
Abstract
Neutrophil extracellular traps (NETs) as special release products of neutrophils have received extensive attention. They are composed of decondensed chromatin and coated with nucleoproteins, including histones and some granulosa proteins. NETs can form a network structure to effectively capture and eliminate pathogens and prevent their spread. Not only that, recent studies have shown that NETs also play an important role in venous thrombosis. This review provides the most important updated evidence regarding the mechanism of NETs formation and the role of NETs in the process of venous thrombosis. The potential prophylactic and therapeutic value of NETs in venous thrombotic disease will also be discussed.
Similar content being viewed by others


Introduction
More recently, it has been recognized that the release of neutrophil extracellular traps (NETs) may lead to venous thrombosis. NETs are primarily caused by stimulated neutrophils and were first described by Brinkmann et al. in 2004 [1]. They are composed of decondensed chromatin and coated with nucleoproteins, including histones and granulosa proteins such as neutrophil elastase (NE) and myeloperoxidase (MPO), forming a network DNA structure [2, 3]. This review focuses on the role of NETs and their main components in venous thromboembolism (VTE), and explores the potential therapeutic significance of NETs.
Formation of NETs
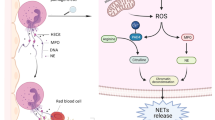
In contrast to normal apoptosis and necrosis, neutrophils undergo a carefully planned cell death pattern to release NETs, a process known as NETosis [4]. The formation of NETs can be divided into suicidal NETosis and vital NETosis depending on nature of the stimulation, activation of signaling pathways, and cell membrane integrity [5, 6] (Fig. 1). Suicidal NETosis was primarily discovered following in the context of phorbol 12-myristate 13-acetate (PMA) chemical stimulation. And then, activation of neutrophils can lead to an increase in intracellular calcium concentration. Some kinases activated downstream of calcium influx [7] and cell cycle regulators, such as PKC [8], Raf-MEK-ERK [9, 10] pathways, are all associated with suicidal NETosis. The production of reactive oxygen species (ROS) from nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria in neutrophils are also integral to suicidal NETosis induction. Unlike suicidal NETosis, vital NETosis occured without NADPH oxidase activity. Vital NETosis can be stimulated by activated platelets, microorganisms, and complement proteins. It has been shown that during vital NETosis, neutrophils activate toll-like receptors (TLR) 2 and TLR 4 under the stimulation of lipopolysaccharide and gram-negative bacteria, leading to peptidyl arginine deiminase-4 (PAD4) activation. Calcium-activated small conductance potassium channel member three (SK3) was also involved in the regulation [11]. Through this series of signal conduction events, NETosis will be transformed from signal changes to the following morphological changes [12].

Two types of process of NETosis: Suicidal NETosis (A-C) vs vital NETosis (D-F). A Various substances, including PMA and cytokines, can induce suicidal NETosis. Subsequent increases in cytoplasmic calcium activate PKC or Raf/MEK/ERK kinase pathways, which in turn activate niacinamide adenine dinucleotide phosphate (NADPH) oxidase complexes (NOX) and subsequent reactive oxygen species (ROS) production. B Peptidyl arginine deiminase-4 (PAD4), together with neutrophil elastase (NE) and myeloperoxidase (MPO), induces histone H3 citcitylation (CitH3), which further leads to decondensation of chromatin and loss of the lobular shape of the nucleus. Subsequently, nuclear envelope rupture and chromatin expansion into the cytoplasm. C Plasma membrane rupture and NET release. D In contrast, vital NETosis can be stimulated by activated platelets (PLTs), microorganisms, and complement proteins. And then, toll-like receptors (TLR) 2 and TLR 4 can be activated, leading to the activation of PAD4. Small conductance potassium channel member 3 (SK3) activated by calcium is also involved in the regulation. E Decondensation of chromatin and loss of the lobular shape of the nucleus. The external and internal nuclear membranes are then separated and the vesicles sprout. F NETs are sent out of the neutrophil by vesicles. This process protects the outer membrane of the neutrophils, thus allowing them to continue to function, even to the point of becoming anuclear
Chromatin decondensation is the dominant feature of NETosis. There are two main theories. Most studies suggested that chromatin decondensation in NETosis is mediated by posttranslational modification of histones. PAD4 -mediated citrullination plays a key role in this process [13]. PAD4-mediated citrullination can induce a reduction in histone positive charge. By reducing the affinity between histones and negatively charged DNA, it causes histones to dissociate from DNA, resulting in loss and decondensation of dense chromatin structure [12]. Li et al. [14] showed that PAD4(-/-) neutrophils cannot form NETs when incubated with bacteria, and lack the bactericidal ability of NETs. PAD4 has also been shown to play an important role in NETs formation during aseptic inflammation [15].
