
Abstract
Background
Unexplained recurrent spontaneous abortion (URSA) is one of the most common diseases in pregnancy and is mainly caused by immune disorders. The foetus is similar to semiallogeneic maternal tissue, so the balance of immune tolerance must be dynamically maintained during pregnancy. Decidual natural killer (dNK) cells primarily mediate the immune tolerance microenvironment at the maternal–fetal interface. By using single-cell RNA sequencing (scRNA-seq) and high-throughput transcriptome sequencing analysis, we explored the characteristic distribution of dNK cells in URSA patients.
Methods
Control maternal–fetal interface tissue (from normal pregnant women, n = 3) and case maternal–fetal interface tissue (from patients with URSA, n = 3) samples were analysed by scRNA-seq and high-throughput transcriptome sequencing.
Results
By scRNA-seq, we demonstrated the maturation process of the transition of dNK cells from cytotoxic characteristics to immune tolerance in transcriptome analysis. Moreover, compared with normal pregnant women, serious disturbances in the polarization process of dNK cells were found in URSA. Simultaneously, the transcriptional level of the extracellular matrix (ECM) in URSA patients showed a significant decrease. The dNK cells interacted with extravillous trophoblasts to achieve immune-tolerant polarization.
Conclusions
Insufficient expression of KIRs during dNK cell differentiation might be a key reason why polarized dNK cells still had high cytotoxic reactivity in URSA patients. Abnormal expression of ECM may affect the interaction of dNK cells with EVTs, making dNK cells immature. Both resulted in maternal immune intolerance to the foetus during pregnancy.
Similar content being viewed by others


Introduction
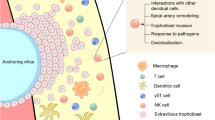
Recurrent spontaneous abortion (RSA) is the occurrence of two or more spontaneous abortions in a row, including consecutive biochemical pregnancies [1]. With the increasing number of women of childbearing age globally, the prevalence of RSA is also increasing. It has become one of the most common diseases afflicting women. According to statistical data, the incidence of RSA is approximately 1%-5% worldwide, and clinical studies have found that the miscarriage rate of the second pregnancy in such patients is as high as 70%-80% [2]. The mechanisms of RSA can be divided into two categories: nonimmune and immune. Approximately 50% of RSA is caused by immunologic factors, known as unexplained recurrent spontaneous abortion (URSA). In reproductive immunology, pregnancy is an analogue of the semiallogeneic transplantation process. Even though the foetus’s paternal antigens are directly exposed to the maternal immune system, the immunological tolerance microenvironment ensures a successful pregnancy at the decidua, also called the maternal–fetal interface. This process is essential for immune tolerance between immune cells, such as NK and T cells and extravillous trophoblasts (EVTs) in the maternal–fetal interface [3, 4].
During pregnancy, the foetus is a semiallogeneic tissue of the mother, and the maternal immune system should maintain both immune tolerance to the foetus and immune defence against infection [5, 6].The number of decidual natural killer (dNK) cells reaches more than 70% of decidual immune cells during the first trimester of pregnancy [7,8,9]. After a certain period, the proportion of dNK cells gradually decreases, though they still act as the most important component of the immune regulatory cells in the maternal–fetal interface. Acquisition of immune tolerance function is accompanied by adequate maturation of dNK cells, and the interaction between dNK cells and EVTs is essential for the process of polarization. Unlike most somatic cells that express classical major histocompatibility complex class I (MHC-I) molecules (HLA-A or HLA-B), invasive EVTs mainly express HLA-C and nonclassical MHC-I molecules (HLA-E and HLA-G) [10,11,12]. The inhibitory receptors of dNK cells, such as killer immunoglobulin-like receptors (KIRs), interact with these special MHC-I molecules, limiting the cytotoxicity of dNK cells and facilitating the maintenance of immune tolerance balance [13, 14]. However, when the intercellular crosstalk in the maternal–fetal interface is broken, inadequate maturation of dNK cells leads to downregulation of immune tolerance and upregulation of cytotoxicity [15]. Immune imbalance induces a series of pregnancy complications, such as insufficient trophoblast invasion and defective decidual vascular remodelling, which may eventually trigger URSA [16]. However, the factors that interfere with the polarization of dNK cells are diverse [17], and the dominant pathological mechanism has still not been clarified.
