
Abstract
Background
Non-obstructive azoospermia (NOA) is the most severe form of male infertility; more than half of the NOA patients are idiopathic. Although many NOA risk genes have been detected, the genetic factors for NOA in majority of the patients are unknown. In addition, it is difficult to retrieve sperm from these patients despite using the microsurgical testicular sperm extraction (microTESE) method. Therefore, we conducted this genetic study to identify the potential genetic factors responsible for NOA and investigate the sperm retrieval rate of microTESE for genetically deficient NOA patients.
Methods
Semen analyses, sex hormone testing, and testicular biopsy were performed to categorize the patients with NOA. The chromosome karyotypes and Y chromosome microdeletion analyses were used to exclude general genetic factors. Whole exome sequencing and Sanger sequencing were performed to identify potential genetic variants in 51 patients with NOA. Hematoxylin and eosin staining (H&E) and anti-phosphorylated H2AX were used to assess the histopathology of spermatogenesis. Quantitative real time-polymerase chain reaction, western blotting, and immunofluorescence were performed to verify the effects of gene variation on expression.
Results
We performed whole exome sequencing in 51 NOA patients and identified homozygous helicase for meiosis 1(HFM1) variants (NM_001017975: c.3490C > T: p.Q1164X; c.3470G > A: p.C1157Y) in two patients (3.9%, 2/51). Histopathology of the testis showed that spermatogenesis was completely blocked at metaphase in these two patients carrying the HFM1 homozygous variants. In comparison with unaffected controls, we found a significant reduction in the levels of HFM1 mRNA and protein expression in the testicular tissues from these two patients. The patients were also subjected to microTESE treatment, but the sperms could not be retrieved.
Conclusions
This study identified novel homozygous variants of HFM1 that are responsible for spermatogenic failure and NOA, and microTESE did not aid in retrieving sperms from these patients.
Similar content being viewed by others


Introduction
Azoospermia is a medical condition characterized by the absence of sperms in the ejaculated semen. Azoospermia is the most severe form of male infertility and accounts for approximately 1% of the total male population and 10% of the infertile males globally [1, 2]. Based on pathological features, azoospermia can be classified into obstructive azoospermia (OA) and non-obstructive azoospermia (NOA). With the development of assisted reproductive technology, techniques such as intracytoplasmic sperm injection have made it possible for most patients with OA to conceive their own offspring [3, 4]. However, it is a challenge to retrieve sperms from patients with NOA, even by microsurgical testicular sperm extraction (microTESE) [5, 6]. Therefore, it is important to investigate the etiology and pathogenesis of NOA, which will facilitate the development of a targeted therapy.
It has been reported that some factors, such as abnormal chromosomes, Y-chromosomal microdeletion, and cryptorchidism, can cause NOA. However, a majority of the NOA cases remain idiopathic, and no medical etiologies have been identified [4]. In recent years, it has been reported that several genes, such as TEX11 (OMIM: 300,311), TEX14 (OMIM: 605,792), FANCM (OMIM: 609,644), SPINK2 (OMIM: 605,753), MEIOB (OMIM: 617,670), and STAG3 (OMIM: 608,489), are associated with spermatogenic defects in patients with NOA [7,8,9,10,11,12,13]. Although the identified genes only account for a small proportion of NOA cases, they strengthen our understanding of the causes of NOA.
Helicase for meiosis 1 (HFM1) plays an important role in crossing over and synapsis during meiosis [14]. Previous studies have reported that variations in human HFM1 are related to female premature ovarian failure (POF) and male NOA or severe oligozoospermia [15, 16]. The phenotypes of POF and NOA are also found in Hfm1−/− female and male mice, respectively [17]. In this study, we identified two novel homozygous variants of HFM1 in two patients with NOA, which strengthens the clinical significance of HFM1 for NOA phenotype. Additionally, we also investigated the results of microTESE in patients with HFM1 variants for the first time, which may provide a clinical reference that microTESE is not the ideal extraction method for such patients.