Although there is much evidence that PAD4 is associated with the formation of NETs, some studies have taken different views. Kenny et al. [8] demonstrated the formation of NETs in the absence of histone H3 citrullination. Some proteases, such as NE, proteinase 3 (PR3), cathepsin G, and calpains, have all been implicated in NETosis and chromatin decondensation [12, 16]. The main mechanism may be that proteases (especially NE) mediate chromatin decondensation through histone cleavage [17]. However, these views have also been challenged. Martinod et al. [18] showed that NE deficiency did not prevent NETosis and deep vein thrombosis (DVT) results in vivo. Neutrophils from NE-deficient mice still developed NETosis under PMA or platelet-activating factor (PAF) stimulation. Some other studies have found that neutrophils from patients with MPO defects fail to release NETs when stimulated by PMA, Candida albicans, or Group B Streptococcus [8]. Although MPO does not decondense chromatin directly, it has been shown to enhance the process of NE-mediated chromatin decondensation [19]. It seems that NE-mediated chromatin decondensation has a more complex pathway.
Nuelear membrane disintegration and cell lysis were often key factors in the release of NETs in suicidal NETosis. Gasdermin D (GSDMD), a pore-forming protein, could form pores in the plasma membrane and chromatin can be released into the environment. Due to electrostatic interactions, the proteins released from the granules bind tightly to the decondensed chromatin to form NETs [20, 21]. However, GSDMD is not the only pore-forming protein involved in the release of NETs. Some studies have reported that the activation of another pore-forming protein, mixed lineage kinase domain‐like (MLKL) can also lead to the release of NETs [22, 23]. Interestingly, vital NETosis was thought to occur in the absence of rupture of the nuclear envelope. It has been demonstrated that DNA vesicles germinate from the nuclear membrane, cross the cytoplasm, and bind to the plasma membrane, thus delivering the NETs from the cell without the need to cross the membrane. In suicidal NETosis, neutrophil cell membranes rupture so that they can no longer perform recruitment, chemotaxis, and phagocytosis, leading to loss of traditional functions. In contrast, the vital NETosis, a non-suicidal pathway of NETosis, allows NETs release and regular host defense to coexist [24]. Thus, it can be seen that NETosis is not a single and simple process, but involves a variety of mechanisms that may overlap and are still poorly understood, which still require further study.
NETs and specific conditions of high risk of thrombosis
The development of NETs also varies with different conditions of high risk of thrombosis. When tissue or organ damage occurs, including trauma and ischemia–reperfusion (IR) induced damage, severe inflammatory responses can be induced, and increasing the risk of sepsis. NETs are produced in the context of neutrophil antimicrobial mechanisms. However, the production of NETs may be a double-edged sword [25]. Su et al. [26] showed that activated platelets could promote the formation of NETs and kill pathogens in the early stages of sepsis. However, with the progression of sepsis and the occurrence of platelet pyroptosis, the excess formation of NETs in turn intensifies the inflammatory response and further induces platelet pyroptosis. The latest study found that activation of platelet-specific STING appears to be a critical driver. In the absence of STING, platelet activation and granule secretion were inhibited, thereby alleviating intravascular thrombosis and NETosis in sepsis mice [27]. Moreover, endogenous mitochondrial DNA (mtDNA) and oxidized mtDNA were observed to induce the formation of NETs and sterile inflammation in acute peripheral tissue trauma models [28].
Recent studies have also reported a strong relationship between NETs and immune thrombosis in patients with COVID-19, and have identified the development of cytokine storms in COVID-19 patients [29, 30]. Dysregulated activation of a variety of leukocytes (including neutrophils, monocytes, macrophages, B-lymphocytes and T-lymphocytes, etc.) and epithelial or endothelial cells induces cytokine storms that release large amounts of pro-inflammatory cytokines. The above environment also plays an important role in the activation of NETosis, and the NETosis process is induced by cytokines, chemokines, antibodies and immune complexes.
In addition to the factors mentioned above, other factors such as pregnancy and malignancy have been reported to be associated with an increased risk of VTE. Giaglis et al. [31] have reported that maternal obstetric neutrophils exhibit a strong pro-NETotic phenotype with increased CitH3, MPO, NE, and ROS plasma levels, compared to non-pregnant and pregnant controls, and are associated with an increased risk of thrombosis. Unlike other NETs formation, NETs formation is primarily driven by G-CSF and finely regulated by sex hormones in the context of the systemic milieu formed during pregnancy [32].