In recent years, some studies [18,19,20] using single-cell RNA-seq(scRNA-seq) technology have analysed the transcriptome regulation codes of cells in the maternal–fetal interface, revealed the distribution and developmental track of the cell subtype in the decidua, and defined the regulation of immune tolerance during the uterine spiral artery recasting process in a successful pregnancy. ScRNA-seq methods provide a viable way to systematically study pregnancy immunity. To deeply reveal the imbalance mechanism of immune tolerance regulation during URSA, we collected decidual tissues from typical URSA patients and normal pregnant women, and dNK cells were used as the main research objects. Through transcriptome analysis following scRNA-seq, the dysregulated characteristics that affected dNK cells in URSA patients during the process of polarization were explored.
Results
Identification the cell atlas at the decidua in scRNA-seq
To obtain maternal–fetal interface tissues from typical URSA patients, we excluded patients who had a previous autoimmune history, uterine anomalies, congenital prethrombotic state, acquired prethrombotic state, endocrine diseases, parental chromosome abnormalities or abnormal chromosomal abnormalities in previous embryos. Finally, we diagnosed these patients with URSA. We obtained 6 human maternal–fetal interface tissues during early pregnancy, of which 3 were URSA patients and 3 were normal control women. The average age of patients in the URSA group was 32.33 years old, and that in the normal pregnancy group was 31.67 years old. The average gestational age of the URSA group was 7.66 weeks, and that of the normal control group was 7.24 weeks. All 3 URSA patients had a history of embryo loss twice, and they underwent electric negative-pressure aspiration within 1 week of the diagnosis of foetal arrest (Table 1). And in URSA group, we did the fetal chromosome examination to exclude the error caused by the fetal chromosomal abnormality.
In the study, the maternal–fetal interface tissues were made into single-cell suspensions and then sorted and analysed using the BD Rhapsody™ system (Fig. 1A). After scRNA-seq, we focused on the changes in dNK cells from URSA patients through transcriptome analysis (Fig. 1B). First, data quality control was performed by assessing the single-cell expression profile matrix, including filtering cells and genes. The cells with more than 25% mitochondrial UMI, less than 500 UMI, or 200 genes were regarded as low-quality cells and were excluded from the downstream analysis, and 63,249 high-quality cell samples were obtained for further analysis (Fig. 1C). According to the first 50 pcs, the analysis scope was preliminarily determined by principal component analysis (PCA) with related gene screening (Fig. 1D). Subsequently, Seurat was used for uniform manifold approval and projection (UMAP) visual analysis, and 29 (0 to 28) cell clusters were identified by bioinformatics analysis of the gene expression characteristics of all single-cell samples (Fig. 1E).

Identification the cell atlas at the decidua in scRNA-seq. A The maternal–fetal interface tissues were made into single-cell suspensions and then sorted and analysed using the BD Rhapsody™ system. B The changes in dNK cells in URSA patients were studied by transcriptome analysis. C A total of 63,249 high-quality cell samples were obtained for further analysis. D The analysis scope was preliminarily determined by principal component analysis (PCA) with related gene screening. E Twenty-nine clusters (0 to 28) of cells were identified by bioinformatics analysis of gene expression characteristics of all single-cell samples by UMAP visual analysis
Changes in proportions of immune cells in URSA decidua
Further analysis by Seurat identified a total of 31,112 marker genes (Supplement Table 1) from all cell samples. The marker genes significantly expressed by each cell cluster were subjected to gene expression difference analysis (Fig. 2A, Supplement Table 2). According to the calculation of Single R software and manual judgements, the cell types were comprehensively identified as 20 cell types, which included NK cells, T cells, B cells, dendritic cells (DCs), macrophages, monocytes, neutrophils, decidual stromal cells, endothelial cells, epithelial gland cells, villous cytotrophoblasts (VCTs), and EVTs. Among them, NK cells highly expressed GNLY, NCAM1, CD7 and KIR3DL1, and T cells highly expressed CD3G, CD8A, TRAC, and LTB (Fig. 2A). Compared with the control samples, some distribution changes in URSA cell samples were clearly observed in UMAP, especially the increasing number of immune cells (Fig. 2B). The proportions of NK cells and proliferating NK cells in the maternal–fetal interface of URSA patients were 9.45% and 1.98%, respectively, while the proportions in the control sample were 5.96% and 1.28%, respectively (Fig. 2C). Similarly, the proportion of T cells in URSA patients compared with normal pregnant women also increased significantly. Due to the critical role of NK cells in maintaining immune tolerance during pregnancy, subsequent transcriptome analyses were conducted on NK cells and their polarization processes.