Methods
Subjects
A cohort of 51 Chinese men with idiopathic NOA were enrolled from the First Affiliated Hospital of Anhui Medical University. The ejaculated semen and urine were centrifuged and analyzed, together with some other tests were performed, included the determination of somatic karyotypes, screening of Y chromosome microdeletions, sex hormone testing, subsequent testicular biopsy, and testicular pathological analysis to identify the NOA phenotype in these patients. Participants with a history of cryptorchidism, testicular torsion, epididymitis, epididymo-orchitis, mumps orchitis and/or ascending sexually transmitted infections were not included in the study because of the causative relationships of such pathologies with obstructive/non-obstructive azoospermia [18,19,20]. The patients with abnormal somatic karyotypes and Y chromosome microdeletions were also excluded from this study.
Ethical approval
All participants signed a written informed consent to participate in the study. The study was approved by the review board committee of the First Affiliated Hospital of Anhui Medical University, and it was conducted in accordance with the Declaration of Helsinki.
Whole exome sequencing (WES), Sanger sequencing, and bioinformatics analysis
DNA was extracted from the whole peripheral blood of the patients. WES, Sanger sequencing, and bioinformatics analysis were performed according to previous studies [18, 19]. In summary, we annotated variants using allele frequency databases (1000G, EXAC, gnomAD), deleterious prediction tools (SIFT, PolyPhen-2, Mutation Taster, and CADD), and Genotype-Tissue Expression database by ANNOVAR, VarCards and dbNSFP [21,22,23]. The variants with allele frequencies > 0.05 were excluded. We retained deleterious missense variants and loss-of-function variants including splicing (≤ 2 bp), stop-gain, stop-loss, and frameshift variants. The variant that was predicted to be deleterious by three of the four tools, namely, SIFT, PolyPhen-2, Mutation Taster, and CADD (score > 20), was defined as a deleterious variant. The pathogenic variants expressing testis-specific genes were used for further analysis. Testis-specific expression genes were defined as those genes having an average expression value ≥ 5 reads per kilobase per million mapped reads in human testis and a value that is two folds higher than the average expression value in other tissues based on the Genotype-Tissue Expression data. For patients from consanguineous families, we focused on autosomal homozygous variants or X-linked variants. Finally, we further assessed the potential function and phenotype of the selected gene using Gene Ontology database, Online Mendelian Inheritance in Man database, Model Organism database, and PubMed based literature review. Sanger sequencing was performed to identify parental origins of pathogenic variants. The primers used are listed in Supplementary Table 2.
Hematoxylin and eosin (H&E) and immunofluorescence assay
H&E staining and immunofluorescence assay were performed as previously described [24, 25]. Histopathological assessment of spermatogenesis was performed by H&E staining. The location of candidate gene expression in seminiferous tubules was determined by immunofluorescence. The following antibodies were used: mouse monoclonal antibody againstγH2AX [(1:1000; Millipore, 05–636) to detect meiotic double-strand breaks (DSBs) and the XY body in pachytene nuclei and apoptotic metaphases in case of pan-chromosomal signal, rabbit polyclonal anti-H3Ser10ph (1:1,000; 06–570, Millipore), and peanut agglutinin (PNA) conjugated to rhodamine (1:500; Vector laboratories, RL-1072). The Probable ATP-dependent DNA helicase HFM1 antibody was used as the primary anti-HFM1 antibody (1:50; Abbexa, abx124216).
Quantitative real-time PCR (qPCR) and western blotting
The mRNA and protein expression levels of the candidate gene were determined by qPCR and western blotting, respectively, according to previously described procedures [25, 26]. The primary anti-HFM1 antibody (Abbexa) was used for both immunofluorescence (1:50) and western blotting (1:1000); the PCR primers used are listed in Supplementary Table 3.
MicroTESE
As no sperm was detected in routine testicular biopsy, microTESE was performed to retrieve testicular sperms, according to a previous study [27].
Results
Homozygous variants in HFM1 were identified in two Chinese men with NOA
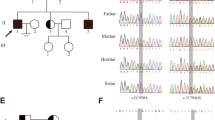
We performed bioinformatics analysis of the WES data of one patient with NOA from a consanguineous family (F1 II-1) and found a homozygous HFM1 loss-of-function variant (NM_001017975: c.3490C > T: p.Q1164X), which was absent in our 500 in-house Chinese unrelated cohort controls in the general population. For other variants that passed the filter threshold, no potential NOA related variants in F1 II-1 were observed (Supplementary Table 1). Sanger sequencing confirmed that the patient was homozygous for the variant and his parents were identified as heterozygous carriers (Fig. 1A). In addition, we also found a pathogenic homozygous missense variant of the HFM1 gene (NM_001017975: c.3470G > A: p.C1157Y) in one of the remaining 50 patients from non-consanguineous families (F2 II-1) (Fig. 1B). This variant was rare in our 500 in-house Chinese unrelated cohort controls in the general population. Moreover, this variant was defined as deleterious by the four prediction tools (Table 1). Similarly, no potential NOA related variants in F2 II-1 were observed (Supplementary Table 1).

