
Abstract
Background
Pyroptosis is a recently identified pathway of caspase-mediated cell death in response to microbes, lipopolysaccharide, or chemotherapy in certain types of cells. However, the mechanism of how pyroptosis is regulated is not well-established.
Methods
Herein, the intracellular bacteria were detected by staining and laser confocal microscopy and TEM. Live/dead cell imaging assay was used to examine macrophage death. Western blot and immunohistochemical staining were used to examine the protein changes. IFA was used to identify typical budding vesicles of pyroptosis and the STAT3 nuclear localization. SEM was used to observe the morphological characteristics of pyroptosis. ELISA was used to detect the level of inflammatory cytokines. Pyroptosis was filmed in macrophages by LSCM.
Results
S. aureus was internalized by human macrophages. Intracellular S. aureus induced macrophage death. S. aureus invasion increased the expression of NLRP3, Caspase1 (Casp-1 p20) and the accumulation of GSDMD-NT, GSDMD-NT pore structures, and the release of IL-1β and IL-18 in macrophages. Macrophages pyroptosis induced by S. aureus can be abrogated by blockage of S. aureus phagocytosis. The pyroptosic effect by S. aureus infection was promoted by either rapamycin or Stattic, a specific inhibitor for mTORC1 or STAT3. Inhibition of mTORC1 or STAT3 induced pyroptosis. mTORC1 regulated the pyroptosic gene expression through governing the nuclear localization of STAT3. mTORC1/STAT3 axis may play a regulatory role in pyroptosis within macrophages.
Conclusions
S. aureus infection induces human macrophage pyroptosis, inhibition of mTORC1/STAT3 axis facilitates S. aureus-induced pyroptosis. mTORC1 and STAT3 are associated with pyroptosis. Our findings demonstrate a regulatory function of the mTORC1/STAT3 axis in macrophage pyroptosis, constituting a novel mechanism by which pyroptosis is regulated in macrophages.
Video Abstract Macrophages were infected with S. aureus for 3 h (MOI 25:1), and pyroptosis was filmed in macrophages by laser confocal microscopy. A representative field was recorded. Arrow indicates lysing dead cell.
Similar content being viewed by others

Background
Pyroptosis is a lytic form of caspase-dependent cell death, which encompasses caspase-1, -4, -5, and -11. Caspase-1 is activated by various canonical inflammasomes, and caspase-4/5/11 recognizes cytosolic bacterial lipopolysaccharide directly, both of which trigger pyroptosis [1]. In the canonical pathway, intracellular bacteria upregulate caspase-1, which then cleaves gasdermin D to produce N-terminal GSDMD fragment (GSDMD-NT) [2]. GSDMD-NT forms pores in the membrane that drive swelling and membrane rupture, and the cytokines IL-1β and IL-18 are released through GSDMD-NT pore structures [3]. Pyroptosis can defend against microbial challenges and endogenous threats by eliminating such compromised cells [4]. Many factors cause pyroptosis, including lipopolysaccharide, chemotherapy drugs, TNF-a, 17β-estradiol (E2), and bacteria [5,6,7].
As extracellular pathogens, certain types of bacteria can invade a variety of mammalian nonprofessional phagocytes and can be engulfed by professional phagocytes, such as neutrophils and monocytes, and survive in them [8, 9]. The uptake of bacteria by nonprofessional phagocytes is mediated by adhesins [10]. Fibronectin-binding proteins, a major type of adhesin, mediate the internalization of adherent bacteria [11]. Professional phagocytic cells, such as neutrophils and macrophages, are designed to engulf microorganisms and clear debris—a process that can be divided conceptually into formation of the phagosome and subsequent evolution into a degradative compartment [12]. The monocyte-derived macrophages (MDMs) are separated into two specific phenotypes, classically activated macrophages (M1) and alternatively activated ones (M2) [13], Macrophages differentiate into M1 induced by IFN-γ and LPS, and the M1macrophages participate in the removal of pathogens during infection, induce M1 phenotype polarization which have main roles in host defense against various microbial pathogens and increased tumoricidal activity [14, 15]. Therefore, M1-polarized human MDMs were used as a cell model for macrophages function [16, 17].
Several pathogens can invade cells and survive intracellularly for various periods [18]. The success of intracellular pathogens is attributed to their ability to inhibit phagocytosis by preventing opsonophagocytosis and blocking specific signaling pathways [19], such as avoiding delivery to the lysosome and release into the cytoplasm and arresting phagosome maturation, creating an optimal niche for replication [20].
Staphylococcus aureus, a gram-positive bacterium and human pathogen that causes a wide range of illnesses, from skin infections to severe pneumonia and sepsis, can invade cells and trigger pyroptosis in human macrophages [7]. However, the mechanism by which it is regulated in professional phagocytes is unknown.
Mammalian target of rapamycin complex 1 (mTORC1) is a central coordinator in eukaryotic cells with regard to cell growth and metabolism with environmental inputs [21], autophagy, apoptosis, necroptosis, and other forms of cell death [22]. mTORC1 regulates autophagy by inhibiting ULK and the nuclear translocation of transcription factor EB (TFEB) [23, 24] and suppresses apoptosis in pterygium by controlling Beclin1-dependent autophagy by targeting Bcl-2 [25]. Among the intrinsic forms of cell death, pyroptosis has received increased attention recently, by the function of mTORC1 in pyroptosis is unkonwn.
Signal transducer and activator of transcription 3 (STAT3) is a latent transcription factor that mediates extracellular signals, such as those from cytokines and growth factors [26]. STAT3 plays an important role in programmed cell death, and inhibition of STAT3 leads directly to apoptosis [27]. In addition, autophagy is governed by STAT3 activation by upregulating or downregulating essential autophagy genes [28]. However, the relationship between STAT3 and pyroptosis has not been reported. Moreover, mTOR has been implicated in the regulation of STAT3 activation [29], and mTORC1 stimulates STAT3 to restrain proinflammatory responses [30]. Nevertheless, the regulatory function of mTORC1 in the expression of pyroptosic genes via STAT3 and pyroptosis is unknown.
Pyroptosic macrophages are important in the defense against microbial infections, removing pathogens and rendering them susceptible to phagocytosis and killing by a secondary phagocyte [31, 32]. As mentioned previously, mTORC1 and STAT3 are associated with autophagy and apoptosis. Thus, in this study, we hypothesized that mTORC1/STAT3 axis mediates pyroptosis via regulating expression of pyroptosic genes in human macrophages. We examined whether mTORC1 signaling regulates S. aureus-induced pyroptosis and whether the inhibition of the mTORC1/STAT3 axis causes pyroptosis in human macrophages. Collectively, our study demonstrates that mTORC1 and STAT3 have critical functions in the regulation of S. aureus-induced pyroptosis and that inactivation of the mTORC1/STAT3 axis causes pyroptosis in human macrophages. Our results implicate a novel function of the mTORC1/STAT3 axis in regulating cell death and provide insights into the mechanism by which pyroptosis is governed in professional phagocytic cells.
Methods
Reagents and antibodies
The anti-GSDMD (Cat# ab215191), anti-caspase-1 (Cat# ab1872), anti-S6 (Cat# ab184551), anti-4EBP1 (Cat# ab2606), anti-p-mTOR (Ser2448) (Cat# ab32028), anti-mTOR (Cat# ab10926), goat anti-rabbit (Cat# ab136817), and anti-mouse (Cat# ab205719) secondary antibodies were purchased from Abcam (Abcam plc 330 Cambridge Science Park, Cambridge, UK.) The anti-p-S6 (Ser240/244) (Cat# 5346s), anti-STAT3 (Cat# 4904s) anti-p-4EBP1 (Thr37/46) (Cat# 2855s), and anti-p-STAT3 (Cat# 4113s) were purchased from Cell Signaling Technology (Cell Signaling Technology, Inc., Beverley, MA, USA). The anti-GSDMD (Cat# abs128820) was purchased from Absin (Absin Co., Ltd. Shanghai, China). The goat FITC-conjugated anti-rabbit IgG (Cat# 115-095-003) and FITC-conjugated anti-mouse IgG (Cat# 115-095-146) were purchased from Jackson (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). The anti-β-actin (Cat# A5441) was purchased from Sigma (Sigma-Aldrich, Inc. St. Louis, MO, USA).