Changes in the proportions of immune cells in URSA decidua. A The cells were comprehensively identified as 20 cell types according to highly expressed genes. B The number of immune cells in URSA cell samples (case) was increased compared with that in control samples (control). C The proportions of NK cells and proliferating NK cells in the maternal–fetal interface of URSA patients and the control sample
Different states of dNK cells in URSA
Through further clustering with UMAP, NK cells in the decidua were divided into 10 clusters (0 to 9) of characteristic cells according to differences in transcription (Fig. 3A). Comparing the differences in the distribution of URSA and control NK cells in UMAP, the degree of increase in the number of NK cells in each cluster of URSA was different. Meanwhile, the proportion of each NK-cell cluster in the total cells was calculated, and the changes were compared between the URSA and control groups. There were three groups of NK-cell subsets whose numbers increased more than twofold in the URSA sample. The fold changes of Clusters 3, 4 and 8 were 2.03, 2.54, and 2.04, respectively (Fig. 3B). Cluster 8 was similar to Clusters 5 and 6, and all of them were distinguished from other cell types due to differences of their proliferative levels. Interestingly, we found not only increased proportions of Clusters 3 and 4 in the decidua of URSA patients, but also comparable transcriptional characteristics between the two clusters of cells. Clusters 3 and 4 might be associated with the sequential differentiation of Cluster 1 because of the close spatial distribution of UMAP. Therefore, the molecular mechanisms and pathways of these clusters were defined through KEGG enrichment analysis. The results showed that the genes expressed by these three groups of cells could be enriched in NK-cell-mediated cytotoxicity and were associated with pathways such as autoimmune response and graft rejection (Fig. 3C). Thus, these three clusters of cells could be used as target cell subsets for our further study of URSA.

Different states of dNK cells in URSA. A NK cells in the decidua were divided into 10 clusters (0 to 9) of characteristic cells according to differences in transcription. B The proportion of each NK-cell cluster in total cells was calculated, and the changes were compared between the URSA and control groups. C The molecular mechanism and pathway of NK Clusters 3, 4, and 1 were defined through KEGG enrichment analysis. D The differences in transcriptional levels between URSA and normal pregnant women were analysed in NK Clusters 3, 4, and 1. E The expression levels of differentially expressed genes in Clusters 3, 4, and 1 in URSA patients and normal pregnant women were compared by heatmap
The differences in transcriptional levels between URSA and normal pregnant women were analysed in these three groups of cells. Interestingly, the numbers of differentially expressed genes in these three groups that were significantly upregulated and downregulated were also higher than those in other NK-cell clusters (Fig. 3D). The associated differentially expressed genes were enriched into three groups, and the expression levels of these genes in Clusters 3, 4, and 1 were compared by heatmap (Fig. 3E). The first heatmap mainly enriched genes associated with NK-cell cytotoxicity, such as NFKBIA, FOS, TNFAIP3, and REL. The expression of this group of genes decreased in the order of cell Clusters 3, 4, and 1, but the NK-cell reactivity of URSA patients increased significantly. The second heatmap enriched NK-cell characteristic genes, including granule protein genes such as GNLY, GZMB, and GZMA, and KIR genes such as LILRB1, KIR2DL3, and KIR2DL1. Compared with the control, granule proteins were generally upregulated, while KIRs were downregulated, especially in the Cluster 4 stage. KIR2DL3 and KIR2DL1 transcriptional levels decreased significantly in URSA dNK cells. The third set of heatmaps enriched ECM-related genes, a group of genes that were significantly downregulated in URSA patients, in which collagen expression was significantly downregulated, such as COL1A1 and COL1A2. Additionally, glycodelin (PAEP), which is an immunomodulator in pregnancy, also decreased significantly in URSA samples.