Identification of bi-allelic HFM1 variants in two Chinese azoospermic men. A-B The two families affected by the variants in HFM1. The red frames indicate mutated positions in the Sanger sequencing results. C The affected amino acid residues of HFM1 are conserved among species. The red arrows indicate the locations of HFM1 variants occurring in the domains of HFM1 protein. M, HFM1 mutation; WT, wild type
HFM1 (the human homologue of yeast Mer3), a meiotic gene, is located on human chromosome 1p22.2 and contains 39 exons encoding a predicted 1435-amino acid protein, highly expressed in the testis and ovaries. HFM1 protein is mainly composed of helicase ATP-binding domain, helicase C-terminal domain, and SEC63 domain. The two homozygous variations found in this study were located in the zinc finger motif of HFM1 protein. F1 II-1, harboring the stop-gain variant p.Q1164X, introduces a premature stop codon and thus is expected to produce either no protein or truncated non-functional proteins, while the variant p.C1157Y might lead to a change at splice donor site 3 bp downstream in exon 31. These two mutated residues are conserved in many organisms and may probably lead to HFM1 protein dysfunction (Fig. 1C).
Meiotic metaphase arrest in the testis of patients
To determine the potential effects of the HFM1 homozygous variants on human spermatogenesis, testicular histological tissues were collected from F1 II-1 and F2 II-1 and analyzed using H&E staining. Compared to the normal control, the seminiferous tubules from the testis of patients carrying the HFM1 variants showed reduced diameters and the absence of spermatid (Fig. 2A). To further assess meiotic progression of testis in patients carrying the homozygous variants, we stained the histological sections using anti-phosphorylated H2AX (γH2AX) alone or with a combination of γH2AX and anti-phosphorylated H3 (H3Ser10ph) antibodies. As a marker of meiotic DSBs and the XY body, γH2AX was used to verify the meiotic phase, the progression of DSB repair, and the formation of XY body. We found that a portion of the tubule section was marked by the presence of multiple γH2AX patches which indicated the stage of spermatocytes, including leptotene and zygotene (Fig. 2B). Moreover, we observed two (or more) γH2AX-positive XY body like structures in the testis of the F1 II-1 patient and extremely reduced XY body positive tubules in the testis of the F2 II-1 patient (Fig. 2B). These results indicate that the cells of the testis rarely reach the pachytene stage in the HFM1 variants. In addition, H3Ser10ph immunostaining was used to identify M-phase cells; the metaphases also displayed intense γH2AX signals along the condensed chromosomes. Since this type of pan-chromosomal γH2AX staining has been described as a hallmark of cells entering apoptosis, for HFM1 variants subjects, most of the metaphase cells were apoptotic. The organization of the apoptotic metaphases appeared to be more dispersed in the testis of the F1 II-1 patient than that in the F2 II-1 patient. In F2 II-1 patient, rare metaphases were observed and all the metaphases displayed an aberrant pattern of γH2AX spots, where the γH2AX patches covered the entire nucleus (Fig. 3). Moreover, round (green arrow), elongating (red arrow), and condensing spermatids (brown arrow) could be observed in the control sample using the combined label of acrosome antibody and Hoechst. However, spermatid was absent in the testis of men harboring homozygous HFM1 variants (Fig. 4). All these findings suggested that the deficiency of HFM1 largely resulted in the blocking of spermatogenesis at the pachytene stage; some spermatocytes that developed further exhibited dramatic apoptotic metaphases and complete metaphase arrest.

