetic activity in breast cancer and metastasis: partners in crime. Bone research. 2021;9(1):9.
Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84.
Ren H, Zuo S, Hou Y, Shang W, Liu N, Yin Z. Inhibition of α1-adrenoceptor reduces TGF-β1-induced epithelial-to-mesenchymal transition and attenuates UUO-induced renal fibrosis in mice. FASEB J. 2020;34(11):14892–904.
Kainuma K, Kobayashi T, D’Alessandro-Gabazza CN, Toda M, Yasuma T, Nishihama K, et al. β(2) adrenergic agonist suppresses eosinophil-induced epithelial-to-mesenchymal transition of bronchial epithelial cells. Respir Res. 2017;18(1):79.
Strell C, Niggemann B, Voss MJ, Powe DG, Zänker KS, Entschladen F. Norepinephrine promotes the β1-integrin-mediated adhesion of MDA-MB-231 cells to vascular endothelium by the induction of a GROα release. Mol Cancer Res. 2012;10(2):197–207.
Kim TH, Gill NK, Nyberg KD, Nguyen AV, Hohlbauch SV, Geisse NA, et al. Cancer cells become less deformable and more invasive with activation of β-adrenergic signaling. J Cell Sci. 2016;129(24):4563–75.
Moretti S, Massi D, Farini V, Baroni G, Parri M, Innocenti S, et al. β-adrenoceptors are upregulated in human melanoma and their activation releases pro-tumorigenic cytokines and metalloproteases in melanoma cell lines Laboratory investigation. J Tech Methods Pathol. 2013;93(3):279–90.
Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939–44.
Liu J, Qu L, Wan C, Xiao M, Ni W, Jiang F, et al. A novel β2-AR/YB-1/β-catenin axis mediates chronic stress-associated metastasis in hepatocellular carcinoma. Oncogenesis. 2020;9(9):84.
Gruet M, Cotton D, Coveney C, Boocock DJ, Wagner S, Komorowski L, et al. β2-adrenergic signalling promotes cell migration by upregulating expression of the metastasis-associated molecule LYPD3. Biology. 2020;9(2).
Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Can Res. 2010;70(18):7042–52.
Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell. 2018;33(1):75-90.e7.
Nagata Y, Kawahara T, Goto T, Inoue S, Teramoto Y, Jiang G, et al. Effects of α(1)-adrenergic receptor antagonists on the development and progression of urothelial cancer. Am J Cancer Res. 2020;10(12):4386–98.
Cao Z, Koochekpour S, Strup SE, Kyprianou N. Reversion of epithelial-mesenchymal transition by a novel agent DZ-50 via IGF binding protein-3 in prostate cancer cells. Oncotarget. 2017;8(45):78507–19.
Qin JF, Jin FJ, Li N, Guan HT, Lan L, Ni H, et al. Adrenergic receptor β2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep. 2015;48(5):295–300.
Bravo-Calderón DM, Oliveira DT, Marana AN, Nonogaki S, Carvalho AL, Kowalski LP. Prognostic significance of beta-2 adrenergic receptor in oral squamous cell carcinoma. Cancer Biomark. 2011;10(1):51–9.
Liu H, Wang C, Xie N, Zhuang Z, Liu X, Hou J, et al. Activation of adrenergic receptor β2 promotes tumor progression and epithelial mesenchymal transition in tongue squamous cell carcinoma. Int J Mol Med. 2018;41(1):147–54.
Shan T, Cui X, Li W, Lin W, Li Y, Chen X, et al. Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci. 2014;105(7):847–56.
Hensley PJ, Cao Z, Pu H, Dicken H, He D, Zhou Z, et al. Predictive and targeting value of IGFBP-3 in therapeutically resistant prostate cancer. Am J Clin Exp Urol. 2019;7(3):188–202.
Huang Z, Li G, Zhang Z, Gu R, Wang W, Lai X, et al. β2AR-HIF-1α-CXCL12 signaling of osteoblasts activated by isoproterenol promotes migration and invasion of prostate cancer cells. BMC Cancer. 2019;19(1):1142.
Carie AE, Sebti SM. A chemical biology approach identifies a beta-2 adrenergic receptor agonist that causes human tumor regression by blocking the Raf-1/Mek-1/Erk1/2 pathway. Oncogene. 2007;26(26):3777–88.
Pérez Piñero C, Bruzzone A, Sarappa MG, Castillo LF, Lüthy IA. Involvement of α2- and β2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation. Br J Pharmacol. 2012;166(2):721–36.