Pseudotime analysis of dNK cells in URSA
To reveal the interrelationships between target NK cells, Clusters 3 (A), 4 (B, E, F) and 1 (C, D) were further grouped (Fig. 4A). We used Monocle 2 (http://cole-trapnell-lab.github.io/monocle-release/) to perform pseudotime trajectory analysis. The maturation of dNK cells is thought to be a transition from cytotoxicity to immune tolerance. In the pseudotime analysis, Cluster 3 (A) and Cluster 1 (C, D) were located at both ends of the differentiation of dNK cells. Cluster 1 (C, D) was considered to be mature dNK cells because of its immune tolerance, while Cluster 3 (A) was immature dNK cells with strong cytotoxicity, and Cluster 4 (B, E, F) was a stage of transition (Fig. 4B). The dNK cell differentiation status of URSA patients and normal pregnant women was compared in a single pseudotime analysis. The analysis results showed that the differentiation of dNK cells in URSA patients was disordered, and Clusters A, B, E, and F all stagnated during the differentiation stage, resulting in insufficient polarization of Clusters C and D, which should be mature (Fig. 4C). Simultaneously, the marker genes in the differentiation process of target dNK cells were obtained from feature selection based on pseudotime analysis, such as CYP26A1, LILRB1, and B4GALNT1 in the polarization stage and C1orf162, RASGEF1B, and ZNF683 in the transition stage (Fig. 4D).

Pseudotime analysis of dNK cells in URSA. A Cluster 3 (A), 4 (B, E, F) and Cluster 1 (C, D) were further grouped. B Cluster 3 (A) and Cluster 1 (C, D) were located at both ends of the differentiation of dNK cells. C The dNK cell differentiation status of URSA patients and normal pregnant women. D The marker genes in the differentiation process of target dNK cells were obtained from the feature selection based on pseudotime analysis
Characteristics of abnormally differentiated dNK cells in URSA
In this study, polarization disorders of dNK cells could be the key mechanism leading to URSA, where the phenotypic changes in intermediate cells, Cluster 4, directly reflected the degree of disorder of differentiation of dNK cells. C1orf162, RASGEF1B, and ZNF683 could be used as Cluster 4 markers by tSNE clustering and difference analysis, while C1orf162 and RASGEF1B would be of value for the diagnosis of URSA (Fig. 5A). ECM was an indispensable factor for dNK cell maturation, and the low expression of COL1A1, COL3A1, and PAEP at the maternal–fetal interface led to dNK cell differentiation disorders (Fig. 5B). Decreased expression of KIR2DL1 and KIR2DL3 in Cluster 4 directly affected the state of mature dNK cells (Fig. 5C), and even though KIRs could eventually be upregulated to normal expression levels, the mature dNK cells in URSA still had high cytotoxicity and immunoreactivity (Fig. 5D). All these pathological effects might stem from inadequate interaction of dNK cells with EVTs due to ECM deficiency (Fig. 5E).

Characteristics of abnormally differentiated dNK cells in URSA. A Cluster 4 reflected the degree of disordered differentiation of dNK cells. B ECM was an indispensable factor for dNK cell maturation. C Decreased expression levels of KIR2DL1 and KIR2DL3 in Cluster 4 directly affected the state of mature dNK cells. D The mature dNK cells in URSA still had high cytotoxicity and immunoreactivity. E Diagram of the inadequate interaction of dNK cells with EVTs due to ECM deficiency
Discussion
Decidualization can be considered as a biological solution for adapting to maternal–fetal genetic differences during human evolution, and the invasiveness of embryos is designed to ensure maximum expansion of offspring genes [A], while also ensuring a high-quality fetal adaptive response during a longer pregnancy [B]. The decidualization process involves multiple immune cell types, transcriptional regulation, and cytokine interactions, while immune dysregulation at the maternal–fetal interface can trigger pregnancy failure. Research evidence suggests that IL-2, IFN-γ, TNF-α, IL-6, IL-8, IL-17, and IL-23 increase in URSA decidual tissue, whereas IL-4, IL-10, IL-22, IL-27, TGF-β, LIF, and MIF were reduced, and the shift in cytokines balance indicated a maintenance disorder of immune tolerance [C].