Investigation of patients harboring the HFM1 variants and the controls. A Testicular histological sections from an OA patient (control-i) and patients harboring the HFM1 variants (F1 II-1-i, F2 II-1-ii) were stained with hematoxylin and eosin (H&E). Scale bar represents 20 μm. B Immunofluorescent staining of histological sections from the testis biopsy of the control and patients carrying the HFM1 variants usingγH2AX (green, marker for DSBs and XY body) and Hoechst (blue). Many aberrant γH2AX spots in samples of F1 II-1 were observed, such as small patches of γH2AX staining or two (or more) γH2AX-positive XY body–like structures. Extremely reduced XY body positive tubules were observed in samples of F2 II-1, indicating that the cells rarely reach the pachytene stage. Scale bar represents 5 µm

Immunofluorescent staining of histological sections from the testis biopsy of the patient carrying the HFM1 variants using γH2AX (green), H3S10p (red), and Hoechst (blue). Scale bar represents 5 μm. A high density of metaphases was observed and almost all the metaphases were apoptotic. The organization of the apoptotic metaphases appeared to be more dispersed in samples of F1 II-1. In F2 II-1 patient rare metaphases were observed and all the metaphases displayed an aberrant pattern of γH2AX spots whereby γH2AX patches covered the entire nucleus

Immunofluorescent staining of histological sections from the testis biopsy of the patient carrying the HFM1 variants using peanut agglutinin (PNA, brown) conjugated to rhodamine to locate the acrosome of spermatid. Compared with the control, the spermatid was absent in testis from men harboring biallelic HFM1 variants, indicating a premetaphase arrest. Hoechst (blue) was stained as a nuclear marker. Scale bar represents 20 μm
After an exhaustive understanding of microTESE, two patients consented to undergo the procedure. However, using this technique, sperms were not found in any of the multiple testis fragments from the two patients carrying the homozygous variants, although spermatocytes were observed. Thus, microTESE further confirmed our histological observations.
HFM1 expression were reduced in the testicular tissues of patients
To investigate the pathogenicity of the HFM1 variants identified in this study, we examined the expression levels of HFM1 mRNA and protein in the testis from the patients harboring the homozygous HFM1 variants and the controls. QPCR results suggested that the abundance of HFM1 mRNA was significantly reduced in the testis of men harboring the homozygous HFM1 variants (Fig. 5A). Consistently, the expression of HFM1 protein was also dramatically reduced in the testicular tissues of F1 II-1 and F2 II-1, compared to the control (Fig. 5B).

Expression and location analysis of HFM1 protein in the testis from the control patient and men harboring bi-allelic HFM1 variants. A qPCR analysis indicated that the abundance of HFM1 mRNA decreased significantly in the testis of men harboring homozygous HFM1 variants when compared to that of a normal control male. Data represents the means ± SEM. (standard error of measurement) of three independent experiments. Two-tailed Student’s paired or unpaired t tests were used as appropriate (*** P < 0.001). B Western blotting assay revealed that HFM1 protein levels reduced significantly in the testis from men harboring HFM1 mutations. β-actin was used as a loading control. C HFM1 localization in the testis from a control individual and men harboring bi-allelic HFM1 variants. HFM1 immunostaining (red) was primarily concentrated in cytoplasm of spermatogonia and spermatocytes in seminiferous tubules in normal control. The immunostaining was decreased in the testicular tissues of F1 II-1 and F2 II-1. Hoechst (blue) was stained as a nuclear marker. The γH2AX (green) was stained as a marker of spermatocyte. Scale bars: 20 μm
Furthermore, we performed immunofluorescence analysis of HFM1 expression in the testicular tissues of affected individuals in comparison with OA control. Immunostaining signal of the testicular tissues from OA patient revealed that HFM1 was mainly concentrated in the cytoplasm of spermatogonia and spermatocytes in the seminiferous tubules. In contrast, HFM1 signal was significantly reduced in the testicular tissues of both F1 II-1 and F2 II-1 (Fig. 5C).
Discussion
In this study, two homozygous HFM1 variants were identified as the genetic factor responsible for the impairment of spermatogenesis in two patients with NOA. The deficiency of HFM1 resulted in spermatogenesis being blocked at the pachytene stage. However, some spermatocytes apparently developed further, and a dramatic increase in the number of apoptotic metaphases indicated a complete metaphase arrest. Additionally, HFM1 protein was mainly concentrated in the cytoplasm of spermatogonia and spermatocytes in the seminiferous tubules of the control (OA) patient. The expression levels of HFM1 mRNA and protein significantly decreased in men carrying the homozygous HFM1 variants. Moreover, no sperms were recovered by microTESE from HFM1 mutated subjects. These results indicate that HFM1 variants are novel causative variants of NOA in humans.