LPS (Cat# L2630), Stattic (Cat# S7947), CFSE (Cat# 21888), and PMA (Cat# P1585) were purchased from Sigma-Aldrich, Inc. (St. Louis, MO, USA). Rapamycin (Cat# 53123-88-9) was purchased from (Gene Operation, Ann Arbor, MI, USA). IFN-γ (Cat# 121527) was purchased from PEPROTECH (PEPROTECH Inc., Rocky Hill, USA). β-mercaptoethanol (Cat# M8211) was obtained from Solarbio (Solarbio Science and Technology, Co., Ltd. Beijing, China). Dil (Cat# KGMP002) was purchased from KeyGEN (KeyGEN BioTECH, Co., Ltd. Jiangsu, China). DAPI (Cat# C1005) was acquired from Beyotime (Beyotime Biotechnology, Co., Ltd. Shanghai, China). Alexa Fluor® 594 Phalloidin (Cat# A12381) was purchased from Invitrogen (Invitrogen, Carlsbad, New Mexico, USA). Cytochalasin B (Cat# HY-16928) was purchased from MCE (MedChemExpress, New Jersey, Monmouth, USA).
Cell culture
Human myeloid leukemia mononuclear (THP-1) cells and THP-1-derived macrophages were cultured in RPMI-1640 medium (Hyclone Laboratories, Inc. Logan, UT, USA) that was supplemented with 10% fetal bovine serum (BI Biological Industries, Beit Haemek Israel), 100 U/mL penicillin G, 100 mg/mL streptomycin (Sigma-Aldrich, Inc. St. Louis, MO, USA), and 0.1% β-mercaptoethanol (Solarbio Science and Technology, Co., Ltd. Beijing, China). To induce the classical (M1) polarization program, the medium was replaced with fresh medium without 10% fetal bovine serum and with PMA (100 ng/mL) for 24 h and then supplemented with 2.5 ng/mL IFN-γ and 100 ng/mL LPS for 24 h. Cells were cultured at 37 °C in humidified air with 5% CO2. Macrophages were infected by S. aureus (ATCC25923) at a multiplicity of infection (MOI) of 25:1 (bacteria to macrophages).
Spread plate method
Macrophages were infected with S. aureus for 3 h at an MOI of 25, and the extracellular bacteria were killed and lysed by antibiotics and lysozyme for 2 h. Monolayer macrophages were lysed, and the number of intracellular bacteria was determined by spread plate method.
Bacteria and macrophage staining
The macrophages were seeded onto a slide and incubated overnight. Bacteria (S. aureus) were washed with PBS and then incubated with CFSE (5(6)-carboxyfluorescein diacetate N-succinimidyl ester) at 4 °C for 15 min. The stained bacteria were centrifuged for 10 min at 3000×g at 4 °C 3 times. Macrophages were infected by the stained bacteria at an MOI of 25, washed with PBS 3 times, and then fixed with 4% paraformaldehyde for 20 min. After being treated with 1% Triton X-100 for 5 min, the macrophages were stained with Alexa Fluor® 594 Phalloidin for 1 h in the dark, washed with PBS 3 times, and counterstained with 100 μL DAPI for 3 min to assess the nuclear morphology. Finally, the slide was mounted with glycerinum for examination under a laser scanning confocal microscope (LSCM) (NIKON AIR, Nikon Corp., Tokyo, Japan).
Immunofluorescence assay (IFA)
The cells were seeded onto a slide, incubated overnight, washed with PBS, and fixed with 4% paraformaldehyde for 15 min. After treatment with 1% Triton X-100 for 10 min, the cells were blocked with 1% BSA for 1 h, stained with GSDMD/Caspase-1 primary antibody overnight at 4 °C, and incubated with FITC-labeled goat anti-rabbit/mouse IgG for 1 h at room temperature. Dil was added at 37 °C for 12 min to stain the membrane, and nuclear was stained with 100 μL DAPI for 3 min. Finally, the slide was mounted with glycerinum and examined under an LSCM (NIKON AIR, Nikon Corp., Tokyo, Japan).
TEM and SEM
Macrophages were infected by bacteria (S. aureus) for 3 h at an MOI of 25, and the extracellular bacteria were killed and lysed by antibiotics and lysozyme for 2 h. The infected macrophages were washed with PBS 3 times centrifuged for 10 min at 3000×g at 4 °C, and fixed with 2.5% glutaraldehyde overnight. Finally, the cells were embedded in 4% AGAR and fixed with 2.5% glutaraldehyde overnight. The samples were examined by TEM (Hitachi HT7700, Hitachi, Ltd., Tokyo, Japan) to detect intracellular bacteria. For SEM, macrophages were seeded onto a slide and incubated overnight. The macrophages were infected by bacteria or treated with rapamycin, washed with PBS, and fixed with 2.5% glutaraldehyde for 4 h. After being air-dried, the slides were examined by SEM (Hitachi S-4800, Hitachi, Ltd., Tokyo, Japan) to detect GSDMD-NT pores on the cytomembrane.
Western blot
Macrophages were washed with cold PBS and lysed in cell lysis buffer. The lysis buffer comprised 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1% PMSF, and phosphatase inhibitors. Equal amounts (40 μg) of protein were electrophoresed on 10% (w/v) sodium dodecyl sulfate–polyacrylamide gels, transferred to polyvinylidene fluoride membranes, and incubated with the primary antibody. Peroxidase-conjugated secondary antibody and enhanced chemiluminescence (ECL) reagent were used to detect the signals with the Western Blotting System (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA).
Immunohistochemical staining
Cells were seeded onto slides, incubated overnight, washed with PBS, and fixed with 4% paraformaldehyde for 15 min. For immunohistochemical detection of proteins (1000× magnification), antigens were detected using UltraSensitive™ S-P Immunohistochemistry hypersensitive kits (mouse/rabbit) (MXB Biotechnology Co. Ltd., Fujian, China) with hematoxylin (MXB Biotechnology Co. Ltd., Fujian, China) for counterstaining. A biotin-streptavidin peroxidase-based method was used to detect the primary antibodies.
ELISA
Levels of IL-1β and IL-18 in the supernatant was quantified by enzyme linked immunosorbent assay (ELISA) using NeoBioscience ELISA kits (Neobioscience Technology Co. Ltd., Shenzhen, China). The amount of culture media and intracellular α-hemolysin were measured via standard Mbbiolog ELISA kit (Mbbiology Biological Technology Co. Ltd., Jiangsu, China) according to the manufacturer’s guidelines. Absorbance at 450 nm and 630 nm was read on a Varioskan Flash Multimode Reader (Thermo Fisher Scientific, Pittsburgh, PA, U.S.A.). All measurements were performed in triplicate, and the mean value of 3 independent measurements was used for statistical analysis.