Imbalances in maternal–fetal interface immune tolerance are thought to be the main cause of URSA with immune disorder. NK cells are the main immune cells in the decidua, and in our analysis results, the proportions of NK and T cells in the decidua of URSA patients increased, and the increase in NK cells was more obvious. By comparing the differences between the cell-type composition and the immune cell transcriptome in the decidua of URSA patients and normal pregnant women by scRNA-seq, it was possible to comprehensively and systematically identify the features of immune dysregulation in URSA. Since the interaction between NK cells and EVTs formed the core link in the establishment of immune tolerance at the maternal–fetal interface, we focused on the changing characteristics of dNK cells in URSA patients through bioinformatics analysis.
In a successful pregnancy, different from peripheral blood CD16+CD56dim NK cells, dNK cells typically have a CD16−CD56bright surface phenotype with limited cytotoxicity [13, 21]. The acquisition of immune tolerance function by dNK cells is a process of maturation and differentiation that requires adequate interaction with EVTs [5, 7]. In our study, the three clusters of interrelated NK-cell subsets were annotated in UMAP clustering, and they were speculated to be 3 characteristic differentiation stages during the process of immune tolerance acquisition through pseudotime analysis. Interestingly, the results of our analysis were very similar to the three subsets of dNK cells identified by Tormo et al. [18]. The analysis showed that Cluster 1 cells, with ENTPD1, CYP26A1, and B4GALNT1 as marker genes, highly expressed KIRs, including KIR2D1, KIR2D3, KIR3D1, and LILRB1, and contained more cytoplasmic granules than the other two groups of NK cells, including GNLY, GZMA, GZMB and GZMK. Therefore, Cluster 1 cells were deemed to be mature dNK cells that interact with EVTs, and this type of dNK cell maintained the balance between immune tolerance and immune effects during pregnancy. In the analysis of differences with normal pregnant women, the immune tolerance characteristics of cluster-1 cells in URSA patients decreased, while the propensity of cytotoxicity increased significantly. In addition to cytoplasmic granules, the levels of FOSB, TNFAIP3, NFKBIA and MAP3K8 were enhanced. The imbalance of immune regulation might be a direct cause of miscarriage. In contrast to the immune tolerance of Cluster 1 cells, Cluster 3 cells with ANXA1 and ITGB2 marker genes had a pronounced propensity for cytotoxicity and expressed the lowest levels of KIRs among the 3 groups of dNK cells. The Cluster 3 cells might be the initial NK cells that had not yet interacted with EVTs and did not yet have property of immune tolerance. Finally, Cluster 4 NK cells, which were distributed between Cluster 3 and Cluster 1, were marked by C1orf162, RASGEF1B, and ZNF683 and might be the essential differentiation process for the transfer of dNK cells to immune tolerance. It is worth noting that the transcriptional level of KIRs in URSA patients at the Cluster 4 stage was significantly downregulated, among which KIR2DL1 and KIR2DL3 differed significantly from the expression levels in normal pregnant women. Insufficient expression of KIRs during dNK cell differentiation might be a key reason why the polarized dNK cells still had high cytotoxic reactivity in URSA patients. The activation and cytotoxicity of dNK cells are affected by a variety of factors such as the cytokines environment and post-transcriptional regulation. Studies have focused on the role of miRNAs in pregnancy, in which miR-30e was found to be expressed in both peripheral blood and decidual tissues in RSA patients, while its downstream target gene PRF1 (Perforin 1) was upregulated, suggesting that miR-30e may be associated with the activation of dNK cells by negatively regulating PRF1. In addition, the miR-30e mimetic has also been shown to upregulate KIR2DL1 and downregulate the expression of the activated receptor NKp44, thereby regulating cytotoxicity in dNK cells [D].