Sakakitani S, Podyma-Inoue KA, Takayama R, Takahashi K, Ishigami-Yuasa M, Kagechika H, et al. Activation of β2-adrenergic receptor signals suppresses mesenchymal phenotypes of oral squamous cell carcinoma cells. Cancer Sci. 2021;112(1):155–67.
Dawes RP, Burke KA, Byun DK, Xu Z, Stastka P, Chan L, et al. Chronic stress exposure suppresses mammary tumor growth and reduces circulating exosome TGF-β content via β-adrenergic receptor signaling in MMTV-PyMT mice. Breast Cancer. 2020;14:1178223420931511.
Wang W, Guo X, Dan H. α2A-adrenergic receptor inhibits the progression of cervical cancer through blocking PI3K/AKT/mTOR pathway. Onco Targets Ther. 2020;13:10535–46.
Powe DG, Voss MJ, Zänker KS, Habashy HO, Green AR, Ellis IO, et al. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1(7):628–38.
Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population-based study. J Clin Oncol. 2011;29(19):2635–44.
Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, et al. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol. 2011;29(19):2645–52.
Choy C, Raytis JL, Smith DD, Duenas M, Neman J, Jandial R, et al. Inhibition of β2-adrenergic receptor reduces triple-negative breast cancer brain metastases: the potential benefit of perioperative β-blockade. Oncol Rep. 2016;35(6):3135–42.
Cardwell CR, Coleman HG, Murray LJ, Entschladen F, Powe DG. Beta-blocker usage and breast cancer survival: a nested case-control study within a UK clinical practice research datalink cohort. Int J Epidemiol. 2013;42(6):1852–61.
Gillis RD, Botteri E, Chang A, Ziegler AI, Chung NC, Pon CK, et al. Carvedilol blocks neural regulation of breast cancer progression in vivo and is associated with reduced breast cancer mortality in patients. Eur J Cancer (Oxford, England: 1990). 2021;147:106–16.
Hiller JG, Cole SW, Crone EM, Byrne DJ, Shackleford DM, Pang JB, et al. Preoperative β-blockade with propranolol reduces biomarkers of metastasis in breast cancer: a phase II randomized trial. Clin Cancer Res. 2020;26(8):1803–11.
Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. 2008;19(11):4875–87.
Robinson D, Garmo H, Bill-Axelson A, Mucci L, Holmberg L, Stattin P. Use of 5α-reductase inhibitors for lower urinary tract symptoms and risk of prostate cancer in Swedish men: nationwide, population based case-control study. BMJ (Clinical research ed). 2013;346:f3406.
Ørsted DD, Bojesen SE, Nielsen SF, Nordestgaard BG. Association of clinical benign prostate hyperplasia with prostate cancer incidence and mortality revisited: a nationwide cohort study of 3,009,258 men. Eur Urol. 2011;60(4):691–8.
Murtola TJ, Tammela TL, Määttänen L, Ala-Opas M, Stenman UH, Auvinen A. Prostate cancer incidence among finasteride and alpha-blocker users in the Finnish Prostate Cancer Screening Trial. Br J Cancer. 2009;101(5):843–8.
Van Rompay MI, Curtis Nickel J, Ranganathan G, Kantoff PW, Solomon KR, Lund JL, et al. Impact of 5α-reductase inhibitor and α-blocker therapy for benign prostatic hyperplasia on prostate cancer incidence and mortality. BJU Int. 2019;123(3):511–8.
Sarkar RR, Parsons JK, Bryant AK, Ryan ST, Kader AK, McKay RR, et al. Association of treatment with 5α-reductase inhibitors with time to diagnosis and mortality in prostate cancer. JAMA Intern Med. 2019;179(6):812–9.
Scardino PT, Weaver R, Hudson MA. Early detection of prostate cancer. Hum Pathol. 1992;23(3):211–22.
Martin FM, Harris AM, Rowland RG, Conner W, Lane M, Durbin E, et al. Decreased risk of bladder cancer in men treated with quinazoline-based α1-adrenoceptor antagonists. Gene Therapy Mol Biol. 2008;12(2):253–8.
Tahmatzopoulos A, Lagrange CA, Zeng L, Mitchell BL, Conner WT, Kyprianou N. Effect of terazosin on tissue vascularity and apoptosis in transitional cell carcinoma of bladder. Urology. 2005;65(5):1019–23.
Siddiqui EJ, Shabbir M, Thompson CS, Mumtaz FH, Mikhailidis DP. Growth inhibitory effect of doxazosin on prostate and bladder cancer cells. Is the serotonin receptor pathway involved? Anti Res. 2005;25(6b):4281–6.