Confocal microscopy and light microscopy
Mounted IFA slides were observed under an LSCM (NIKON AIR, Nikon Corp., Tokyo, Japan). The macrophages phenotype, IHC slides, and large bubbles on the macrophages were observed under a light microscope (NIKON Eclipse80i, Nikon Corp., Tokyo, Japan).
qRT-PCR analysis
Quantitative real-time PCR (qPCR) was performed to determine the levels of CASP1, and GSDMD in macrophages in the treatment and control groups. For the bacterial infection, macrophages were infected with S. aureus for 3 h at an MOI of 25, and the extracellular bacteria were killed and lysed by antibiotics and lysozyme for 2 h. For the inhibitor treatment, macrophages were treated with 100 nM rapamycin for 6 h or 100 μM Stattic for 1 h, and the total RNA was extracted from untreated and treated cells. Total RNA was prepared with the RNAiso Plus reagent according the manufacturer's instructions (Takara Co. Ltd., Dalian China). Briefly, the cells were washed with PBS and lysed in RNAiso Plus, and chloroform was added to the cell lysates for homogenization. The top aqueous layer was transferred to a new tube after centrifugation, and isopropanol was added to the supernatant and mixed well. Total RNA was precipitated by centrifugation, and the pellet was dissolved in RNase-free water. RNA quantities over 600 ng/μL and a purity of 1.90–2.0, based on the 260/280 ratio, were used to synthesize cDNA.
mRNA was reverse-transcribed with oligo (dT)12–18 primer using the AMV 1st Strand cDNA Synthesis Kit (Takara Co. Ltd., Dalian China). ACTB was selected as the internal control gene. The primer sequences were as follows (5′–3′):
-
CASP1: forward, CGACAAGGTCCTGAAGGAGAAGAG
reverse, CGTGTGCGGCTTGACTTGTCCATT
-
GSDMD: forward, TGTACGTGGTGACTGAGGTGCTGC
reverse, CTGAGGGGATGGTGACCGTCTTCT
-
ACTB: forward, TCACCAACTGGGACGACAT
reverse, GCACAGCCTGGATAGCAAC
The KAPA SYBP® FAST qPCR Kit Optimized for LightCycler® 480 (KAPA BIOSYSTEMS, Inc., Boston, MA, U.S.A.) was used for the PCR according to the manufacturer’s instructions. The program comprised an initial denaturation step at 95 °C for 5 min; 40 cycles of 95 °C for 5 s, 54 °C for 30 s, and 72 °C for 20 s; and a final extension of 72 °C for 10 min. Three technical replicates were run in each experiment. 2−ΔΔCT values were calculated to determine expression levels, and the qPCR results were analyzed by student’s t-test to compare the expression between untreated and treated groups. Three independent experiments were performed.
Caspase-1 activity assay
The caspase-1 activity was measured using a Caspase1 Activity Assay kit (Beijing solarbio science and technology co., ltd. Beijing, China). According to the manufacturer’s instructions, about 50 μg protein from cells was mixed with synthetic tetrapeptide Ac-YVAD-pNA and incubated at 37 °C for 2 h. A standard curve was prepared using the pNA standard. The absorbance was determined at 405 nm with a 96-well plate reader and the caspase1 activity was normalized for total proteins of cell lysates.
Live/dead cell imaging assay
It is performed using the Live & Dead® Viability/Cytotoxicity Assay Kit (US Everbright, Inc., Suzhou, China). Macrophages were infected with S. aureus and incubated with 2 μM Calcein AM and 4 μM PI solution. Incubation at room temperature for 15–20 min and observe labeled cells under Live cell Imaging System (NIKON TI-E, Nikon Corp., Tokyo, Japan.)
Phagocytosis assays
Macrophages were treated with 100 nM rapamycin for 6 h or 10 μg/mL Cytochalasin B for 30 min, respectively, cells and S. aureus were co-cultured at an MOI of 25 for 3 h to enable phagocytosis. Phagocytosis was stopped by placing on ice and washing the macrophages cultures twice to remove non-phagocytosed bacteria. The extracellular bacteria were killed and lysed by antibiotics and lysozyme. Monolayer macrophages were lysed, and the number of intracellular bacteria was determined by spread plate method.
Statistical analyses
Statistical analyses were conducted using SPSS PASW Statistics for Windows, v18.0 (SPSS Inc.: Chicago, IL, USA). Data were analyzed using standard parametric statistics and one-way ANOVA, followed by Tukey’s method. Data are expressed as mean ± SD. The results are presented as the average of at least 3 independent experiments. Statistical significance was accepted when p ≤ 0.05.
Results
Phagocytosis of S. aureus by macrophages
M1-polarized human MDMs increase their caspase-1 activity, release of IL-1β, and loss of cell membrane integrity compared with the M2 program [7, 33]. To characterize S. aureus-induced pyroptosis in macrophages, THP-1 cells were differentiated into macrophages in culture with 100 ng/mL PMA (phorbol myristate acetate) for 24 h. Then, the macrophages were treated in differentiation medium with 2.5 ng/mL IFN-γ and 100 ng/mL LPS for 24 h to transform them into M1-polarized macrophages (Additional file 1: Figure S1a). The M1 macrophages released more TFN-α and IL-1β than the Mϕ phenotype (Additional file 1: Figure S1b), indicating that M1-polarized macrophages were successfully induced by IFN-γ and LPS.
To measure the internalization of S. aureus by macrophages, we infected M1 phenotype macrophages with S. aureus for 3 h and then killed and lysed the extracellular bacteria with antibiotics and lysozyme. The bacteria were evaluated intracellularly and extracellularly by bacterial colony count—7.5 ± 0.12 × 103 CFU/mL were counted in whole-cell lysates, whereas extracellular bacteria were not found in the culture medium (Additional file 2: Table S1). Further, intracellularly stained bacteria were observed under a laser scanning confocal microscope (LSCM) (Fig. 1a), and under a transmission electron microscope (TEM), S. aureus adhered to the cell membrane and invaded the macrophages (Fig. 1b). These data indicate that S. aureus was internalized by the macrophages.

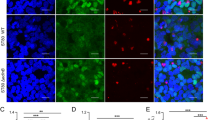
Staphylococcus aureus invades macrophages. S. aureus infected macrophages for 3 h, and then, the macrophages were cultured in medium supplemented with antibiotics and lysozyme to kill and lyse the extracellular bacteria. The intracellular bacteria were detected by staining and laser confocal microscopy and TEM. a The intracellular S. aureus (green) stained with CFSE was observed by laser confocal microscopy; macrophages nuclei were co-stained with DAPI (blue), and actin was stained with phalloidin (red). Scale bars represent 50 μm. b Bacteria were attached to the cell membrane and engulfed, based on micrographs obtained by TEM; several important observations are magnified. Red arrows indicate S. aureus. Scale bars represent 2 μm. a′ Control, non-infected human macrophages. b′ The bacteria were located predominantly in the plasma membrane; macrophages were allowed to engulf bacteria and initiate phagocytosis. c′ S. aureus was engulfed by macrophages via the formation of typical phagocytic cups
Intracellular S. aureus induces pyroptosis of macrophages
Considering S. aureus can induce NLRP3-mediated signaling triggering caspase-1 activation and programmed necrosis through the production of a pore-forming toxin in macrophages [7, 34], to eliminate the possibility of pore-forming toxin induces NLRP3 inflammasome-dependent pyroptosis during S. aureus infection period, α-hemolysin as the most prominent [35], we first evaluated the levels of α-hemolysin (Hla) in cell medium and in macrophages. The results showed that α-hemolysin was not detectable both in cell medium and macrophages (Additional file 3: Figure S2a), indicating α-hemolysin was not accumulated during the 3 h infection period. To determine whether inhibition of phagocytosis was specifically restricted to the engulfment of S. aureus and has effect on pyroptosis, macrophages were treated with Cytochalasin B, an inbibitor of phagocytosis [36], and intracellular bacteria were detected. The results showed that the number of intracellular bacteria were statistically decreased in Cytochalasin B treated cells (Additional file 3: Figure S2b). Furthermore, the activation of NLRP3 and Caspase-1, the expression of GSDMD-NT, IL-1β, and IL-18 triggered by S. aureus were abrogated by Cytochalasin B (Fig. 2a, b). These data indicate that blockage of S. aureus phagocytosis could abrogate the pyroptosis induction.