Additionally, complex interactions between malignancy, inflammation, and hemostatic systems are increasingly being extensively studied. Abdol et al. [33] found that rapid formation of NETs was induced when human pancreatic cancer cells (AsPC-1) were co-cultured with neutrophils isolated from healthy individuals. Moreover, ASPC-1-activated platelets promoted the release of NETs compared with unstimulated platelets. Yang et al. [34] found that neutrophils isolated from patients with gastric cancer had a greater ability to spontaneously form NETs than neutrophils isolated from healthy individuals. Li et al. [35] found similar results that NETs were more likely to form in blood and tissue samples from patients with gastric cancer than in healthy individuals. These data suggest that malignancy-specific environments may be associated with systemic changes in neutrophils and NETs.
Recent studies have raised interest in low-density neutrophils (LDNs). As a subpopulation of neutrophils, LDNs are increased in severe infections [36, 37], COVID-19 [38] and malignancy [39], etc. Although LDNs display decreased ability to phagocytose bacteria, their ability to form NETs was significantly enhanced [40]. LDNs have proved to be the subpopulation of neutrophils prone to NETs production compared to other cell types. All in all, the results of various studies show that the formation of NETs exhibits different characteristics under specific condition or tissue, which has important research significance.
The role of NETs in promoting thrombosis
The thrombogenic effect of NETs was first proposed by Fuchs et al. in 2010 [41]. NETs can form a histone-DNA reticular scaffold to capture platelets, erythrocytes and other substances, such as fibrinogen, von Willebrand factor (VWF), tissue factor (TF) and coagulation factor XII (FXII), which could conducive to thrombosis [42]. The network structure of NETs not only forms the structural basis of thrombus, but also can increase the stability of fibrin scaffold in thrombus. Some studies have reported that citrullinated fibrinogen can decrease fiber diameter and porosity, increase the density of fibrin, and reduce the solubility of fibrin clots [43]. And fibrous proteins containing histone-DNA complexes have thicker fibrin fibers, greater stability and rigidity. This combination significantly prolongs the clot dissolution time [44].
In addition, some other components of NETs can also promote platelet activation and aggregation, facilitating the formation of thrombus. Histones in NETs can activate platelets by utilizing TLR 2 and TLR 4, and further enhance platelet aggregation and activation through recruitment of fibrinogen to promote thrombin production [45, 46]. Histones can also stimulate the release of VWF from vascular endothelial cells, further mediating platelet adhesion and aggregation [47]. Meanwhile, histones can promote the expression of TF in vascular endothelial cells and monocytes, and initiate activating external coagulation pathways. The coagulation pathways induced by FXII and TF can also be further enhanced by NE and cathepsin G through hydrolyzed tissue factor pathway inhibitor (TFPI) [45, 48]. NETs can also directly bind FXII and further enhance the activation of FXIIa in coordination with platelets [49, 50].
Some studies suggested that NETs play a role primarily in the early stages of thrombosis. de Boer et al. [51] found that NETs were mainly present in fresh and lytic, but not in organized thrombus. Savchenko et al. [52] analyzed thrombus samples obtained during surgery or at autopsy and showed that NETosis occurs in the organizing stage of thrombus development. Moreover, NETs markers such as CitH3 (citrullinated histones 3) were different at each stage of thrombosis, and they were replaced by collagen fibers later in thrombus formation. Sharma et al. [53] suggested that neutrophil inflammation and NETs also play an important role in chronic thrombosis. NETs and their components mediate fibrotic remodeling of thrombus by enhancing transforming growth factor-β (TGF-β) signaling and promoting differentiation of monocytes into activated fibroblasts. There are indications that NETs appear to play different roles in different stages of thrombus formation.
Many studies have described the direct and indirect mechanisms by which NETs promote thrombosis. However, the vast majority of these studies researched components of NETs rather than intact NETs. Noubouossie et al. [54] questioned the ability of intact NETs to directly activate coagulation. Compared with intact NETs and nucleosomes, purified DNA and histones were observed to induce thrombin production more effectively. It is possible that the tight packing of histones and DNA in the nucleosome partially reduces their ability to interact with the clotting system. Although the relative contribution of intact NETs and their individual NETs components to activating coagulation remains controversial, it is clear that NETs can influence the development of thrombosis in a variety of ways, and the evidences are also emerging about how NETs promote thrombosis.
Biomarkers of NETs in venous thrombosis related diseases
The presence of NETs or their key components such as extracellular DNA, nucleosomes, MPO, CitH3, or NE in human venous thrombosis related diseases has also been further investigated. Mauracher et al. [55] showed that elevated plasma levels of cell-free DNA (cfDNA) and nucleosomes appeared to predict VTE in the short term. In other venous thrombotic diseases, such as DVT [56,57,58], pulmonary embolism (PE) [59], portal vein thrombosis (PVT) [60, 61], and cerebral venous sinus thrombosis (CVST) [62], high plasma levels of NETs have been shown to be associated with increased risks of thrombosis. By analyzing organizing thrombi obtained during surgery or autopsy, several studies supported the role of key components of NETs in venous thrombosis and stabilization [52, 62, 63]. Moreover, Ząbczyk et al. [59] observed that elevated circulating CitH3 may be linked to a higher early mortality risk in patients with acute PE. NETs or their key components showed potential diagnostic and prognostic value as biomarkers in venous thrombosis related diseases.
