
Abstract
Among the pathways and mediators that may be dysregulated in COVID-19 infection, there are proinflammatory cytokines, lymphocyte apoptosis, and the coagulation cascade. Venous and arterial thromboembolisms also are frequent in COVID-19 patients with the increased risk of some life-threatening complications such as pulmonary embolism, myocardial infarction, and ischemic stroke. In this regard, overproduction of proinflammatory cytokines such as IL-6, IL-1β, and TNF-α induce cytokine storms, increase the risk of clot formation, platelet activation, and multiorgan failure that may eventually lead to death among these patients. Surface S protein of SARS-CoV-2 binds to its target transmembrane receptor, named as angiotensin converting enzyme 2 (ACE2(, on various cells such as lymphocyte, alveolar cells, monocytes/macrophages, and platelets. Notably, the activation of the coagulation cascade occurs through tissue factor (TF)/FVIIa-initiated hemostasis. Accordingly, TF plays the major role in the activation of coagulation system during viral infection. In viral infections, the related coagulopathy multiple factors such as inflammatory cytokines and viral specific TLRs are involved, which consequently induce TF expression aberrantly. SARS-COV-2 may directly infect monocytes/ macrophages. In addition, TF expression/release from these cells may play a critical role in the development of COVID-19 coagulopathy. In this regard, the use of TF- VIIa complex inhibitor may reduce the cytokine storm and mortality among COVID-19 patients.
Similar content being viewed by others

Introduction
Although the mechanisms that activate coagulation cascade in SARS-CoV-2 infection are still unknown, they are evident to be linked to inflammatory responses [1].
inflammatory response of COVID-19 infection may be self-limited in those patients experiencing mild symptoms; however, in a smaller fraction of patients with COVID-19 infection, it is associated with the inductions of coagulopathy, disseminated intravascular coagulation (DIC) [2], cerebrovascular accidents [3], pulmonary thromboembolism, and multiorgan failure [4]. COVID-19 coagulopathy was also indicated to be associated with an increase in procoagulant factors like fibrinogen as well as a strong increase of D-dimers that are linked with a higher mortality rate [5, 6] .
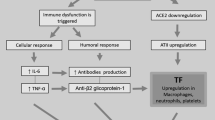
Enhancement of D-dimer above 1000 ng/ml is known as a risk factor of death in COVID-19 patients [5]. Moreover, SARS-CoV-2 infected cells down-regulate the expression of ACE2 protein. As a result, the accumulation of angiotensin II (AT-II) occurs secondary to the reduced ACE2 in COVID-19 infection. In addition, the increased AT-II may promote clot formation via having interactions with platelets and endothelial cells [7]. In promoting microvascular damage associated with AT-II, IL-6 and TNF-α may play direct roles. Correspondingly, AT-II and TNF-α have been implicated in promoting the overexpression or release of TF in platelets and macrophages [8]. Additionally, promoting an increase in TF due to antiphospholipid antibodies, may also be associated with COVID-19 coagulopathies [9]. Therefore, TF may play a role in coagulation cascade irreregulations in COVID-19 patients. Besides, some additional pro thrombotic events observed in COVID-19 patients, including an increase in serum IL-6 that has potentially characterized activities platelets [10] and coagulation factors [11], T lymphocytes [12] as well as IL-6 and TNF-α may play direct roles in promoting micro vascular damage associated with AT-II [13].
Tissue factor (TF) is the physiological activator of extrinsic coagulation pathway playing the central role in hemostatic protection of vital organs such as lung, brain, and heart [14].
TF is a transmembrane glycoprotein that is mostly restricted to the cells of the sub endothelial vessel wall under physiologic conditions (i.e., pericytes, smooth muscle cells and fibroblasts), which are not in direct contact with the blood [15].
In healthy individuals, by the exclusion of 1–2% of blood circulating monocytes that expresses intracellular TF in a low levels, blood leukocytes cannot express it. It is noteworthy that blood is not thought to contain functional TF because of its high procoagulant activity [16].
After the disruption of the vessel wall or after the upregulation of TF on monocytes under inflammatory situation transmembrane, TF would have a contact to FVII and FVIIa with a high-affinity. Moreover, TF stabilizes the catalytic site of FVIIa on plasma membrane to provide an optimal interaction with FIX and FX, similar to its substrates. The activation of FX and FIX proceeds coagulation protease cascade, which consequently produces fibrin clot.