HFM1 (also known as Mer3 in yeast) is highly expressed in tissues of the testis and ovaries, contains 39 exons, and encodes a predicted 1435-amino acid protein, which is required in many organisms for crossover formation and the complete synapsis of homologous chromosomes during meiosis [14, 17]. Two variants found in this study were located in the zinc finger motif of HFM1 protein; F1 II-1, harboring a stop-gain variant p.Q1164X, introduced a premature stop codon resulting in the absence of protein expression, while the homozygous missense variant p.C1157Y, reported previously by Zhang et al., might lead to a change at splice donor site 3 bp downstream in exon 31 resulting in a significant decrease in the expression of HFM1 protein [16]. These two mutated residues are conserved in many organisms and probably lead to HFM1 protein dysfunction, thus blocking spermatogenesis at the pachytene stage. In addition, Zhang et al. found that HFM1 variations were associated with idiopathic azoospermia or severe oligozoospermia in Chinese men, which further confirmed our results [16].
Similarly, knockout of Hfm1 in male mice showed that spermatogenesis is blocked at diakinesis of meiosis I while apoptosis of spermatocytes at diakinesis is common in the seminiferous tubules of Hfm1−/− mice. Majority of the initial recombination events (homologous recognition, pairing, and initial synapsis) in Hfm1−/− spermatocytes were normal compared to that in wild-type mice. In addition, synapsis was incomplete for most of the chromosomes in Hfm1−/− spermatocytes, suggesting that HFM1 participates in a major crossover pathway [17]. However, these results were different from those in budding yeast or C. cinereus, where Mer3 deletion results in mid prophase I arrest, or in S. macrospora, where Mer3 mutants show a delay in the leptotene-zygotene transition [28,29,30]. These differences among species indicate that the biological functions that require HFM1/Mer3 and/or the responses to its absence have diverged along with other features of meiotic chromosome metabolism in these organisms.
Furthermore, Hfm1−/− adult female mice presented a significant reduction in ovary size, reduced number of follicles, and increase in stromal cells and corpora lutea. The phenotypes were similar to those observed for primary ovarian insufficiency [15, 31, 32]. Hence HFM1 is also known as POF9.
No sperms were observed in routine testicular biopsy of these two patients. Considering the high sperm retrieval rate by microTESE in NOA patients [33, 34], F1 II-1 and F2 II-1 consented to undergo the procedure. However, no sperms were retrieved from either of the patients. This is the first study to report the outcomes of microTESE in NOA patients with variations in HFM1. Although only two patients were enrolled, it may provide a clinical reference that microTESE is not beneficial for these patients.
However, this study had a few limitations. Firstly, since sequencing of HFM1 was not performed in healthy fertile males, we cannot comment on the variant frequency of the gene in this population. Secondly, the chromosome spread experiment, which can detect possible defects during synapsis and recombination, was not performed due to shortage of testicular sample. Thirdly, only two cases with homozygous variants in HFM1 underwent microTESE, further research is needed to verify the chances of successful sperm retrieval by microTESE in NOA patients with bi-allelic HFM1 variants.
Conclusions
Our results provide further evidence that HFM1 is a candidate gene responsible for NOA in humans, and that homozygous variants in HFM1 can cause autosomal recessive male infertility due to NOA. In addition, it is likely that microTESE cannot be used for sperm retrieval in these patients.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- NOA:
-
Non-obstructive azoospermia
- microTESE:
-
Microsurgical testicular sperm extraction
- WES:
-
Whole exome sequencing
- H&E:
-
Hematoxylin and eosin staining
- OA:
-
Obstructive azoospermia
- HFM1:
-
Helicase for meiosis 1
- POF:
-
Premature ovarian failure
- qPCR:
-
Quantitative real-time PCR
- ACMG:
-
American College of Medical Genetics and Genomics
- SIFT:
-
Sorting Intolerant From Tolerant
References
Tournaye H, Krausz C, Oates RD. Concepts in diagnosis and therapy for male reproductive impairment. The lancet Diabetes & Endocrinol. 2017;5(7):554–64.