NETs can be visualized at high resolution by a scanning or transmission electron microscope [64], or tissue immunoassay of granule proteins bound to DNA or histones by a conventional microscope. In order to exclude interference from extracellular DNA and granulosa proteins sources other than NETs, multiple staining of NETs components, such as extracellular DNA, nucleosomes, MPO, CitH3 and NE, is recommended for colocalization [65, 66]. Of course, morphological standards of microscopic observation are not quantitative [67]. Nevertheless, the clinical application of NETs or their key components as biomarkers of thrombosis remains problematic. As tissue samples are often difficult to obtain in clinical practice, key NETs components are usually tested using blood samples, such as enzyme-linked immunosorbent assays (Elisa), which has gradually become one of the most widely used clinical methods for NETs. But unfortunately, Elisa provides relatively limited information, due to lack of specificity and standardized testing [68, 69]. Other methods, such as the use of flow cytometry to detect NETs components on cells [70] or extracellular vesicles (EVs) [71], are also being considered as novel methods to assess NETs formation in vivo. But again, the lack of objectivity and specificity of these methods remains the problems that need to be addressed. Therefore, appropriate assays specifically for the measurement of NETs or their components will be worthy of further exploration in future studies.
Potential prophylactic and therapeutic value of NETs in venous thrombosis
Since NETs play an important role in thrombosis, they provide a potential target for the prophylaxis and treatment of venous thrombotic diseases. At present, most researches focus on promoting the degradation and inhibiting the formation of NETs.
Promotion of NETs degradation
Due to the essence of NETs, which are high degree of decondensed chromatin in the extracellular environment, deoxyribonuclease (DNase) seems to become a promising targeted prophylaxis and treatment strategy for NETs. Two major DNases had been identified, including DNase 1 and DNase 1-like 3 (DNase 1-L3) [72]. Since DNase1 and DNase 1-L3 are expressed independently, they can provide dual host protection and are effective against harmful effects of intravascular NETs [72]. At present, most research directions were focused on the application of DNase 1 in NETs. Treatment of plasma with DNase 1 has been shown to reverse the increase in thrombin generation and protects from venous thrombosis during aging [73]. Jimenez-Alcazar et al. [74] reported that plasma levels of DNase 1 were reduced in patients with acute thrombotic microangiopathy (TMA), and NETs degradation activity was restored by recombinant human DNase 1 supplementation. This suggests that the restoration of plasma DNase 1 activity may be the potential prevention and treatment measures.
DNase 1 in combination with other medicines seems to have unexpected effects. In vitro experiments, the combination of DNase 1 with tissue plasminogen activator (tPA) could accelerate thrombolysis when the DNase 1 alone was ineffective [75]. Other studies have also shown that DNase 1 combined with tPA can achieve complete thrombolysis, making up for the defects of incomplete and inefficient thrombolysis of tPA alone, and may improve the therapeutic window of thrombotic disease [63, 76]. Manfredi et al. [77] reported that the treatment of low molecular weight heparins (LMWH) could abrogate the ability of neutrophils to generate NETs and neutralize the harmful effects of histones. Heparins combination with DNase 1 may further reduce the risk of future thrombotic events. The interaction between NETs and VWF also contributes to venous thrombosis. Recombinant ADAMTS13 (rADAMTS13) has been shown to prevent thrombosis by reducing initial platelet accumulation and neutrophil recruitment [78]. Targeting the NET-VWF axis via a combination of DNase 1 and ADAMTS13 has also been shown to be a potential therapeutic approach [78].
Although DNase has shown strong potential in the prophylaxis and treatment of venous thrombotic disease alone or in combination with other medicines, there are still some concerns in this regard. In some cases, treatments that use DNase 1 to lysate NETs that have been formed and released may result in the rapid release of the contents of the NETs (such as histones and MPO) or the microorganisms that have accumulated in the NETs, which then promote further inflammation and thrombosis. Since the effects between NETs and thrombosis may be reciprocal, this may put patients with thrombosis at an increased risk of thrombosis after infection [79]. The efficacy and safety of DNase 1 still remain to be tested and further studies are needed.