TF procoagulant state in viral infections
The activation of coagulation cascade during viral infections may be considered as a protective response and a limit spread of the pathogen [17].
Endosomal Toll Like Receptors (TLRs) detected viral infections mainly are specific for nucleic acids. TLR3 sensing viral double strand RNA (ds RNA), and TLR7 and TLR8 recognize viral single strand RNA (ssRNA) and TLR9 is triggered by viral DNA [18]. The TLR3 agonist polyinosinic polycytidylic acid (poly I:C) induces TF expression in the cultured endothelial cells and finally activates the coagulation cascade in mice [19].
TF appears to be the major activator of the coagulation system during viral infection. For instance, Ebola, Denge, HIV, HSV, Marburg, Hantavirus, and SARS-COV-2 were all shown to be associated with coagulation activation and thrombosis [20,21,22,23,24,25,26].
In addition, respiratory viruses such as influenza A, influenza B, parainfluenza-1, respiratory syncytial virus, adenovirus, and cytomegalovirus induce infection in human endothelial cells during culturing. Accordingly, these viruses induce TF expression in various cells, including monocytes and ECs that are associated with the activation of coagulation and thrombosis. Moreover, the pronounced activation of coagulation in elderly patients with an influenza virus infection was previously described [27].
TF-dependent activation of the coagulation cascade in HSV1 infection of both of the cultured endothelial cells and in mice leads to the increased infection [28].
TF-VIIa complex inhibitors may also reduce the cytokine storm and mortality in COVID-19 patients [29].
Under the procoagulant or proinflammatory conditions, both platelets and monocytes have been shown to express TF. TF expression in monocytes and platelets obtained from the HIV infected patients played a major role in HIV-associated coagulopathy.
In severe cases of COVID-19, TF expression was described from the activated monocytes. The induced TF expression in monocytes during COVID-19 were also indicated to be associated with severity and mortality in severe COVID-19 patients [30].
AT-II accumulation and TF overexpression
SARS-CoV-2 attaches to and enters those cells that express ACE-2 protein. This receptor has been identified on pneumocytes, macrophages, and monocytes, as well as on the cardiomyocytes, trachea, and bronchi [31, 32]. It is noteworthy that SARS-CoV-2 binding induces an immediate down-regulation of ACE-2 transmembrane receptor [33], and as a result, leads to a secondary increase in its endogenous substrate levels, as AT-II. This acute accumulation in levels of AT-II may have direct implications for the vascular endothelial, coagulation, and immune responses [34,35,36].
In some situations like systemic sepsis, monocytes / macrophages respond with the increased synthesis and release of TF, and these findings have been linked to coagulation cascade dysegulations [37]. Similarly, macrophage activation and the resulting increases in TF production may also occur in the ACE-2 down-regulation and secondary accumulation of AT-II, in order to promote deregulated thrombosiss. Platelets also express TF in response to infectious situation, which can interact with monocytes /macrophages cells [38]. Correspondingly, such findings have been reported in platelet activation response to AT-II-mediator as well [39, 40].
In case of dengue hemorrhagic fever, Wills et al. [41] described that dengue infection may activate fibrinolysis pathway by directly degrading fibrinogen, thus promoting the secondary activation of pro-coagulant state including increases in plasma levels of TF. Geisbert et al. in their study postulated that the expression of TF was only observed in monocytes /macrophages with morphological changes of Ebola virus replication, suggesting that the expression of TF may be directly induced by Ebola infection [24].
Bautista-Vargas have described that the secondary accumulation AT-II in SARS-COV-2 infection may also induce overexpression of TF protein. It is likely that the overproduction of TF can be considered as the primary initiating factor for the observed coagulopathy in COVID-19 patients [8].
Conclusion
Venous and arterial thromboembolisms are frequent among critically ill COVID-19 patients with the increased risk of some life-threatening complications such as pulmonary embolism, myocardial infarction, and ischemic stroke [42, 43].
Although the mechanisms of hypercoagulopathy in SARS-CoV-2 infection are still unknown, they appear to be associated with inflammatory responses [44].