Staphylococcus aureus triggers pyroptosis in human macrophages. Cells were infected with S. aureus for 3 h (MOI 25:1), and the pyroptotic characteristics were examined, including pyroptosic protein markers, inflammatory cytokines release, and morphology. a Expression of pyroptosis-related proteins in response to S. aureus invasion and Cytochalasin B was assessed by western blot. NLRP3, caspase1-p20 and GSDMD-NT proteins were less prominent than S. aureus invasion after Cytochalasin B treated. b The levels of IL-18 and IL-1β were quantified by an ELISA. The levels of IL-18 and IL-1β were less prominent than S. aureus invasion after Cytochalasin B treated. c Western blot analysis showed S. aureus enhanced NLRP3 and caspase1-p20 expression in infected macrophages. d Immunofluorescence assay showed greater caspase 1 expression in infected macrophages than in the control. Representative confocal microscopy images of caspase 1 expression (Green) in cells that were co-stained with DAPI (blue). Scale bars represent 10 μm. e GSDMD-NT expression was examined in S. aureus-infected macrophages and controls by western blot. S. aureus invasion induced greater GSDMD-NT expression in infected macrophages. f Immunofluorescence assay showed greater GSDMD-NT expression in infected macrophages than in the control. Representative confocal microscopy images of GSDMD-NT expression (green) in cells that were co-stained with DAPI (blue). Scale bars represent 10 μm. g IL-18 and IL-1β levels were determined by ELISA. S. aureus invasion induced more release of IL-18 and IL-1β in infected macrophages. h Scanning electron microscopy of GSDMD-NT pores on plasma membrane in S. aureus-infected macrophages. Red arrow indicates GSDMD-NT pore. Scale bar, 500 nm. i GSDMD-NT (green) in cells co-stained with Dil (red) as a membrane marker. Representative confocal microscopy images of S. aureus-infected macrophages and control cells by immunofluorescence assay. The purple arrows indicate the necks of budding vesicles, indicating shedding of wounded plasma membrane in S. aureus-infected macrophages. Scale bars represent 1 μm. The resolved bands were quantified using Gel-Pro Analyzer 4.0 (Media Cybernetics, Inc., Rockville, MD, USA). Fluorescence intensity of the immunofluorescent was measured by imaging analysis software (NIS-Elements Viewer, Nikon Instruments Inc. Shanghai, China). *p < 0.05; **p < 0.01. n = 3 independent experiments. Error bar indicates SD
Intracellular bacteria may lead to cell pyroptosis, which is accompanied by caspase-1 activation, following the generation of GSDMD-NT fragment to form membrane pores and the release of IL-18 and IL-1β [37]. To determine whether pyroptosis is induced by S. aureus infection in human macrophages, we first measured the expression of several markers by western blot, immunofluorescence, and immunohistochemistry staining. NLRP3, Caspase-1 (Casp-1 p20) (Fig. 2c, d) and GSDMD-NT (Fig. 2e, f and Figure S2c), were upregulated in cells that harboured intracellular S. aureus. By ELISA, compared with control, IL-18 and IL-1β levels were significantly increased (p < 0.05) in the medium of cells that contained bacteria (Fig. 2g).
Next, to examine the morphological characteristics of macrophages that were infected by S. aureus, a scanning electron microscope (SEM) was used to observe the GSDMD-NT pore structures on the plasma membrane. Pore structures with ~ 30-nm diameters were observed (Fig. 2h). Moreover, when the cell membrane was damaged, the exocytic patching mechanism was initiated to repair the membranes by removing the pores in cells with few gasdermin pores [38]. We examined the typical budding vesicles that act during the shedding of damaged plasma membranes using LSCM, interesting, we found that budding vesicles were present in macrophages with intracellular S. aureus but not control cells (Fig. 2i). Further, once GSDMD pores form at levels that exceed the cell’s compensatory abilities, cells begin to swell, with characteristic large bubbles that protrude from the plasma membrane [39]. Thus, a light microscope was used to examine the bubbles that formed on the macrophages—large bubbles were observed on cells with intracellular S. aureus versus control cells (Additional file 3: Figure S2d). By video, cell swelling with characteristic large bubbles and lytic cells were seen (Video Abstract). Notably, by live/dead cell imaging assay, S. aureus was found to induce macrophage death (Additional file 4: Video 1). Taken together, these results suggest that intracellular S. aureus induces pyroptosis in human macrophages.
Rapamycin promotes S. aureus-induced pyroptosis in macrophages
As discussed, mTORC1 regulates autophagy and apoptosis. Thus, we hypothesized that S. aureus-induced pyroptosis is associated with mTORC1 signaling. We treated S. aureus-invaded cells with rapamycin and then examined the expression of caspase-1 (Casp-1 p20), NLRP3 and GSDMD-NT by western blot, immunofluorescence, and immunohistochemistry staining and the levels of IL-18 IL-1β in the cell medium by ELISA. Rapamycin upregulated the S. aureus-induced expression of caspase-1 (Casp-1 p20) and NLRP3 (Fig. 3a, b), GSDMD-NT (Fig. 3c, d and Additional file 5: Figure S3a) and the release of IL-18 and IL-1β Fig. 3e) (p < 0.01). Further, rapamycin promoted the formation of S. aureus-induced GSDMD-NT membrane pores (Fig. 3f) and bubbles (Fig. 3g). Moreover, to assess the effect of rapamycin on macrophage uptake of S. aureus, macrophages that had engulfed bacteria were analysed by bacterial colony count. Notably, rapamycin did not influence the ability of macrophages to phagocytose bacteria (Additional file 5: Figure S3b). These data suggest that rapamycin accelerates S. aureus-induced pyroptosis in macrophages and that mTORC1 is involved in pyroptosis.

Rapamycin promotes Staphylococcus aureus-induced pyroptosis in macrophages. Macrophages were pretreated with rapamycin (100 nM) for 6 h and then infected with S. aureus for 3 h. The pyroptotic characteristics were determined, including pyroptosic protein markers, inflammatory cytokine release, and morphology. (a Western blot analysis of S. aureus-induced NLRP3 and caspase1-p20 expression. b Immunofluorescence assay of S. aureus-induced caspase 1 expression. Representative confocal microscopy images of caspase 1 (green) in cells that were co-stained with DAPI (blue). Scale bars represent 10 μm. c GSDMD-NT expression by western blot. d GSDMD-NT expression by immunofluorescence assay. Representative confocal microscopy images of GSDMD-NT (green) in cells that were co-stained with DAPI (blue). Scale bars represent 10 μm. e Macrophages were pretreated with rapamycin for 6 h and then infected with S. aureus for 3 h. The levels of IL-18 and IL-1β in cell culture medium were determined by ELISA. f GSDMD-NT pores on plasma membrane by scanning electron microscopy. Red arrow indicates GSDMD-NT pore. Scale bar, 500 nm. g Large bubbles by light microscopy, as indicated by red arrows in pyroptotic cells. Scale bars represent 1 μm. The resolved bands were quantified using Gel-Pro Analyzer 4.0 (Media Cybernetics, Inc., Rockville, MD, USA). Fluorescence intensity of the immunofluorescent was measured by imaging analysis software (NIS-Elements Viewer, Nikon Instruments Inc. Shanghai, China). *p < 0.05; **p < 0.01. n = 3 independent experiments. Error bar indicates SD
mTORC1 inhibition causes macrophage pyroptosis
Based on the data above, we speculated that rapamycin induces pyroptosis in macrophages. To this end, we treated cells with 100 nM rapamycin for 6 h. We first examined mTORC1 signaling and found the phosphorylation of mTOR, S6, and 4EBP1 decreased (Additional file 6: Figure S4a), indicating that the mTORC1 pathway is inhibited by rapamycin. By western blot, immunofluorescence, and immunohistochemistry, NLRP3, caspase-1 (Casp-1 p20), (Fig. 4a, b) and GSDMD-NT (Fig. 4c, d and Additional file 6: Fig. S4b) were upregulated by rapamycin, and the release of IL-18 and IL-1β was enhanced by rapamycin (Fig. 4e) (p < 0.01). Meanwhile, using a colorimetric assay to assess caspase-1 activity during rapamycin treatment, we found that caspase-1 activity was significantly increased (Fig. 4f). As in S. aureus-invaded cells, rapamycin induced the formation of GSDMD-NT membrane pores (Fig. 4g) by SEM. Budding vesicles from the plasma membrane were observed under an LSCM (Fig. 4h), acting as a membrane damage repair mechanism and appearing early during pyroptosis. Bubbles were also observed under a light microscope (Fig. 4i). Further, by video, cell swelling with characteristic large bubbles and lytic cells were seen (Additional file 7: Video 2). These data indicate that inhibition of mTORC1 causes pyroptosis in macrophages and that mTORC1 signaling has inhibitory effects on pyroptosis in macrophages.