Inhibition of the formation of NETs
The PADs are a family of enzymes (including PAD1, 2, 3, 4, and 6) that can convert protein arginine residues to produce citrulline. Among them, PAD4 is considered to play an essential role in the formation of NETs [80, 81]. PAD4 deficiency has been shown to reduce the occurrence of thrombosis in a mouse model of venous thrombosis. And importantly,in this model, PAD4 deficiency did not lead to increased susceptibility to bacterial infection, which may indicate that NET deficiency due to PAD4 inhibition does not result in a significantly impaired host immune function [82]. Several PAD inhibitors, such as F- and Cl-amidine [83], have been shown to be effective against PAD4 under different conditions. However, because they inhibit several other subtypes of PAD at the same time, these PAD inhibitors lack the specificity required for clinical treatment. Recently, reversible PAD inhibitors of GSK199 and GSK484 have been developed that show high specificity against PAD4 and can effectively inhibit the formation of NETs in mice and human neutrophils [84]. But further studies are needed to verify its therapeutic potential for vein thrombosis.
ROS also plays an important role in the formation of NETs. Currently, N-acetylcysteine (NAC) has been shown the efficient antioxidant activity in a variety of ROS-related diseases [85], and can inhibit formation of NETs by inhibiting the production of ROS [86]. Other antioxidants, such as vitamin C, also known as ascorbic acid, have also been shown to reduce the formation of NETs in sepsis [87] and can effectively reduce the mortality of patients with sepsis [88]. Recent studies have suggested that molecular hydrogen (H2) therapy, by inhaling H2, can effectively inhibit neutrophils activation and excess NETs formation without inhibiting the essential function of neutrophils, and H2 appears to be more effective than other antioxidants, such as NAC or ascorbic acid [89].
As the key components of NETs, NE and CitH3 have also become potential prophylactic and therapeutic targets for NET-related anti-thrombotic therapy. Several studies have shown that small molecule inhibitors of NE, such as sivelestat [90] and CitH3 inhibitors, such as therapeutic anti-citrullinated protein antibody (tACPA) [91], can effectively inhibit the formation of NETs in mice and humans. More recently, neonatal NET inhibitory factor (nNIF), a novel NETosis inhibitor, has been shown to inhibit key terminal events in NETs formation, including histone H3 citrullination and the activity of PAD4 [92]. In addition to the above drugs, there are some other substances that may inhibit the formation of NETs, such as anti-P-selectin aptamer or anti-P-selectin glycoprotein ligand-1 (PSGL-1) [93], aspirin [94], vitamin D [95], chloroquine [96, 97], metformin [98], etc. But the mechanisms of these findings may remain unclear.
In conclusion, promotion of NETs degradation or inhibition of the formation of NETs have the potential to prevent and treat venous thrombosis in diseases with high risk of thrombosis such as trauma, sepsis, pregnancy, COVID-19 and malignancy, etc. Although the specific effects on venous thrombosis need to be further validated, they represent possible different directions for future research.
Conclusion
At present, NETs or their key components, such as extracellular DNA, nucleosome, MPO, CitH3 and NE, have been shown to play a role in venous thrombosis in a large number of clinical samples and animal experiments, showing their potential as biomarkers to provide evidence for the diagnosis or prognosis of venous thrombosis related diseases. However, it should be noted that the sensitivity of NETs or their components still needs to be further improved and better and more accurate measurements are needed. Meanwhile, according to the significant role of NETs in promoting thrombosis, it also provides various potential directions for the prophylaxis and treatment of venous thrombotic diseases, which is worthy of further extensive research. In conclusion, as a unique immune mechanism after neutrophil death, the unique role of NETs in promoting venous thrombosis is of great significance for the further diagnosis and treatment of venous thrombotic diseases.
Availability of data and materials
Not applicable.
References
Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5.
Pires RH, Felix SB, Delcea M. The architecture of neutrophil extracellular traps investigated by atomic force microscopy. Nanoscale. 2016;8(29):14193–202.
Petretto A, Bruschi M, Pratesi F, et al. Neutrophil extracellular traps (NET) induced by different stimuli: a comparative proteomic analysis. PLoS One. 2019;14(7):e0218946.
Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE. 2007;2007(379):pe11.
Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, et al. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. 2017;8:81.
Huang J, Hong W, Wan M, et al. Molecular mechanisms and therapeutic target of NETosis in diseases. MedComm (2020). 2022;3(3):e162.
Schappe MS, Szteyn K, Stremska ME, et al. Chanzyme TRPM7 Mediates the Ca(2+) influx essential for lipopolysaccharide-induced toll-like receptor 4 endocytosis and macrophage activation. Immunity. 2018;48(1):59-74.e55.
Kenny EF, Herzig A, Krüger R, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6:e24437.