In Monocle pseudotime analysis, we found that 3 clusters of dNK cells in normal pregnant women showed a pronounced polarized distribution, which suggested the process of smooth immune tolerance transfer. However, the NK cells in URSA patients revealed an inadequate and abnormal differentiation process. Many possible mechanisms lead to imbalances in the immune regulation of dNK cells during pregnancy. In our study, the expression of ECM genes decreased significantly in URSA patients, of which the downregulations of COL1A1 (collagen type I) and COL3A1 (collagen type III) were the most obvious. During pregnancy, the endometrium undergoes a series of changes, including collagen breakdown and remodelling. Collagen induces the expression of KIR2DL1 in dNK cells [22]and inhibits STAT1 and STAT4 signalling through LAIR-1, reducing the cytotoxic effects of dNK cells [23]. It also plays a critical role in the immune tolerance of dNK cells. Additionally, glycodelin (PAEP) was reported to have immunosuppressive properties, which are indispensable for the maintenance of pregnancy [24]. In our analysis, the PAEP transcriptional level was significantly downregulated during dNK cell differentiation in URSA patients. Therefore, abnormal expression of ECM may affect the interaction of dNK cells with EVTs, making dNK cells immature and resulting in maternal immune intolerance to the foetus during pregnancy. However, the causes of URSA decidual immune tolerance disorders are diverse, such as insufficient progesterone secretion, which can lead to decreased expression of COL1A1 and COL3A1. Therefore, the clinical treatment of URSA should still consider the specific condition of the patient.
Methods
Clinical samples
The maternal–fetal interface tissues were collected after obtaining written informed consent from 3 patients diagnosed with URSA and 3 normal pregnant women at Shuguang Hospital of Shanghai University of Traditional Chinese Medicine. This study has passed the ethical review (Ethics number: 2020–860-69–01). Certified pathologists with extensive experience performed the surgical resections of endometrial tissue for patients in this study.
Single-cell solution preparation
The maternal–fetal interface tissues were cut into approximately 1–2 mm3 pieces and digested in digestion solution with type I collagenase, neutral protease and DNase 1 at 37 °C for 15–30 min. Then, the enzymatic digestion was stopped with excess RPMI-1640 medium, and the cells were filtered with a 40-μm cell strainer. Single-cell solution samples were kept on ice before loading to a BD Rhapsody™ cartridge for single-cell transcriptome capture.
Single-cell transcriptome capturation, library construction and sequencing
Cells were first stained with two fluorescent dyes, Calcein-AM and DRAQ7, for precise determination of cell concentration and viability via a BD Rhapsody™ Scanner (BD Biosciences). Cells were loaded in one BD Rhapsody™ microwell cartridge based on Fan et al. [25]. Cell capture beads were then loaded excessively to ensure that nearly every microwell contained one bead, and the excess beads were washed away from the cartridge. After lysing the cells with lysis buffer, the cell capture beads were retrieved and washed prior to performing reverse transcription. The microbead-captured single-cell transcriptome was generated into a cDNA library containing cell labels and unique molecular identifier (UMI) information. All procedures were performed with a BD Rhapsody cDNA Kit (BD Biosciences, Cat. No. 633773) and a BD Rhapsody Targeted mRNA & AbSeq Amplification Kit (BD Biosciences, Cat No. 633801) strictly following the manufacturers’ protocol. All the libraries were sequenced in PE150 mode (paired-end, 150-bp reads) on the NovaSeq platform (Illumina).
Sequencing data processing
Raw sequencing reads of the cDNA library were processed through the BD Rhapsody Whole Transcriptome Assay Analysis Pipeline (v1.8), which included filtering by read quality, annotating reads, annotating molecules, determining putative cells and generating a single-cell expression matrix. Among all output files, the matrix of UMI counts for each gene per cell was used for downstream analysis. Genome Reference Consortium Human Build 38 (GRCh38) was used as a reference for the BD pipeline.
Dimensionality reduction, clustering and visualization
Seurat was used for subsequent clustering analysis and visualization [26, 27]. Gene expression matrices for each sample were read and converted to Seurat objects. Cells with more than 25% mitochondrial UMI, less than 500 UMI or 200 genes were excluded from the downstream analysis. After log normalization according to the total cellular UMI count, PCA was performed based on the top 2000 highly variable features after scaling the data with respect to UMI counts. Then, we performed clustering at a resolution of 0.6 and visualized the data using either t-distributed stochastic neighbour embedding (t-SNE) or uniform manifold approximation and projection (UMAP). Feature plots, violin plots and heatmaps were used to visualize the expression of the indicated genes in each cluster.