Rapamycin causes pyroptosis in macrophages. Macrophages were treated with rapamycin (100 nM) for 6 h, and the pyroptotic characteristics were examined. a Western blot of NLRP3 and caspase1-p20 expression. b Immunofluorescence assay of caspase 1 expression. Representative confocal microscopy images of caspase 1 expression (green) in cells that were co-stained with DAPI (blue). Scale bars represent 10 μm. c Rapamycin upregulates GSDMD-NT expression. d Rapamycin upregulates GSDMD-NT expression in cells. Representative confocal microscopy images of caspase 1 expression (green) in cells that were co-stained with DAPI (blue) by immunofluorescence assay. Scale bars represent 10 μm. e Rapamycin increases the levels of IL-18 and IL-1β in cell culture medium. f Whole cell lysates were extracted from macrophages, Caspase-1 activity was determined by colorimetric assay and induced by rapamycin treatments. g Scanning electron microscopy of GSDMD-NT pores on plasma membrane in rapamycin-treated macrophages. Red arrow indicates GSDMD-NT pore. Scale bar, 500 nm. h GSDMD-NT (green) in cells co-stained with Dil (red) as membrane marker. Representative confocal microscopy images of rapamycin-treated macrophages and control cells by immunofluorescence assay. The purple arrows indicate the necks of budding vesicles in rapamycin-treated macrophages. Scale bars represent 1 μm. i Large bubbles by light microscopy, indicated by red arrows in pyroptotic cells. Scale bars represent 1 μm. The resolved bands were quantified using Gel-Pro Analyzer 4.0 (Media Cybernetics, Inc., Rockville, MD, USA). Fluorescence intensity of the immunofluorescent was measured by imaging analysis software (NIS-Elements Viewer, Nikon Instruments Inc. Shanghai, China). *p < 0.05; **p < 0.01. n = 3 independent experiments. Error bar indicates SD
mTORC1 regulates the expression of CASP1 and GSDMD through STAT3 in macrophages
The experiments above demonstrated that rapamycin increased the protein expression levels of caspase-1 (Casp-1 p20) and GSDMD-NT; previous study has shown that STAT3 is associated with apoptosis and autophagy and thus [28], we hypothesized that mTORC1 may regulate these proteins through STAT3 in macrophages.
To determine whether mTORC1 regulates the expression of CASP1 and GSDMD via STAT3, we first treated macrophages with 100 nM rapamycin for 6 h and found that repamycin reduced the phosphorylation of STAT3, indicating STAT3 activation was inhibited (Fig. 5a). Then, we measured the mRNA levels of these pyroptosic genes by qRT-PCR, demonstrating that rapamycin upregulates CASP1 and GSDMD (Fig. 5b) (p < 0.01). These data suggest that mTORC1 regulates the expression of pyroptosic genes, likely through STAT3 in macrophages.

mTORC1 regulates the expression of CASP1 and GSDMD through STAT3 in macrophages. Macrophages were treated with rapamycin (100 nM) for 6 h, and the activity of mTORC1 and STAT3 and the expression of pyroptosic genes were examined. a Western blot analysis of mTORC1 activation and STAT3 activation. Phosphorylation of STAT3 was inhibited. b Expression of CASP1 and GSDMD, mRNA upregulated by rapamycin. The resolved bands were quantified using Gel-Pro Analyzer 4.0 (Media Cybernetics, Inc., Rockville, MD, USA). *p < 0.05; **p < 0.01. n = 3 independent experiments. Error bar indicates SD
STAT3 inhibition causes macrophage pyroptosis through upregulations of CASP1 and GSDMD
To determine whether STAT3 affects the expression of CASP1, and GSDMD, we treated cells with Stattic, a selective inhibitor of STAT3 activation and dimerization. Stattic inhibited STAT3 phosphorylation in macrophages (Fig. 6a). Stattic increased the mRNA levels of CASP1 and GSDMD (Fig. 6b), indicating that their expression was governed by STAT3 in macrophages. Further, cells were pretreated with Stattic and exposed to S. aureus produced more NLRP3, Casp-1 p20 and GSDMD-NT comparing to only Stattic treated and untreated cells (Fig. 6c). By ELISA, the levels IL-18 and IL-1β in the cell medium increased in treated cells (Fig. 6d) (p < 0.01). In addition, Stattic treatment significantly induced caspase-1 activation (Fig. 6e). These data suggest that inhibition of STAT3 induces pyroptosis in macrophages. By video, pseudopod contracting and cell rounding (Additional file 8: Video 3), cell swelling with characteristic large bubbles and lytic cells were observed (Fig. 6f and Additional file 9: Video 4). Thus, STAT3 is critical in pyroptosis, controlling the expression of pyroptosic genes in human macrophages.

Inhibition of STAT3 by Stattic upregulates expression of pyroptosic genes and causes pyroptosis in macrophages. Macrophages were treated with Stattic (100 μM) for 1 h to inhibite STAT3 activation. The expression of pyroptosic genes and pyroptosic protein markers and the release of inflammatory cytokines were examined. a Stattic inhibits STAT3 phosphorylation in cells. b Stattic enhances the expression of CASP1 and GSDMD mRNA in macrophages. c Stattic enhances NLRP3, caspase1-p20 and GSDMD expression, pyroptosis-related proteins were severally elevated in S. aureus infected cells which pretreated with Stattic. d Stattic increases the production of IL-18 and IL-1β in cell culture medium. e Caspase-1 activity was enhanced by Stattic treatment. f Stattic triggers pyroptosis. Red arrows indicate large bubbles in pyroptic cells. Scale bars represent 100 μm. The resolved bands were quantified using Gel-Pro Analyzer 4.0 (Media Cybernetics, Inc., Rockville, MD, USA). *p < 0.05; **p < 0.01. n = 3 independent experiments. Error bar indicates SD
Inactive mTORC1 prevents the nuclear localization of STAT3 in macrophages
Based on the results above, we reasoned that mTORC1 governs the nuclear localization of STAT3 to regulate the expression of pyroptosic genes. To this end, immunofluorescence was performed to confirm whether mTORC1 promotes the nuclear localization of STAT3. Cells were treated with rapamycin or Stattic as a positive control. The nuclear localization of STAT3 was prevented in cells that were treated with either compound, compared with control (Fig. 7a, b), indicating that the nuclear localization of STAT3 is controlled by mTORC1. These results demonstrate that mTORC1 signaling negatively regulates human macrophage pyroptosis by regulating the nuclear localization of STAT3 and pyroptosic gene expression (Fig. 7c).