In this regard, rapid investigation is required to determine which coagulopathy pathways mostly contribute to morbidity and mortality in COVID-19 infection. Among these critical mechanisms, F overexpression may play a critical role. Previous studies have shown that inflammatory cytokines and agonist of viral specific TLRs could induce TF expression. In addition, the presence of ACE-2, as SARS-COV-2 receptor on monocyte/macrophage cells, may directly induce TF overexpression in these cells.
Further studies are required to determine the exact role of TF that might be helpful in describing the pathogeneses of thrombosis and coagulopathies associated with COVID-19 infection. In addition, we suggest that the inhibition of TF-FVIIa complex may reduce the cytokine storm and mortality in COVID-19 patients.
Availability of data and materials
Not applicable.
Abbreviations
- SARS-CoV-2 :
-
Severe acute respiratory syndrome coronavirus 2
- COVID-19 :
-
Coronavirus disease
- ACE-2 :
-
Angiotensin converting enzyme-2
- AT-II :
-
Angiotensin II
- TF :
-
Tissue factor
- IL-6 :
-
Interleukin 6
- TNF-α:
-
Tumor necrosis factor-α
References
Becker RC. COVID-19 update: Covid-19-associated coagulopathy. J Thromb Thrombolysis. 2020;50(1):54–67.
Lillicrap D. Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J Thromb Haemost. 2020;18(4):786–7.
Zhang Y, Cao W, Xiao M, Li YJ, Yang Y, Zhao J, et al. Clinical and coagulation characteristics in 7 patients with critical COVID-2019 pneumonia and acro-ischemia. Zhonghua Xue Ye Xue Za Zhi. 2020;41(4):302–7.
Danzi GB, Loffi M, Galeazzi G, Gherbesi E. Acute pulmonary embolism and COVID-19 pneumonia: a random association? Eur Heart J. 2020;41(19):1858.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020:1052–62.
Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–7.
Dmitrieva NI, Burg MB. Elevated sodium and dehydration stimulate inflammatory signaling in endothelial cells and promote atherosclerosis. PLoS One. 2015;10(6):e0128870.
Bautista-Vargas M, Bonilla-Abadía F, Cañas CA. Potential role for tissue factor 242 in the pathogenesis of hypercoagulability associated with in COVID-19. J Thromb Thrombolysis. 2020:1–5.
Mammen EF. Disseminated intravascular coagulation (DIC). Clin Lab Sci. 2000;13(4):239–45.
Ishibashi T, Kimura H, Shikama Y, Uchida T, Kariyone S, Hirano T, Kishimoto T, Takatsuki F, Akiyama Y. Interleukin-6 is a potent thrombopoietic factor in vivo in mice. Blood Coagul Fibrinolysis. 1989;74(4):4.
Hou T, Tieu BC, Ray S, Recinos Iii A, Cui R, Tilton RG, et al. Roles of IL-6-gp130 signaling in vascular inflammation. Curr Cardiol Rev. 2008;4(3):179–92.
Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–60.
Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19(2):149–54.
Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005;25(11):2273–81.
Drake TA, Morrissey J, Edgington T. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134(5):1087.
Egorina EM, Sovershaev MA, Bjørkøy G, Gruber FX, Olsen JO, Parhami-Seren B, et al. Intracellular and surface distribution of monocyte tissue factor: application to intersubject variability. Arterioscler Thromb Vasc Biol. 2005;25(7):1493–8.
Antoniak S, Mackman N. Multiple roles of the coagulation protease cascade during virus infection. Blood. 2014;123(17):2605–13.
Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449(7164):819–26.
Shibamiya A, Hersemeyer K, Schmidt Wöll T, Sedding D, Daniel J-M, Bauer S, et al. A key role for toll-like receptor-3 in disrupting the hemostasis balance on endothelial cells. Blood J Am Soc Hematol. 2009;113(3):714–22.
Goeijenbier M, Meijers J, Anfasa F, Roose JM, van de Weg CA, Bakhtiari K, et al. Effect of Puumala hantavirus infection on human umbilical vein endothelial cell hemostatic function: platelet interactions, increased tissue factor expression and fibrinolysis regulator release. Front Microbiol. 2015;6:220.
Debuc B, Smadja DM. Is COVID-19 a new hematologic disease? Stem Cell Rev Rep. 2020:1–5.
Geisbert TW, Young HA, Jahrling PB, Davis KJ, Larsen T, Kagan E, et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol. 2003;163(6):2371–82.