Amulic B, Knackstedt SL, Abu Abed U, et al. Cell-cycle proteins control production of neutrophil extracellular traps. Dev Cell. 2017;43(4):449-462.e445.
Hakkim A, Fuchs TA, Martinez NE, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7(2):75–7.
Douda DN, Khan MA, Grasemann H, et al. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A. 2015;112(9):2817–22.
Thiam HR, Wong SL, Wagner DD, et al. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218.
Thiam HR, Wong SL, Qiu R, et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117(13):7326–37.
Li P, Li M, Lindberg MR, et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–62.
Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. 2013;110(21):8674–9.
Gößwein S, Lindemann A, Mahajan A, et al. Citrullination licenses calpain to decondense nuclei in neutrophil extracellular trap formation. Front Immunol. 2019;10:2481.
Papayannopoulos V, Metzler KD, Hakkim A, et al. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–91.
Martinod K, Witsch T, Farley K, et al. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost. 2016;14(3):551–8.
Metzler KD, Goosmann C, Lubojemska A, et al. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8(3):883–96.
Chen KW, Monteleone M, Boucher D, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. 2018;3(26):eaar6676.
Sollberger G, Choidas A, Burn GL, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26):eaar6689.
Desai J, Kumar SV, Mulay SR, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol. 2016;46(1):223–9.
Amini P, Stojkov D, Wang X, et al. NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol. 2016;46(1):178–84.
Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–94.
Liu FC, Chuang YH, Tsai YF, et al. Role of neutrophil extracellular traps following injury. Shock. 2014;41(6):491–8.
Su M, Chen C, Li S, et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res. 2022;1(8):732–47.
Yang M, Jiang H, Ding C, et al. STING activation in platelets aggravates septic thrombosis by enhancing platelet activation and granule secretion. Immunity. 2023;56(5):1013-1026.e1016.
Liu L, Mao Y, Xu B, et al. Induction of neutrophil extracellular traps during tissue injury: involvement of STING and toll-like receptor 9 pathways. Cell Prolif. 2019;52(3):e12579.
Bautista-Becerril B, Campi-Caballero R, Sevilla-Fuentes S, et al. Immunothrombosis in COVID-19: implications of neutrophil extracellular traps. Biomolecules. 2021;11(5):694.
Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–79.
Giaglis S, Sur Chowdhury C, van Breda SV, et al. Circulatory neutrophils exhibit enhanced neutrophil extracellular trap formation in early puerperium: NETs at the nexus of thrombosis and immunity. Int J Mol Sci. 2021;22(24):13646.
Giaglis S, Stoikou M, Sur Chowdhury C, et al. Multimodal regulation of NET formation in pregnancy: progesterone antagonizes the Pro-NETotic effect of estrogen and G-CSF. Front Immunol. 2016;7:565.
Abdol Razak N, Elaskalani O, Metharom P. Pancreatic cancer-induced neutrophil extracellular traps: a potential contributor to cancer-associated thrombosis. Int J Mol Sci. 2017;18(3):487.
Yang C, Sun W, Cui W, et al. Procoagulant role of neutrophil extracellular traps in patients with gastric cancer. Int J Clin Exp Pathol. 2015;8(11):14075–86.
Li JC, Zou XM, Yang SF, et al. Neutrophil extracellular traps participate in the development of cancer-associated thrombosis in patients with gastric cancer. World J Gastroenterol. 2022;28(26):3132–49.
Cohen TS, Takahashi V, Bonnell J, et al. Staphylococcus aureus drives expansion of low-density neutrophils in diabetic mice. J Clin Invest. 2019;129(5):2133–44.
Cho Y, Bukong TN, Tornai D, et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. 2023;78(1):28–44.
Morrissey SM, Geller AE, Hu X, et al. A specific low-density neutrophil population correlates with hypercoagulation and disease severity in hospitalized COVID-19 patients. JCI Insight. 2021;6(9):e148435.
Mauracher LM, Hell L, Moik F, et al. Neutrophils in lung cancer patients: activation potential and neutrophil extracellular trap formation. Res Pract Thromb Haemost. 2023;7(2):100126.
Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol. 2013;35(4):455–63.
Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107(36):15880–5.
Wang Y, Luo L, Braun O, et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci Rep. 2018;8(1):4020.
Varjú I, Tóth E, Farkas ÁZ, et al. Citrullinated fibrinogen forms densely packed clots with decreased permeability. J Thromb Haemost. 2022;20(12):2862–72.
Longstaff C, Varjú I, Sótonyi P, et al. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J Biol Chem. 2013;288(10):6946–56.
Yang X, Li L, Liu J, et al. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-κB and AP-1. Thromb Res. 2016;137:211–8.