Tüttelmann F, Werny F, Cooper TG, Kliesch S, Simoni M, Nieschlag E. Clinical experience with azoospermia: aetiology and chances for spermatozoa detection upon biopsy. Int J Androl. 2011;34(4pt1):291–8.
Jashnani K, Gundawar R, Kavishwar V, Parameshwar V. Fine-Needle aspiration cytology of the testes for the classification of azoospermia and its value in the assessment of male infertility. Acta Cytol. 2020;64(3):216–23.
Jungwirth A, Giwercman A, Tournaye H, Diemer T, Kopa Z, Dohle G, et al. European Association of Urology guidelines on male infertility: the 2012 update. Eur Urol. 2012;62(2):324–32.
Caroppo E, Colpi EM, Gazzano G, Vaccalluzzo L, Piatti E, D’Amato G, et al. The seminiferous tubule caliber pattern as evaluated at high magnification during microdissection testicular sperm extraction predicts sperm retrieval in patients with non-obstructive azoospermia. Andrology. 2019;7(1):8–14.
Li P, Yao CC, Zhi EL, Xu Y, Wan Z, Jiang YC, et al. Modified stepwise mini-incision microdissection testicular sperm extraction: a useful technique for patients with a history of orchidopexy affected by non-obstructive azoospermia. J Zhejiang Univ Sci B. 2020;21(1):87–92.
Cerván-Martín M, Castilla JA, Palomino-Morales RJ, Carmona FD. Genetic landscape of nonobstructive azoospermia and new perspectives for the clinic. J Clin Med. 2020;9(2):300.
Yatsenko AN, Georgiadis AP, Röpke A, Berman AJ, Jaffe T, Olszewska M, et al. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med. 2015;372(22):2097–107.
Kim HJ, Yoon J, Matsuura A, Na JH, Lee WK, Kim H, et al. Structural and biochemical insights into the role of testis-expressed gene 14 (TEX14) in forming the stable intercellular bridges of germ cells. Proc Natl Acad Sci USA. 2015;112(40):12372–7.
Kasak L, Punab M, Nagirnaja L, Grigorova M, Minajeva A, Lopes AM, et al. Bi-allelic recessive loss-of-function variants in FANCM cause non-obstructive azoospermia. Am J Hum Genet. 2018;103(2):200–12.
Kherraf ZE, Christou-Kent M, Karaouzene T, Amiri-Yekta A, Martinez G, Vargas AS, et al. SPINK2 deficiency causes infertility by inducing sperm defects in heterozygotes and azoospermia in homozygotes. EMBO Mol Med. 2017;9(8):1132–49.
Van Der Bijl N, Röpke A, Biswas U, Wöste M, Jessberger R, Kliesch S, et al. Mutations in the stromal antigen 3 (STAG3) gene cause male infertility due to meiotic arrest. Hum Reprod. 2019;34(11):2112–9.
Gershoni M, Hauser R, Barda S, Lehavi O, Arama E, Pietrokovski S, et al. A new MEIOB mutation is a recurrent cause for azoospermia and testicular meiotic arrest. Hum Reprod. 2019;34(4):666–71.
Tanaka K, Miyamoto N, Shouguchi-Miyata J, Ikeda JE. HFM1, the human homologue of yeast Mer3, encodes a putative DNA helicase expressed specifically in germ-line cells. DNA Seq. 2006;17(3):242–6.
Wang J, Zhang W, Jiang H, Wu BL. Mutations in HFM1 in recessive primary ovarian insufficiency. N Engl J Med. 2014;370(10):972–4.
Zhang W, Song X, Ni F, Cheng J, Wu BL, Jiang H. Association analysis between HFM1 variations and idiopathic azoospermia or severe oligozoospermia in Chinese Men. Sci China Life Sci. 2017;60(3):315–8.
Guiraldelli MF, Eyster C, Wilkerson JL, Dresser ME, Pezza RJ. Mouse HFM1/Mer3 is required for crossover formation and complete synapsis of homologous chromosomes during meiosis. PLoS Genet. 2013;9(3):e1003383.
Cocuzza M, Alvarenga C, Pagani R. The epidemiology and etiology of azoospermia. Clinics. 2013;68(Suppl 1):15–26.
Schuppe HC, Pilatz A, Hossain H, Diemer T, Wagenlehner F, Weidner W. Urogenital infection as a risk factor for male infertility. Dtsch Arztebl Int. 2017;114(19):339–46.