Inactive mTORC1 prevents the nuclear localization of STAT3 in macrophages. a Nuclear localization of STAT3 was prevented in cells treated with rapamycin. Scale bars represent 10 μm. The nuclear fluorescence intensity of STAT3 was shown. b Nuclear localization of STAT3 was prevented in cells treated with Stattic. Scale bars represent 10 μm. The fluorescence intensity of nuclear STAT3 was graphed. c The model of mTORC1 regulating pyroptosis through nuclear localization of STAT3 and pyroptosic gene expression in human macrophage. STAT3 is likely a negative regulator of pyroptosic gene expression. mTORC1 inhibition by rapamycin or STAT3 inhibition by Stattic prevents the nuclear localization of STAT3, upregulates pyrotosic gene expression, triggering human macrophage pyroptosis
Discussion
During cell death, cell-intrinsic effector functions are coordinated to restrict infection and resolve innate and adaptive immune responses. The most widely used classification of cell death had consisted of 2 types: apoptosis and necrosis [40], but in recent years, necroptosis and pyroptosis have been confirmed [41]. Apoptotic cells retain plasma membrane integrity, versus pyroptosis or necroptosis is the rupture of cell lytic, which weakens the integrity of the plasma membrane and allows the influx of extracellular ions and fluid, leading to cell swelling [42].
In lytic pyroptosis, variety of cytokines are released, rendering it an inflammatory event [43]. Pyroptosis is a form of caspase-mediated cell death, and its regulatory mechanism is poorly understood. Studies have demonstrated that rapamycin induces apoptosis in peritoneal carcinomatosis [44] and necrosis in cardiac cells [45], and mTORC1 negatively regulates autophagy by inhibiting ULK [24]. mTORC1 is a central regulator of apoptosis, necrosis, and autophagy. However, the regulatory function of mTORC1 in pyroptosis has not been reported. In this study, we found that S. aureus induces pyroptosis in human macrophages and that the inhibition of mTORC1 by rapamycin promotes S. aureus-induced pyroptosis. These data suggest that mTORC1 regulates pyroptosis in macrophages.
Pyroptosis was initially observed in macrophages that were infected with Salmonella typhimurium or Shigella flexneri [46, 47]. Cytosol-invasive bacteria, such as Listeria monocytogenes, induce pyroptosis [48]. Of note, S. aureus pore-forming toxins (PFTs) induce NLRP3-mediated signaling, triggering caspase-1 activation and pyroptosis in human and mouse monocytic cells [49]. In our study, the effect of PFTs on pyroptosis have been eliminated and pyroptosis can be abrogated by blockage of S. aureus phagocytosis. These data indicate that S. aureus internalized macrophages, and intracellular S. aureus induced pyroptosis in professional phagocytic cells. Furthermore, inactive mTORC1 did not affect the ability of macrophages to phagocytose bacteria, our data may reveal the biological role of pyroptosis in S. aureus invasion.
In general, plasma membrane damage can be repaired efficiently in macrophages, for which endocytosis, membrane patching, and extracellular budding can be used [50]. Once gasdermin pores are present in numbers that exceed the cell’s compensatory abilities, the cells swell and the plasma membrane separates from the cytoskeleton in large fluid-filled bubbles and cell lytic dying [39]. In the present study, GSDMD-NT pore, swelling, and membrane rupture during pyroptosis—were observed. Further, our video showed pyroptosic cells swelling with characteristic large bubbles and eventually lysing with rapamycin or Stattic treatment. This morphological evidence demonstrates that S. aureus infection, mTORC1 inhibition, and STAT3 inhibition induce macrophage pyroptosis.
In our previous work, mTORC1 signaling was initiated by peptidoglycan (PGN) from S. aureus in mouse macrophages [51]. In the present study, we treated S. aureus-infected macrophages with rapamycin to inhibit mTORC1 and examine how S. aureus-induced pyroptosis is altered. We were surprised to find that S. aureus-induced pyroptosis was promoted by rapamycin in macrophages, for which there are 2 possibilities: rapamycin enhances S. aureus to induce pyroptosis and mTORC1 inhibition causes pyroptosis, in which case mTORC1 inactivation is an independent factor for macrophage pyroptosis. In rapamycin-treated cells, mTORC1 inactivation upregulated pyroptosic proteins, including CASP1 and GSDMD, release of IL-1β, and induced macrophage pyroptosis. Several studies in cells of mice and human reported that the negative regulation of inflammasome by rapamycin. Muhamuda et al. (2017) showed that rapamycin treatment (10 μM, 24 h) of lethal Ixodes ovatus ehrlichia (IOE) -infected WT-BMM attenuated production of IL-1β, which is the gold standard readout of inflammasome activity [52]. In addition, another group have observed that mTORC1 signaling inhibition by rapamycin (100 nM, 24 h) suppressed IL-1β secretion in these cells monocyte-derived macrophages (MDMs) [53]. In contrast, Rojas Márquez et al. [54] showed that mTORC1 inhibition by rapamycin (100 nM, 90 min) upregulation of IL-1β production in macrophages; Chimin et al. [55] reported that adipocyte mTORC1 deficiency promotes adipose tissue inflammation and NLRP3 inflammasome activation in mice with raptor deletion, which are in line with our findings of mTORC1 inhibition by rapamycin (100 nM, 6 h) upregulation of IL-1β and caspase-1 productions in the present study.
As a latent transcription factor, STAT3 modulates a range of target genes, including induced and repressed target genes that mediate cellular and organismal functions [56]. Moreover, STAT3 can be activated by LPS in human monocytes [30] and by PGN in mouse macrophages [51]. STAT3 regulates programmed cell death. Constitutive STAT3 activation leads directly to the induction of BCL-X, which inhibits of apoptosis [25]. STAT3 executes anti-autophagic functions by upregulating negative regulators of autophagy, such as MCL1, PIK3R1/p55a, and PIK3R1/p50a [26], and by downregulating essential autophagy genes, such as BECN1 and PIK3C3 [57]. Moreover, STAT3 negatively regulates gene expression in MEFs and cancer lines to control type I IFN-mediated antiviral response [58,59,60]. In fact, a whole-transcriptome profiling study showed that STAT3 acts as a transcriptional activator and suppressor, with a comparable number of upregulated and downregulated genes in diffuse large B cell lymphoma (DLBCL) cells [61]. In our cases, STAT3 was inhibited by the selective inhibitor Stattic, resulting in the upregulation of pyroptosic genes, including CASP1, GSDMD and triggering pyroptosis in human macrophages.
The nuclear localization of STAT3 comprises phosphorylation, dimerization, and nuclear localization, can be blocked by Stattic [62, 63]. In addition, phosphorylation of the Ser727 residue in the carboxyl transactivation domain might positively regulate STAT3 transcriptional activation; Dodd et al. demonstrated that STAT3 is phosphorylated directly by mTORC1 on Ser727 [64,65,66], confirming that mTORC1 functions in STAT3 activation. In a previous study, we found that rapamycin inhibits STAT3 phosphorylation [67], indicating that mTORC1 can regulate STAT3 activation. In the present study, mTORC1 was inhibited by rapamycin, accompanied by STAT3 inactivation and upregulation of pyroptosic gene. In the regulation of pyroptosis, STAT3 consistently had inhibitory functions, in accordance with the function of mTORC1 in pyroptosis in human macrophages. Further, we found that rapamycin prevents the nuclear localization of STAT3. Thus, mTORC1 combines with STAT3 to form the mTORC1/STAT3 axis to control pyroptosis in human macrophages.