Kawahara T, Ide H, Kashiwagi E, Patterson JD, Inoue S, Shareef HK, et al. Silodosin inhibits the growth of bladder cancer cells and enhances the cytotoxic activity of cisplatin via ELK1 inactivation. Am J Cancer Res. 2015;5(10):2959–68.
Mauffrey P, Tchitchek N, Barroca V, Bemelmans A-P, Firlej V, Allory Y, et al. Progenitors from the central nervous system drive neurogenesis in cancer. Nature. 2019;569(7758):672–8.
Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR, et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature. 2020;578(7795):449–54.
Flint MS, Baum A, Chambers WH, Jenkins FJ. Induction of DNA damage, alteration of DNA repair and transcriptional activation by stress hormones. Psychoneuroendocrinology. 2007;32(5):470–9.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Pantziarka P, Bouche G, Sukhatme V, Meheus L, Rooman I, Sukhatme VP. Repurposing Drugs in Oncology (ReDO)-Propranolol as an anti-cancer agent. Ecancermedicalscience. 2016;10:680.
Iwamoto Y, Ishii K, Sasaki T, Kato M, Kanda H, Yamada Y, et al. Oral naftopidil suppresses human renal-cell carcinoma by inducing G(1) cell-cycle arrest in tumor and vascular endothelial cells. Cancer Prev Res (Phila). 2013;6(9):1000–6.
Ravaud A, Motzer RJ, Pandha HS, George DJ, Pantuck AJ, Patel A, et al. Adjuvant sunitinib in high-risk renal-cell carcinoma after nephrectomy. N Engl J Med. 2016;375(23):2246–54.
Karabacak OR, Yilmazer D, Ozturk U, Sener NC, Saltas H, Karabacak Y, et al. The presence and distribution of alpha adrenergic receptors in human renal pelvis and calyces. Urolithiasis. 2013;41(5):385–8.
Albiñana V, Gallardo-Vara E, de Rojas PI, Recio-Poveda L, Aguado T, Canto-Cano A, et al. Targeting β2-adrenergic receptors shows therapeutical benefits in clear cell renal cell carcinoma from Von Hippel–Lindau Disease. J Clin Med. 2020;9(9).
Shepard MJ, Bugarini A, Edwards NA, Lu J, Zhang Q, Wu T, et al. Repurposing propranolol as an antitumor agent in von Hippel–Lindau disease. J Neurosurg. 2018:1–9.
Boivin V, Jahns R, Gambaryan S, Ness W, Boege F, Lohse MJ. Immunofluorescent imaging of β1- and β2-adrenergic receptors in rat kidney. Kidney Int. 2001;59(2):515–31.
Meister B, Dagerlind A, Nicholas AP, Hökfelt T. Patterns of messenger RNA expression for adrenergic receptor subtypes in the rat kidney. J Pharmacol Exp Ther. 1994;268(3):1605–11.
Sprick JD, Morison DL, Stein CM, Li Y, Paranjape S, Fonkoue IT, et al. Vascular α(1)-adrenergic sensitivity is enhanced in chronic kidney disease. Am J Physiol Regul Integr Comp Physiol. 2019;317(3):R485–90.
Arif E, Nihalani D. Beta2-adrenergic receptor in kidney biology: A current prospective. Nephrology (Carlton). 2019;24(5):497–503.
Rains SL, Amaya CN, Bryan BA. Beta-adrenergic receptors are expressed across diverse cancers. Oncoscience. 2017;4(7–8):95–105.
Weikert S, Boeing H, Pischon T, Weikert C, Olsen A, Tjonneland A, et al. Blood pressure and risk of renal cell carcinoma in the European prospective investigation into cancer and nutrition. Am J Epidemiol. 2008;167(4):438–46.
Sung SY, Le TTH, Chen JH, Hsieh TF, Hsieh CL. Alpha-1 blocker use increased risk of subsequent renal cell carcinoma: A nationwide population-based study in Taiwan. PLoS ONE. 2020;15(11):e0242429.
Colt JS, Hofmann JN, Schwartz K, Chow WH, Graubard BI, Davis F, et al. Antihypertensive medication use and risk of renal cell carcinoma. Cancer Causes Control. 2017;28(4):289–97.
Neumann E, Klaiber P, Freitag K, Schwab M, Schaeffeler E, Hennenlotter J, et al. Assessment of concomitant non-oncologic medication in patients with surgically treated renal cell carcinoma: impact on prognosis, cell-cycle progression and proliferation. J Cancer Res Clin Oncol. 2019;145(7):1835–43.