Banyra O, Nikitin O, Ventskivska I. Acute epididymo-orchitis: relevance of local classification and partner’s follow-up. Cent European J Urol. 2019;72(3):324–9.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Li J, Shi L, Zhang K, Zhang Y, Hu S, Zhao T, et al. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018;46(D1):D1039–48.
Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020;12(1):1–8.
Tang D, Sha Y, Gao Y, Zhang J, Cheng H, Zhang J, et al. Novel variants in DNAH9 lead to nonsyndromic severe asthenozoospermia. Reprod Biol Endocrinol. 2021;19(1):1–9.
Ni X, Wang J, Lv M, Liu C, Zhong Y, Tian S, et al. A novel homozygous mutation in WDR19 induces disorganization of microtubules in sperm flagella and nonsyndromic asthenoteratospermia. J Assist Reprod Genet. 2020;37(6):1431–9.
Yang Y, Zhu J, Zhang N, Zhao Y, Li WY, Zhao FY, et al. Impaired gap junctions in human hepatocellular carcinoma limit intrinsic oxaliplatin chemosensitivity: A key role of connexin 26. Int J Oncol. 2016;48(2):703–13.
Schlegel PN. Testicular sperm extraction: microdissection improves sperm yield with minimal tissue excision. Hum Reprod. 1999;14(1):131–5.
Abdu U, Brodsky M, Schüpbach T. Activation of a meiotic checkpoint during Drosophila oogenesis regulates the translation of Gurken through Chk2/Mnk. Curr Biol. 2002;12(19):1645–51.
Bhalla N, Dernburg AF. A conserved checkpoint monitors meiotic chromosome synapsis in Caenorhabditis elegans. Science. 2005;310(5754):1683–6.
Ashley T, Gaeth AP, Creemers LB, Hack AM, de Rooij DG. Correlation of meiotic events in testis sections and microspreads of mouse spermatocytes relative to the mid-pachytene checkpoint. Chromosoma. 2004;113(3):126–36.
Pu D, Wang C, Cao J, Shen Y, Jiang H, Liu J, et al. Association analysis between HFM1 variation and primary ovarian insufficiency in Chinese women. Clin Genet. 2016;89(5):597–602.
Liu H, Wei X, Sha Y, Liu W, Gao H, Lin J, et al. Whole-exome sequencing in patients with premature ovarian insufficiency: early detection and early intervention. J Ovarian Res. 2020;13(1):1–8.
Bernie AM, Mata DA, Ramasamy R, Schlegel PN. Comparison of microdissection testicular sperm extraction, conventional testicular sperm extraction, and testicular sperm aspiration for nonobstructive azoospermia: a systematic review and meta-analysis. Fertil Steril. 2015;104(5):1099–103.
Deruyver Y, Vanderschueren D, Van der Aa F. Outcome of microdissection TESE compared with conventional TESE in non-obstructive azoospermia: a systematic review. Andrology. 2014;2(1):20–4.
Acknowledgements
We would like to thank all the individuals who participated in and supported this research.
Funding
This study was supported by Natural Science Foundation of Anhui Province in China (No. 1908085QH355) and the National Natural Science Foundation of China (No. 81901541 and 82071705).
Author information
Authors and Affiliations
Contributions
YC, XH, DT and ML designed the study. DT, CW, CX and HG collected the data. YG, and KL analyzed the data of WES and Sanger sequencing. DT, HC, GL and QS performed the experiments of qPCR and immunofluorescence staining. DT, ML and YG wrote the paper. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved and consented by the Ethics Committee of the First Affiliated Hospital of Anhui Medical University.
Consent for publication
All the individuals and their family members, as well as two controls signed written informed consents after having received complete information about the research.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Predicted as pathogenic variants indetified in two patients carrying HFM1 variants
Additional file 2: Supplementary Table 2.
Primers used for verification of HFM1 variants.
Additional file 3: Supplementary Table 3.
Primers used for QRT-PCR assay of HFM1 and β-actin.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tang, D., Lv, M., Gao, Y. et al. Novel variants in helicase for meiosis 1 lead to male infertility due to non-obstructive azoospermia. Reprod Biol Endocrinol 19, 129 (2021). https://doi.org/10.1186/s12958-021-00815-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-021-00815-z