Differentially expressed gene analysis
To identify specific markers between different identities, we used Find-Markers (Seurat R Package) with the Wilcoxon Test under the following criteria: min. pct > 0.25. Genes were regarded as upregulated when their log2 fold change > 0.25 and p value adjustment < 0.05, and genes were regarded as downregulated when their log2 fold change < -0.25 and p value adjustment < 0.05.
Cell type annotation
Specific markers were calculated for each cluster using the Find-All-Markers function with the Wilcoxon Test under the following criteria: log2 fold change > 0.25; min. pct > 0.25. To unbiasedly identify cell type in filtered sample datasets and the combined dataset, we used the R package SingleR (v1.4.1) [28], a computational framework by reference to bulk transcriptomes helping us annotate the cell types for each cluster.
Pseudotime trajectory analysis
After cell type annotation, single-cell pseudotime analysis was performed for each cell type separately using Monocle 2 with the DDR-Tree reduction method [29]. Single-cell pseudotime analysis was performed with default parameters. Briefly, a Monocle object was first created according to the expression matrix and metadata information stored in the Seurat object. During feature selection, the top marker genes of Seurat clusters were set as the ordering genes for downstream analysis. Batch effects were also eliminated during dimensionality reduction. Trajectory plots and heatmaps were used to show the pseudotime results.
Availability of data and materials
The datasets used and/or analysed during the current study are available in the additional files.
Abbreviations
- URSA:
-
Unexplained recurrent spontaneous abortion
- dNK:
-
Decidual natural killer
- scRNA-seq:
-
Single-cell RNA sequencing
- ECM:
-
Extracellular matrix
- EVTs:
-
Extravillous trophoblasts
- MHC-I:
-
Major histocompatibility complex class I
- KIRs:
-
Killer immunoglobulin-like receptors
- UMAP:
-
Uniform manifold approval and projection
- DCs:
-
Dendritic cells
- VCTs:
-
Villous cytotrophoblasts
References
RPL EGGo, Bender Atik R, Christiansen OB, Elson J, Kolte AM, Lewis S, et al. ESHREguideline: recurrent pregnancy loss. Hum Reprod Open. 2018;2018(2):1–18.
Clark DA, Chaouat G, Wong K. Tolerance mechanisms in pregnancy: a reappraisal of the role of class I paternal MHC antigens. Am J Reprod Immunol. 2010;63(2):93–103.
Adib Rad H, Basirat Z, Mostafazadeh A, Faramarzi M, Bijani A, Nouri HR, et al. Evaluation of peripheral blood NK cell subsets and cytokines in unexplained recurrent miscarriage. J Chin Med Assoc. 2018;81(12):1065–70.
Than NG, Hahn S, Rossi SW, Szekeres-Bartho J. Editorial:fetal-maternal immune interactions in pregnancy. Front Immunol. 2019;10:2729.
Le Bouteiller P, Bensussan A. Up-and-down immunity of pregnancy in humans. F1000. 2017;6:1216.
Schumacher A, Sharkey DJ, Robertson SA, Zenclussen AC. Immune cells at the fetomaternal interface: how the microenvironment modulates immune cells to foster fetal development. J Immunol. 2018;201(2):325–34.
Gaynor LM, Colucci F. Uterine natural killer cells: functional distinctions and influence on pregnancy in humans and mice. Front Immunol. 2017;8:467.
Taylor EB, Sasser JM. Natural killer cells and T lymphocytes in pregnancy and pre-eclampsia. Clin Sci (Lond). 2017;131(24):2911–7.
Gong H, Chen Y, Xu J, Xie X, Yu D, Yang B, et al. The regulation of ovary and conceptus on the uterine natural killer cells during early pregnancy. Reprod Biol Endocrinol. 2017;15(1):73.
Ferreira LMR, Meissner TB, Tilburgs T, Strominger JL. HLA-G: at the interface of maternal-fetal tolerance. Trends Immunol. 2017;38(4):272–86.
Sharkey AM, Xiong S, Kennedy PR, Gardner L, Farrell LE, Chazara O, et al. Tissue-specific education of decidual NK Cells. J Immunol. 2015;195(7):3026–32.