Pérez-Cremades D, Bueno-Betí C, García-Giménez JL, et al. Extracellular histones trigger oxidative stress-dependent induction of the NF-kB/CAM pathway via TLR4 in endothelial cells. J Physiol Biochem. 2022. https://doi.org/10.1007/s13105-022-00935-z.
Michels A, Albánez S, Mewburn J, et al. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J Thromb Haemost. 2016;14(11):2274–86.
Folco EJ, Mawson TL, Vromman A, et al. Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1α and cathepsin G. Arterioscler Thromb Vasc Biol. 2018;38(8):1901–12.
Döring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res. 2020;126(9):1228–41.
Noubouossie DF, Reeves BN, Strahl BD, et al. Neutrophils: back in the thrombosis spotlight. Blood. 2019;133(20):2186–97.
de Boer OJ, Li X, Teeling P, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost. 2013;109(2):290–7.
Savchenko AS, Martinod K, Seidman MA, et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost. 2014;12(6):860–70.
Sharma S, Hofbauer TM, Ondracek AS, et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood. 2021;137(8):1104–16.
Noubouossie DF, Whelihan MF, Yu YB, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood. 2017;129(8):1021–9.
Mauracher LM, Posch F, Martinod K, et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost. 2018;16(3):508–18.
Diaz JA, Fuchs TA, Jackson TO, et al. Plasma DNA is elevated in patients with deep vein thrombosis. J Vasc Surg Venous Lymphat Disord. 2013;1(4):341-348.e341.
van Montfoort ML, Stephan F, Lauw MN, et al. Circulating nucleosomes and neutrophil activation as risk factors for deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2013;33(1):147–51.
Kremers BMM, Birocchi S, van Oerle R, et al. Searching for a common thrombo-inflammatory basis in patients with deep vein thrombosis or peripheral artery disease. Front Cardiovasc Med. 2019;6:33.
Ząbczyk M, Natorska J, Janion-Sadowska A, et al. Prothrombotic fibrin clot properties associated with NETs formation characterize acute pulmonary embolism patients with higher mortality risk. Sci Rep. 2020;10(1):11433.
Xing Y, Jiang Y, Xing S, et al. Neutrophil extracellular traps are associated with enhanced procoagulant activity in liver cirrhosis patients with portal vein thrombosis. J Clin Lab Anal. 2022;36(5):e24433.
Seo JD, Gu JY, Jung HS, et al. Contact system activation and neutrophil extracellular trap markers: risk factors for portal vein thrombosis in patients with hepatocellular carcinoma. Clin Appl Thromb Hemost. 2019;25:1076029618825310.
Jin J, Qiao S, Liu J, et al. Neutrophil extracellular traps promote thrombogenicity in cerebral venous sinus thrombosis. Cell Biosci. 2022;12(1):114.
Mangold A, Alias S, Scherz T, et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res. 2015;116(7):1182–92.
Gupta S, Chan DW, Zaal KJ, et al. A high-throughput real-time imaging technique to quantify NETosis and distinguish mechanisms of cell death in human neutrophils. J Immunol. 2018;200(2):869–79.
van der Meer AJ, Kroeze A, Hoogendijk AJ, et al. Systemic inflammation induces release of cell-free DNA from hematopoietic and parenchymal cells in mice and humans. Blood Adv. 2019;3(5):724–8.
Brinkmann V, Abu Abed U, Goosmann C, et al. Immunodetection of NETs in paraffin-embedded tissue. Front Immunol. 2016;7:513.
Masuda S, Nakazawa D, Shida H, et al. NETosis markers: Quest for specific, objective, and quantitative markers. Clin Chim Acta. 2016;459:89–93.
Hayden H, Ibrahim N, Klopf J, et al. ELISA detection of MPO-DNA complexes in human plasma is error-prone and yields limited information on neutrophil extracellular traps formed in vivo. PLoS One. 2021;16(4):e0250265.
Thålin C, Aguilera K, Hall NW, et al. Quantification of citrullinated histones: development of an improved assay to reliably quantify nucleosomal H3Cit in human plasma. J Thromb Haemost. 2020;18(10):2732–43.
Lee KH, Cavanaugh L, Leung H, et al. Quantification of NETs-associated markers by flow cytometry and serum assays in patients with thrombosis and sepsis. Int J Lab Hematol. 2018;40(4):392–9.
Paues Göranson S, Thålin C, Lundström A, et al. Circulating H3Cit is elevated in a human model of endotoxemia and can be detected bound to microvesicles. Sci Rep. 2018;8(1):12641.
Jiménez-Alcázar M, Rangaswamy C, Panda R, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 2017;358(6367):1202–6.