Key NS, Vercellotti GM, Winkelmann JC, Moldow CF, Goodman JL, Esmon NL, et al. Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc Natl Acad Sci. 1990;87(18):7095–9.
Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis. 2003;188(11):1618–29.
Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. 2010;115(2):161–7.
Huerta-Zepeda A, Cabello-Gutiérrez C, Cime-Castillo J, Monroy-Martínez V, Manjarrez-Zavala ME, Gutiérrez-Rodríguez M, et al. Crosstalk between coagulation and inflammation during dengue virus infection. Thromb Haemost. 2008;99(11):936–43.
Bogomolov B, Barinov V, Deviatkin A, Shvedova L, Arkharova M, Frolova I, et al. Hemostasis in influenza and acute respiratory viral infections in the middle-aged and elderly. Terapevticheskii arkhiv. 1990;62(7):98–102.
Sutherland MR, Simon AY, Shanina I, Horwitz MS, Ruf W, Pryzdial EL. Tissue factor on the herpes simplex virus type 1 surface enhances infection in vivo. Washington, DC: American Society of Hematology; 2013.
Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J, et al. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet. 2003;362(9400):1953–8.
Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CR, et al. Platelet activation and platelet-monocyte aggregates formation trigger tissue factor expression in severe COVID-19 patients. Blood. 2020:1330–41.
Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. 2003;200(3):282–9.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3.
Malha L, Mueller FB, Pecker MS, Mann SJ, August P, Feig PU. COVID-19 and the renin-angiotensin system. Kidney Int Rep. 2020.
Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–40.
Senchenkova EY, Russell J, Kurmaeva E, Ostanin D, Granger DN. Role of T lymphocytes in angiotensin II-mediated microvascular thrombosis. Hypertension. 2011;58(5):959–65.
Senchenkova EY, Russell J, Yildirim A, Granger DN, Gavins FNE. Novel role of T cells and IL-6 (Interleukin-6) in angiotensin II-induced microvascular dysfunction. Hypertension. 2019;73(4):829–38.
Chen X, Lao Y, Yi J, Yang J, He S, Chen Y. SENP3 in monocytes/macrophages up-regulates tissue factor and mediates lipopolysaccharide-induced acute lung injury by enhancing JNK phosphorylation. J Cell Mol Med. 2020;24(10):5454–62.
Mege D, Aubert M, Lacroix R, Dignat-George F, Panicot-Dubois L, Dubois C. Involvement of platelets in cancers. Semin Thromb Hemost. 2019;45(6):569–75.
Brambilla M, Gelosa P, Rossetti L, Castiglioni L, Zara C, Canzano P, et al. Impact of angiotensin-converting enzyme inhibition on platelet tissue factor expression in stroke-prone rats. J Hypertens. 2018;36(6):1360–71.
Ozolina A, Sarkele M, Sabelnikovs O, Skesters A, Jaunalksne I, Serova J, et al. Activation of Coagulation and Fibrinolysis in Acute Respiratory Distress Syndrome: A Prospective Pilot Study. Front Med (Lausanne). 2016;3:64.
Wills BA, Oragui EE, Stephens AC, Daramola OA, Dung NM, Loan HT, et al. Coagulation abnormalities in dengue hemorrhagic fever: serial investigations in 167 Vietnamese children with dengue shock syndrome. Clin Infect Dis. 2002;35(3):277–85.
Klok F, Kruip M, Van der Meer N, Arbous M, Gommers D, Kant K, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020:145–7.
Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Müller MC, 348 et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. 2020:1995-2004.
Becker RC. COVID-19 update: Covid-19-associated coagulopathy. J Thromb Thrombolysis. 2020. p. 1-4.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Mahin behzadi fard and masoud soleimani designed the subject of paper. Zahra eslamifar and Saba Behzadi Fard search data base and write the paper. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
As the current study was a review and hypothesis, there was no ethics committee to approve the study.
Consent for publication
All authors have consent for publication.
Competing interests
the authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Eslamifar, Z., Behzadifard, M., Soleimani, M. et al. Coagulation abnormalities in SARS-CoV-2 infection: overexpression tissue factor. Thrombosis J 18, 38 (2020). https://doi.org/10.1186/s12959-020-00250-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12959-020-00250-x