Conclusion
In summary, mTORC1 and STAT3 regulate human macrophage pyroptosis. The mTORC1/STAT3 axis is critical in pyroptosis in human macrophages. S. aureus invaded macrophages to induce pyroptosis; this process was promoted by rapamycin. mTORC1 regulates the expression of pyroptosic genes, including CASP1 and GSDMD, through STAT3. Inhibition of mTORC1 prevents the nuclear localization of STAT3. The pathogenic effects of the inhibition of the mTORC1/STAT3 axis facilitates S. aureus-induced pyroptosis. This study demonstrates a regulatory function of the mTORC1/STAT3 axis in macrophage pyroptosis, constituting a novel mechanism by which pyroptosis is regulated in macrophages.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- MDMs:
-
Monocyte-derived macrophages
- THP-1:
-
Human myeloid leukemia mononuclear
- M1:
-
Classically activated macrophages
- mTORC1:
-
Mammalian target of rapamycin complex 1
- STAT3:
-
Signal transducer and activator of transcription 3
- PMA:
-
Phorbol myristate acetate
- IFN-γ:
-
Interferon-γ
- LPS:
-
Lipopolysaccharide
- S. aureus :
-
Staphylococcus aureus
- GSDMD:
-
Gasdermin D
- caspase-1:
-
Cysteine-requiring aspartate protease 1
- S6:
-
Ribosomal protein S6
- 4EBP1:
-
The eukaryotic initiation factor 4E binding protein 1
- β-actin:
-
Actin beta
- IL-1β:
-
Interleukin-1 beta
- IL-18:
-
Interleukin-18
- NLRP3:
-
NLR family pyrin domain containing 3
References
Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109.
Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265(1):130–42.
Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17(3):151–64.
Ezquerro S, Mocha F, Frühbeck G, et al. Ghrelin reduces TNF-α-induced human hepatocyte apoptosis, autophagy and pyroptosis: role in obesity-associated NAFLD. J Clin Endocrinol Metab. 2019;104(1):21–37.
Wei Q, Zhu R, Zhu J. E2-Induced Activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC Cells. Oncol Res. 2019;27(7):827–34.
Accarias S, Lugo-Villarino G, Foucras G, et al. Pyroptosis of resident macrophages differentially orchestrates inflammatory responses to Staphylococcus aureus in resistant and susceptible mice. Eur J Immunol. 2015;45(3):794–806.
Molinari G, Rohde M, Guzman CA, et al. Two distinct pathways for the invasion of streptococcus pyogenes in non-phagocytic cells. Cell Microbiol. 2000;2(2):145–54.
Gresham HD, Lowrance JH. Caver TE (2000) Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. 2000;164(7):3713–22.
Foster TJ. The remarkably multifunctional fibronectin binding proteins of Staphylococcus aureus. Eur J Clin Microbiol Infect Dis. 2016;35(12):1923–31.
Garzoni C, Kelley WL. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 2009;17(2):59–65.
Lacoma A, Cano V, Moranta D, et al. Investigating intracellular persistence of Staphylococcus aureus within a murine alveolar macrophage cell line. Virulence. 2017;8(8):1761–75.
Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20.
Shirey KA, Cole LE, Keegan AD, et al. Francisella tularensis live vaccine strain induces macrophage alternative activation as a survival mechanism. J Immunol. 2008;181(6):4159–67.
Moghaddam AS, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization and function in health and disease. J Cell Physiol. 2018;233(9):6425–40.
Zhang H, Lin C, Zeng C, et al. Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through R-spondin-2. Ann Rheum Dis. 2018;77(10):1524–34.
Ortiz-Masiá D, Cosín-Roger J, Calatayud S, et al. M1 macrophages activate Notch Signalling in epithelial cells: relevance in Crohn’s disease. J Crohns Colitis. 2016;10(5):582–92.
Kubica M, Guzik K, Koziel J, et al. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS ONE. 2008;3(1):e1409.
Sarantis H, Grinstein S. Subversion of phagocytosis for pathogen survival. Cell Host Microbe. 2012;12(4):419–31.
Vergne I, Chua J, Lee HH, et al. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2005;102(11):4033–8.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361–71.
Abe K, Yano T, Tanno M, et al. mTORC1 inhibition attenuates necroptosis through RIP1 inhibition-mediated TFEB activation. Biochim Biophys Acta Mol Basis Dis. 2019;1865(12):165552.
Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–14.
Ganley IG, du Lam H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–305.
Liu Y, Xu H, An M. mTORC1 regulates apoptosis and cell proliferation in pterygium via targeting autophagy and FGFR3. Sci Rep. 2017;7(1):7339.
LevyDarnell DEDJ Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–62.
Epling-Burnette PK, Liu JH, Catlett-Falcone R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest. 2001;107(3):351–62.
Pensa S, Lloyd-Lewis B, Sargeant TJ, et al. Signal transducer and activator of transcription 3 and the phosphatidylinositol 3-kinase regulatory subunits p55α and p50α regulate autophagy in vivo. FEBS J. 2014;281(20):4557–67.
Kayleigh MD, Jian Y, Ming HS, et al. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene. 2015;34(17):2239–50.
Weichhart T, Costantino G, Poglitsch M, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29(4):565–77.
Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27(9):673–84.
Orning P, Weng D, Starheim K, et al. Pathogen blockade of TAK1 triggers caspase-8 dependent cleavageof gasdermin D and cell death. Science. 2018;362(6418):1064–9.
Alberto M, Antonio S, Silvano S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–86.
Xiaogang W, William JE, Jean CL, et al. Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicle. Proc Natl Acad Sci USA. 2020;117(6):3174–84.
Ichiro I, Naoko I, Georgia AW, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 2011;17(10):1310–4.
Mattila JT, Munderloh UG, et al. Phagocytosis of the Lyme disease spirochete, Borrelia burgdorferi, by cells from the ticks, Ixodes scapularis and Dermacentor andersoni, infected with an endosymbiont, Rickettsia peacockii. J Insect Sci. 2007;7:58.
Jorgensen I, Lopez JP, Laufer SA, et al. IL-1β, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol. 2016;46(12):2761–6.
Rühl S, Shkarina K, Demarco B, et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362(6417):956–60.
Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–25.
Galluzzi L, Maiuri MC, Vitale I, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14(7):1237–43.
Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541.
Gong YN, Guy C, Olauson H, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169(2):286–300.
Bliss-Moreau M, Chen AA, D’Cruz AA, et al. A motive for killing: effector functions of regulated lytic cell death. Immunol Cell Biol. 2017;95(2):146–51.
Song X, Dilly AK, Kim SY, et al. Rapamycin-enhanced mitomycin C-induced apoptotic death is mediated through the S6K1-Bad-Bak pathway in peritoneal carcinomatosis. Cell Death Dis. 2014;5:e1281.
Lassaletta AD, Elmadhun NY, Zanetti AV, et al. Rapamycin treatment of healthy pigs subjected to acute myocardial ischemia-reperfusion injury attenuates cardiac functions and increases myocardial necrosis. Ann Thorac Surg. 2014;97(3):901–7.
Monack DM, Raupach B, Hromockyj AE, et al. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci USA. 1996;93(18):9833–8.
Hilbi H, Moss JE, Hersh D, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem. 1998;273(49):32895–900.
Jorgensen I, Zhang Y, Krantz BA, et al. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016;213(10):2113–28.
Craven RR, Gao X, Allen IC, et al. Staphylococcus aureus α-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE. 2009;4(10):e7446.
Jimenez AJ, Maiuri P, Lafaurie-Janvore J, et al. ESCRT machinery is required for plasma membrane repair. Science. 2014;343(6174):1247136.