Parker WP, Lohse CM, Zaid HB, Cheville JC, Boorjian SA, Leibovich BC, et al. Evaluation of beta-blockers and survival among hypertensive patients with renal cell carcinoma. Urol Oncol. 2017;35(1):36.e1-.e6.
Patel VG, Oh WK, Galsky MD, Liaw BC-H, Tsao C-K. Effect of concurrent beta-blocker (BB) use in patients receiving immune checkpoint inhibitors for metastatic urothelial (mUC) and renal cell carcinomas (mRCC). 2019;37(7_suppl):467.
Wang W, Cao X. Beta-Adrenergic Signaling in Tumor Immunology and Immunotherapy. Crit Rev Immunol. 2019;39(2):93–103.
Franzen D, Metha A, Seifert N, Braun M, Höpp HW. Effects of beta-blockers on sexual performance in men with coronary heart disease. A prospective, randomized and double blinded study. Int J Impot Res. 2001;13(6):348–51.
Meinhardt W, Kropman RF, Vermeij P, Nijeholt AA, Zwartendijk J. The influence of medication on erectile function. Int J Impot Res. 1997;9(1):17–26.
Adverse reactions to bendrofluazide and propranolol for the treatment of mild hypertension. Report of Medical Research Council Working Party on Mild to Moderate Hypertension. Lancet (London, England). 1981;2(8246):539–43.
Waal HJ. Propranolol-induced depression. BMJ. 1967;2(5543):50.
Petrie WM, Maffucci RJ, Woosley RL. Propranolol and depression. Am J Psychiatry. 1982;139(1):92–4.
Snaith RP, McCoubrie M. Antihypertensive drugs and depression. Psychol Med. 1974;4(4):393–8.
Paykel ES, Fleminger R, Watson JP. Psychiatric side effects of antihypertensive drugs other than reserpine. J Clin Psychopharmacol. 1982;2(1):14–39.
Kirk CA, Cove-Smith R. A comparison between atenolol and metoprolol in respect of central nervous system side effects. Postgrad Med J. 1983;59(689):161–3.
Zacharias FJ. Patient acceptability of propranolol and the occurence of side effects. Postgrad Med J. 1976;52(Suppl 4):87–9.
Kostis JB, Rosen RC. Central nervous system effects of beta-adrenergic-blocking drugs: the role of ancillary properties. Circulation. 1987;75(1):204–12.
Acknowledgements
Funding
This study was supported from the following funding agencies and foundations: National Institutes of Health NHLBI, R01HL148786 (N.D.); the Alzheimer's Disease Research Center at Mount Sinai Health (N.D.); National Institutes of Health/NIDDK R01DK120846 (B.P.); Department of Defense, W81XWH-17-1-0590 Grant (N.M.) and the National Institute of Nursing Research R21 NR016518-01A1 (N.M.); the Deane Prostate Health and The Arthur M. Blank Family Foundation (A.K.T); National Institutes of Health/NCI R01CA232574 Grant (N.K.).
Author information
Authors and Affiliations
Contributions
Conceptualization: M.A., D.J.L., N.D., J.A.K., S.A.K., K.T., A.L., Z.D., F.L., K.B., M.M., N.M., P.W., M.G., B.P., A.K.T., N.K. Research Material Acquisition: J.A.K., N.D., D.J.L., M.A., M.M., J.A.S., A.Z., M.R.N., G.S., T.G., F. L., N.M., B.P. Writing: M.A., T.G., L.B., N.D., H-S.J., J.A.K., F. L., Z.D., M.M., J.A.S., A.Z., A.L., N.M. N.K., Supervision: N.K. All authors have read and approved the final submitted version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Non-applicable.
Consent for publication
All authors have read and approved the final version of the article prior to submission for publication.
Competing interests
Dr. C-K Tsao has financial relationships in consulting with Clovis, Pfizer, and Eisai. Dr. Mantu Gupta receives financial compensation for educational training for Cook Urological Inc., Boston Scientific Inc., Olympus Inc., Lumenis Inc., and Retrophin Inc. Dr. Ashutosh K. Tewari has financial relationships in consulting with Intuitive Surgical, Promaxo, Roivant, Simens, and Kyte Pharma. He serves as an advisor for and owns equity in the form of stock certificates in Promaxo.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Archer, M., Dogra, N., Dovey, Z. et al. Role of α- and β-adrenergic signaling in phenotypic targeting: significance in benign and malignant urologic disease. Cell Commun Signal 19, 78 (2021). https://doi.org/10.1186/s12964-021-00755-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-021-00755-6