Kumar R, Sonkar VK, Swamy J, et al. DNase 1 protects from increased thrombin generation and venous thrombosis during aging: cross-sectional study in mice and humans. J Am Heart Assoc. 2022;11(2):e021188.
Jiménez-Alcázar M, Napirei M, Panda R, et al. Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J Thromb Haemost. 2015;13(5):732–42.
Ducroux C, Di Meglio L, Loyau S, et al. Thrombus neutrophil extracellular traps content impair tPA-induced thrombolysis in acute ischemic stroke. Stroke. 2018;49(3):754–7.
Peña-Martínez C, Durán-Laforet V, García-Culebras A, et al. Pharmacological modulation of neutrophil extracellular traps reverses thrombotic stroke tPA (Tissue-Type Plasminogen Activator) resistance. Stroke. 2019;50(11):3228–37.
Manfredi AA, Rovere-Querini P, D’Angelo A, et al. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol Res. 2017;123:146–56.
Yang J, Wu Z, Long Q, et al. Insights into immunothrombosis: the interplay among neutrophil extracellular trap, von willebrand factor, and ADAMTS13. Front Immunol. 2020;11: 610696.
Shirakawa K, Sano M. Neutrophils and neutrophil extracellular traps in cardiovascular disease: an overview and potential therapeutic approaches. Biomedicines. 2022;10(8):1850.
Wang S, Wang Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta. 2013;1829(10):1126–35.
Mao L, Mostafa R, Ibili E, et al. Role of protein deimination in cardiovascular diseases: potential new avenues for diagnostic and prognostic biomarkers. Expert Rev Proteomics. 2021;18(12):1059–71.
Martinod K, Fuchs TA, Zitomersky NL, et al. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood. 2015;125(12):1948–56.
Biron BM, Chung CS, O’Brien XM, et al. Cl-amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. 2017;9(1):22–32.
Lewis HD, Liddle J, Coote JE, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11(3):189–91.
Aldini G, Altomare A, Baron G, et al. N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. Free Radic Res. 2018;52(7):751–62.
Zawrotniak M, Kozik A, Rapala-Kozik M. Selected mucolytic, anti-inflammatory and cardiovascular drugs change the ability of neutrophils to form extracellular traps (NETs). Acta Biochim Pol. 2015;62(3):465–73.
Mohammed BM, Fisher BJ, Kraskauskas D, et al. Vitamin C: a novel regulator of neutrophil extracellular trap formation. Nutrients. 2013;5(8):3131–51.
Kashiouris MG, L’Heureux M, Cable CA, et al. The emerging role of vitamin C as a treatment for sepsis. Nutrients. 2020;12(2):292.
Shirakawa K, Kobayashi E, Ichihara G, et al. H(2) inhibits the formation of neutrophil extracellular traps. JACC Basic Transl Sci. 2022;7(2):146–61.
Okeke EB, Louttit C, Fry C, et al. Inhibition of neutrophil elastase prevents neutrophil extracellular trap formation and rescues mice from endotoxic shock. Biomaterials. 2020;238:119836.
Chirivi RGS, van Rosmalen JWG, van der Linden M, et al. Therapeutic ACPA inhibits NET formation: a potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol Immunol. 2021;18(6):1528–44.
Yost CC, Schwertz H, Cody MJ, et al. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J Clin Invest. 2016;126(10):3783–98.
Etulain J, Martinod K, Wong SL, et al. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126(2):242–6.
Lapponi MJ, Carestia A, Landoni VI, et al. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J Pharmacol Exp Ther. 2013;345(3):430–7.
Handono K, Sidarta YO, Pradana BA, et al. Vitamin D prevents endothelial damage induced by increased neutrophil extracellular traps formation in patients with systemic lupus erythematosus. Acta Med Indones. 2014;46(3):189–98.
Boone BA, Murthy P, Miller-Ocuin J, et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer. 2018;18(1):678.
Zhang S, Zhang Q, Wang F, et al. Hydroxychloroquine inhibiting neutrophil extracellular trap formation alleviates hepatic ischemia/reperfusion injury by blocking TLR9 in mice. Clin Immunol. 2020;216:108461.
Wang H, Li T, Chen S, et al. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015;67(12):3190–200.
Acknowledgements
Not applicable.
Funding
This work was supported by the Hebei Provincial Key Research Project (21377767D).
Author information
Authors and Affiliations
Contributions
WL, ZW and JW performed the literature search. WL ZL and SF contributed to interpretation of results. WL drafted the manuscript. WL, ZW, CS and JL contributed with the interpretation of the results and the intellectual content of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, W., Wang, Z., Su, C. et al. The effect of neutrophil extracellular traps in venous thrombosis. Thrombosis J 21, 67 (2023). https://doi.org/10.1186/s12959-023-00512-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12959-023-00512-4