Parham P, Moffett A. A Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol. 2013;13(2):133–44.
Faas MM, de Vos P. Uterine NK cells and macrophages in pregnancy. Placenta. 2017;56:44–52.
Li L, Feng T, Zhou W, Liu Y, Li H. miRNAs in decidual NK cells: regulators worthy of attention during pregnancy. Reprod Biol Endocrinol. 2021;19(1):150.
Le Bouteiller P. Human decidual NK cells: unique and tightly regulated effector functions in healthy and pathogen-infected pregnancies. Front Immunol. 2013;4(404):e1-5.
Santos ED, Moindjie H, Serazin V, Arnould L, Rodriguez Y, Fathallah K, et al. Preimplantation factor modulates trophoblastic invasion throughout the decidualization of human endometrial stromal cells. Reprod Biol Endocrinol. 2021;19(1):96.
Cristiani CM, Palella E, Sottile R, Tallerico R, Garofalo C, Carbone E. Human NK cell subsets in pregnancy and disease: toward a new biological complexity. Front Immunol. 2016;7:656.
Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature. 2018;563(7731):347–53.
Suryawanshi H, Morozov P, Straus A. A single-cell survey of the human first-trimester placenta and decidua. Sci Adv. 2018;4(4788):1–12.
Samstein Robert M, Josefowicz Steven Z, Arvey A, Treuting Piper M, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. 2012;150(1):29–38.
Co EC, Gormley M, Kapidzic M, Rosen DB, Scott MA, Stolp HA, McMaster M, Lanier LL, Bárcena A, Fisher SJ. Maternal decidual macrophages inhibit NK cell killing of invasive cytotrophoblasts during human pregnancy. Biol Reprod. 2013;88(6):155 1–9.
Fu Q, Tao Y, Piao H, Du MR, Li DJ. Trophoblasts and decidual stromal cells regulate decidual NK cell functions via interaction between collagen and LAIR-1. Am J Reprod Immunol. 2014;71(4):368–78.
Fu Q, Sun Y, Tao Y, Piao H, Wang X, Luan X, et al. Involvement of the JAK-STAT pathway in collagen regulation of decidual NK cells. Am J Reprod Immunol. 2017;78(6):e1-8.
Dixit A, Balakrishnan B, Karande AA. Immunomodulatory activity of glycodelin: implications in allograft rejection. Clin Exp Immunol. 2018;192(2):213–23.
Fan HC, Fu GK, Fodor SP. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science. 2015;347(6222):1258367.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888-1902 e1821.
Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–72.
Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14(10):979–82.
Acknowledgements
We would like to thank all participants for donating samples. Single Cell preparation, library construction, sequencing and analysis were performed by Sinotech Genomics Co., Ltd. Shanghai, China.
Funding
The present study was supported in part by the Scientific and Technological Committee of Shanghai, China (Grant no. 19ZR1458400) and Shuguang Hospital of Shanghai University of TCM “SiMing Youth Fund”, China (Grant no. SGKJ-202110).
Author information
Authors and Affiliations
Contributions
Dingchen Pan: Data curation, project administration, and design the first draft of article.Qian Liu: Data analysis, the initial literature search, and writing part of the original draft. Le Du: Collection of cases, sample preparation, and writing part of the original draft. Yang Yang: Methodology, supervision, and writing. Guojing Jiang: Conceptualization, funding acquisition, supervision, and review and editing. All the authors have reviewed, commented, and edited the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The patients who donated samples signed an informed consent, and the project was approved by the Ethics Committee of the Shuguang Hospital of Shanghai University of Traditional Chinese Medicine (Ethics number: 2020–860-69–01).
Consent for publication
We obtained consent to publish from the participants to report patient data.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplement Table 1. A total of nearly 30000 marker genes were identified in all cell samples.
Additional file 2:
Supplement Table 2. Marker genes significantly expressed by each cell cluster.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pan, D., Liu, Q., Du, L. et al. Polarization disorder of decidual NK cells in unexplained recurrent spontaneous abortion revealed by single-cell transcriptome analysis. Reprod Biol Endocrinol 20, 108 (2022). https://doi.org/10.1186/s12958-022-00980-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-022-00980-9