Vangan N, Cao Y, Jia X, et al. mTORC1 mediates peptidoglycan induced inflammatory cytokines expression and NF-kB activation in macrophages. Microb Pathog. 2016;99:111–8.
Muhamuda K, Mounia AEA, Jennie V, et al. MyD88-dependent inflammasome activation and autophagy inhibition contributes to Ehrlichia-induced liver injury and toxic shock. PLoS Pathog. 2017;13(10):e1006644.
Ip WKE, Hoshi N, Shouval DS, et al. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–9.
Rojas Márquez JD, Ana Y, Baigorrí RE, et al. Mammalian Target of rapamycin Inhibition in Trypanosoma cruzi-infected macrophages leads to an intracellular profile that is detrimental for infection. Front Immunol. 2018;9:313.
Chimin P, Andrade ML, Belchior T, et al. Adipocyte mTORC1 deficiency promotes adipose tissue inflammation and NLRP3 inflammasome activation via oxidative stress and de novo ceramide synthesis. J Lipid Res. 2017;58(9):1797–807.
You L, Wang Z, Li H, et al. The role of STAT3 in autophagy. Autophagy. 2015;11(5):729–39.
Miao LJ, Huang FX, Sun ZT, et al. Stat3 inhibits Beclin 1 expression through recruitment of HDAC3 in nonsmall cell lung cancer cells. Tumor Biol. 2014;35(7):7097–103.
Wang WB, Levy DE, Lee CK. Stat3 negatively regulates type I IFN-mediated antiviral response. J Immunol. 2011;187(5):2578–85.
Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simões M, et al. Stat3 negatively regulates thyroid tumorigenesis. Proc Natl Acad Sci U S A. 2012;109(35):E2361-2370.
Garrido-Tapia M, Hernández CJ, Ascui G, et al. Stat3 inhibition by sta21 increases cell surface expression of MICB and the release of soluble micb by gastric adenocarcinoma cells. Immunobiology. 2017;222(11):1043–51.
Lu L, Zhu F, Zhang M, et al. Gene regulation and suppression of type I interferon signaling by stat3 in diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2018;115(3):E498–505.
Schust J, Sperl B, Hollis A, et al. Stattic: a small-molecule inhibitor of stat3 activation and dimerization. Chem Biol. 2006;13(11):1235–42.
Souissi I, Najjar I, Ah-Koon L, et al. A stat3-decoy oligonucleotide induces cell death in a human colorectal carcinoma cell line by blocking nuclear transfer of stat3 and stat3-bound NF-κB. BMC Cell Biol. 2011;12:14.
Levy DE, Lee CK. What does stat3 do? J Clin Invest. 2002;109(9):1143–8.
Yokogami K, Wakisaka S, Avruch J, et al. Serine phosphorylation and maximal activation of stat3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. 2000;10(1):47–50.
Dodd KM, Yang J, Shen MH, et al. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene. 2015;34(17):2239–50.
Bao W, Wang Y, Fu Y, et al. mTORC1 regulates Flagellin-induced inflammatory response in macrophages. PLoS ONE. 2015;10(5):e0125910.
Acknowledgements
We thank Wuhan Servicebio Technology Co., Ltd for their support in the TEM studies. The authors thank Ms. Xiaoyang Jia and Dr. Xuan Wang for their generous help in the live cell station assay.
Funding
This work was supported by the Natural Sciences Foundation of China (Nos. 31860309, 31960669, 31760675), the Natural Science Foundation of Inner Mongolia Autonomous Region of China (2019MS03022), the Science and Technology Major Project of Inner Mongolia Autonomous Region of China (No. 2020ZD15) and the Science and Technology Major Project of Inner Mongolia Autonomous Region of China to the Stããate Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock (No. zdzx2018065).
Author information
Authors and Affiliations
Contributions
ZW and HH conceived this study, generated hypotheses, and designed the experiments. RY, YC, and HH performed the experiments. ZG, XC, YM, QJ, XY, and YW analyzed the data. RY, YC, and HH wrote, reviewed, and edited the manuscript. XL supervised the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
No ethics approval was required for this study that did not involve patients or patient data.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
PMA-induced differentiation of THP-1 to macrophages and further induction by LPS and IFN-γ into M1-polarized macrophages. THP-1 cells were treated with PMA (100 ng/mL) for 24 h to differentiate into Mϕ -polarized macrophages and then stimulated with IFN-γ (2.5 ng/mL) and LPS (100 ng/mL) to polarize them into classical M1 macrophages. (a) THP-1 and THP-1-derived macrophages and Mϕ and M1 macrophages. Scale bars represent 100 μm. (b) TFN-α and IL-1β in culture medium were determined by ELISA. M1 macrophages had more TFN-α and IL-1β than Mϕ macrophages. **p < 0.01. n = 3 independent experiments. Error bar indicates SD.
Additional file 2: Table S1.
Number of S. aureus in macrophages.
Additional file 3: Figure S2.
(a) The amount of α-hemolysin in culture media and intracellular were evaluated by ELISA. (b) Effect of Cytochalasin B on macrophage phagocytosis. (c) Immunohistochemical analysis of pyroptosic protein GSDMD-NT in S. aureus-infected human macrophages. Scale bars represent 10 μm. (d) Large bubbles observed by light microscopy in S. aureus-infected macrophages. Large bubbles indicated by red arrows in pyroptotic cells. Scale bars represent 1 μm. *p < 0.05; **p < 0.01; NS = p > 0.05. n = 3 independent experiments. Error bar indicates SD.
Additional file 4: Video 1. The Live/Dead assay was performed by simultaneously monitoring the fluorescence after macrophages were infected with S. aureus for 3 h (MOI 25:1), Green, as live cell indicator and red, as dead cell indicator.
Additional file 5: Figure S3.
(a) Immunohistochemical analysis of GSDMD-NT in human macrophages. Macrophages were preincubated with rapamycin for 6 h and then infected with S. aureus. Scale bars represent 10 μm. (b) Quantification of the number of CFU/ml of S. aureus in cells treated with rapamycin and without rapamycin. NS = p > 0.05. n = 3 independent experiments. Error bar indicates SD.
Additional file 6: Figure S4.
Rapamycin inhibits mTORC1 signaling and upregulates GSDMD-NT expression. (a) Macrophages were treated with 100 nM rapamycin for 6 h. (b) Rapamycin upregulates GSDMD-NT in macrophages. Scale bars represent 10 μm. The resolved bands were quantified using Gel-Pro Analyzer 4.0 (Media Cybernetics, Inc., Rockville, MD, USA). *p < 0.05; **p < 0.01. n = 3 independent experiments. Error bar indicates SD.
Additional file 7: Video 2. Macrophages were treated with 100 nM rapamycin for 6 h, and pyroptosis was filmed in macrophages by laser confocal microscopy. A representative field was recorded. Arrow indicates lysing dead cell.
Additional file 8: Video 3. Macrophages were treated with 100 μM Stattic for 1 h, and pseudopod contracting, cell rounding, and lysing dead cells were filmed in macrophages by laser confocal microscopy. A representative field was recorded. Arrow indicate pseudopod contracting, cell rounding, and lysing dead cells.
Additional file 9: Video 4. Macrophages were treated with 100 μM Stattic for 1 h, and pyroptosis was filmed in macrophages by laser confocal microscopy. A representative field was recorded. Arrow indicate lysing dead cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yao, R., Chen, Y., Hao, H. et al. Pathogenic effects of inhibition of mTORC1/STAT3 axis facilitates Staphylococcus aureus-induced pyroptosis in human macrophages. Cell Commun Signal 18, 187 (2020). https://doi.org/10.1186/s12964-020-00677-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-020-00677-9